
INTRODUCTION
As a leading cause of cancer deaths worldwide, lung cancer remains refractory among the major cancers. The five-year survival rate of lung cancer continues to be the lowest compared with other major cancers, such as colon, breast and prostate cancers [1]. Despite many therapeutic endeavors, the survival rate remains bleak, and staggers at about 15% five years after treatment [2]. The most frequent genetic alterations in lung cancer include mutations in TP53, EGFR (Epidermal growth factor receptor), KRAS, EML4-ALK (echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase) rearrangements, and altered MET (tyrosine-protein kinase Met) signaling, which play critical roles in tumorigenesis of lung cancer, and therefore affecting the clinical sensitivity to targeted therapy [3, 4]. The fact that targeted therapy has been successful in a subset of tumors pleas for a better understanding of the pathological mechanisms by which these oncogenic alterations drive tumorigenesis of lung cancer [5]. Searching for novel molecular targets has thus become imperative for advancing targeted therapy of lung cancer.
SPC24 is a subunit of nuclear division cycle 80 (Ndc80) complex, a dumbbell-like, heterotetrameric structure in the outer kinetochore. Ndc80 is comprised of two heterodimers, CDCA1-KNTC2 and SPC24-SPC25. SPC24/SPC25 anchors the Ndc80 complex to the inner kinetochore, and mediates dynamic interactions between the nuclear spindle microtubules and kinetochores, ensuring faithful and accurate chromosomal segregation during mitosis [6, 7]. Therefore, dysregulation of any of these components in the kinetochore-microtubule interface can lead to genomic instability and disrupted control of cell cycle, which ultimately contribute to tumorigenic transformation.
The role of SPC24 in tumorigenesis awaits clarification. It has been reported that simultaneous disruption of both SPC24 and SPC25 genes renders the cell to be spindle checkpoint defective, which allows the cell to bypass mitosis in the absence of correct chromosomal segregation [8, 9], a process that resembles the features of the onset of tumorigenesis. High levels of CDCA1 (also known as NUF2, NDC80 kinetochore complex component), KNTC2 (kinetochore associated 2), SPC24, and SPC25 have been found to be correlated with colorectal and hepatocellular carcinoma tumors [10, 11]. Intriguingly, our initial investigation in Oncomine datasets indicated that SPC24 is up-regulated in lung adenocarcinoma tumors as well. Therefore, in the current study, we aimed at testing the hypothesis that SPC24 may promote tumorigenesis and progression of human lung cancer.
In this study, Oncomine analysis on various datasets and IHC staining in the clinical samples revealed up-regulation of SPC24 in human lung adenocarcinoma. We knocked down SPC24 in multiple non-small cell lung cancer (NSCLC) cell lines by siRNA and evaluated cell proliferation, apoptosis, and migration of the SPC24-defective cells. We compared in vivo tumor growth of the knockdown and control cells in a mouse xenograft model. We also performed multiple Oncomine expression analyses for SPC24 in various lung cancer datasets with important clinical characteristics and risk factors, including staging, survival, recurrence, and smoking. Our results confirm the oncogenic role of SPC24 in the development and progression of lung cancer. Most importantly, SPC24 may serve as a promising prognostic biomarker for stratifying the lung cancer patients into clinically distinct groups that differ in staging, risk of smoking, recurrence, and survival.
RESULTS
SPC24 is over-expressed in human lung adenocarcinoma tumors
Previous work has suggested that disruption of SPC24 renders the cell to be spindle checkpoint defective and confers uncontrolled mitosis [8]. High SPC24 level has been reported for colorectal and hepatocellular carcinoma tumors [10, 11]. Therefore, we first performed Oncomine analysis in published datasets to examine the SPC24 levels in human lung adenocarcinomas (Figure 1) [12, 13]. Interestingly, Oncomine boxed plot of SPC24 expression levels between lung adenocarcinoma and normal samples in multiple datasets showed that SPC24 is consistently over-expressed in lung adenocarcinomas (i.e. 1.5-fold or higher) compared with normal tissues (p < 10–6, Figure 1A) [14–17], indicating a positive role of SPC24 in tumorigenesis of human lung cancer. To further confirm the Oncomine results, we performed IHC analysis in clinical lung tumor samples. Consistent with the Oncomine analysis, we found positive staining of SPC24 is strongly correlated with tumors but not normal tissues (χ2 test, p value < 0.00001) (Figure 1B).
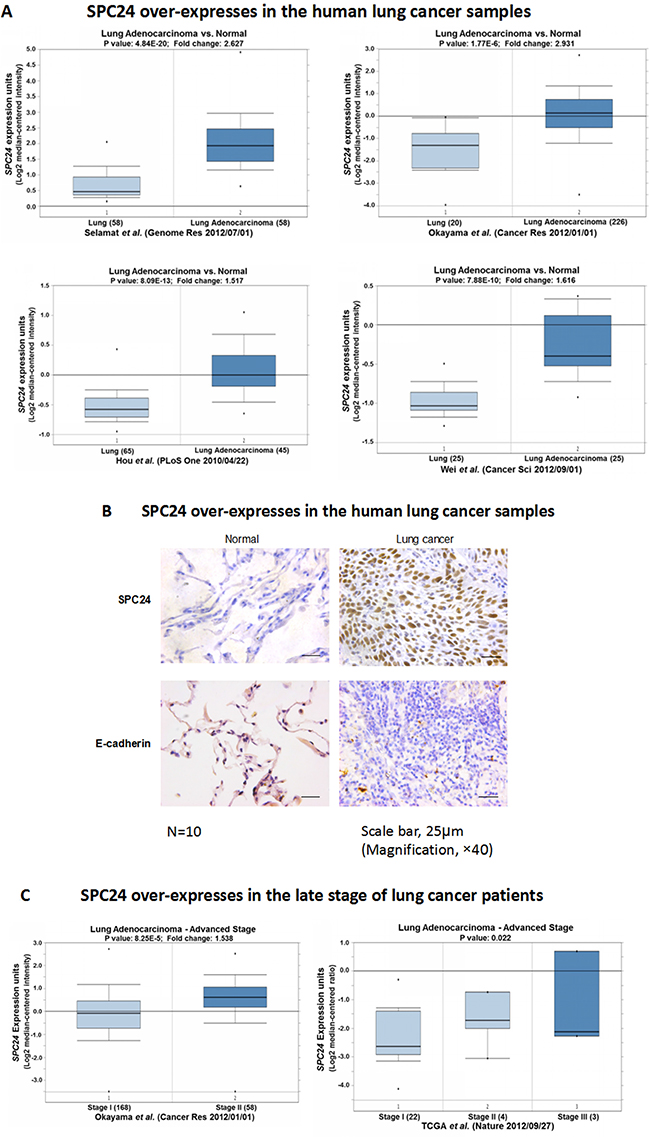
Figure 1: SPC24 is over-expressed in human lung adenocarcinoma tumors. (A) Oncomine boxed plots of SPC24 in human adenocarcinoma and normal lungs. Oncomine data resources are listed under each plot. (B) IHC analysis of SPC24 in clinical normal and lung cancer samples, representing weak staining in the normal tissues, and strong one in the tumors. Ten (N = 10) tumors and 8 normal samples were examined. The staining signals are classified as weak, medium, and strong, and tabulated as the statistics for normal and tumor tissues. The χ2 statistic is 128.808, with p-value < 0.00001. Scale bar, 25 μm. (C) Oncomine boxed plots of SPC24 in lung adenocarcinomas of different stages (stage I, II, and III).
Based on these findings, we wondered if SPC24 levels could further stratify the lung adenocarcinoma tumors according to the staging. Interestingly, in one dataset, SPC24 levels are significantly higher in stage II adenocarcinoma tumors than stage I ones (p < 10-4, Figure 1C) [15]. Consistently, in the TCGA dataset, SPC24 levels are also significantly higher in tumors of advanced stages (stage II&III) compared with the stage I tumors (p < 0.05) [18]. The fact that SPC24 is over-expressed in lung adenocarcinoma, especially in those of the advanced stages, is suggestive of a promoting role of SPC24 in tumorigenesis of human lung cancer. Most importantly, it may also serve as a potential prognostic biomarker for stratifying patients into early and late stages of lung adenocarcinoma.
Knocking down SPC24 represses cell growth and promotes apoptosis in lung cancer cell lines
Dysregulated proliferation and apoptosis are the hallmarks of tumorigenicity of cancer cells. Since SPC24 is up-regulated in lung adenocarcinoma, we wondered if SPC24 regulates cell growth and apoptosis in lung cancer. We knocked down SPC24 in two non-small cell lung cancer (NSCLC) cell lines, PC9 and A549 (Figure 2), and measured proliferation and apoptosis in these cells. It is evident that the SPC24-knockdown (siSPC24) cells grew significantly slower than the control knockdown (siN) ones at all the time points tested (i.e. 24, 72, and 96 h). We also knocked down SPC24 in an immortalized human bronchial epithelial cell line (BEAS 2B), and found that si-SPC24 reduced cell growth with only marginal significance (p < 0.045) (Supplementary Figure 1). On the contrary, over-expression of SPC24 enhanced cell proliferation in both cancer cell lines (Supplementary Figures 2 and 3). Furthermore, flow cytometry analysis of apoptosis using Annexin V-FITC-PI staining showed that siSPC24 cells had more apoptotic cells than siN ones. Notably, for PC9, the populations of both early stage (Annexin V +/PI−) and late stage apoptotic (Annexin V +/PI+) cells in siSPC24 cells were higher than the siN control (Figure 2). Similarly, for A549, siSPC24 had more apoptotic cells (Annexin V +/PI− and Annexin V +/PI+ combined) than siN cells. Taken together, knocking down SPC24 in NSCLC cells suppresses cell growth and promotes apoptosis, suggesting SPC24 positively regulates cellular proliferation and viability in lung cancer.
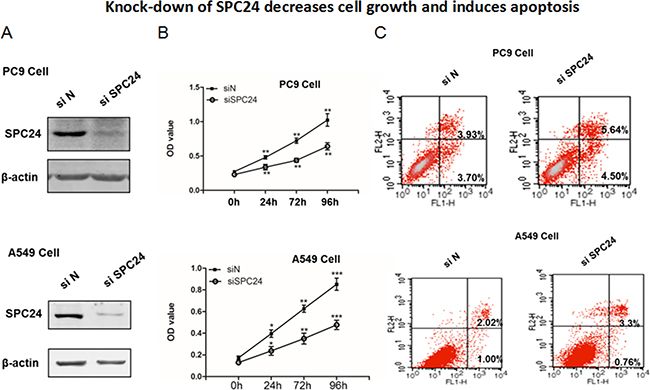
Figure 2: Knocking down SPC24 represses cell growth and promotes apoptosis in lung cancer cell lines. (A) SPC24 was knocked down by siRNA in PC9 and A549 cells. Western blot of SPC24 was analyzed in the knockdown (siSPC24) and control (siN) cells. (B) Cell growth for SPC24-knockdown and control cells was measured as viable cell numbers that was recorded as OD450 at 0, 24, 72, and 96 h. (C) Apoptosis was recorded for knockdown and control cells by flow cytometry analysis of Annexin V-FITC-Propidium Iodide (PI) stained populations.
Knocking down SPC24 suppresses cellular migration in lung cancer cell lines
Given the result that SPC24 positively regulates NSCLC cell proliferation, we further evaluated and compared the invasiveness of siSPC24 and siN cells in vitro. Transwell migration assay showed that knockdown of SPC24 significantly reduced migration of cells by 60% (A549, p < 0.05) to 80% (PC9, p < 0.01) (Figure 3), suggesting a promoting role of SPC24 in lung cancer invasion. Most interestingly, we found E-cadherin level was up-regulated in siSPC24 cells (Figure 3). Concomitantly, the levels of Vimentin and N-cadherin were considerably down-regulated in siSPC24 cells, suggesting a promoting role of SPC24 in the regulation of epithelial-mesenchymal transition (EMT) for lung cancer. Consistently, over-expression of SPC24 could enhance cell migration in both cancer cell lines (Supplementary Figures 2 and 3). This finding and the following IHC staining result of E-cadherin in mouse xenograft model (Figure 4) together support the notion that decreased SPC24 leads to up-regulation of E-cadherin, marking a declined EMT process that is essential for metastasis. Furthermore, disruption of SPC24 in the normal human bronchial epithelial cell line (BEAS 2B) did not affect cell migration, suggesting SPC24 may play a more essential role in cancer progression than in the normal cell (Supplementary Figure 1). Therefore, our current data suggest that SPC24 may positively regulate tumorigenic progression and invasion in NSCLC (see Discussion).
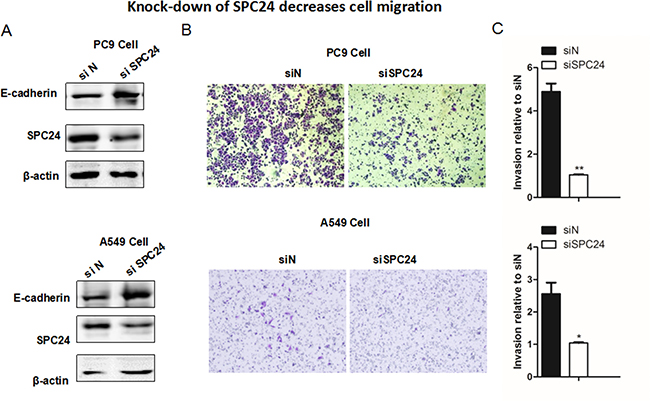
Figure 3: Knocking down SPC24 suppresses cellular migration in lung cancer cell lines. (A) Western blots of the EMT markers, including E-cadherin, Vimentin, N-cadherin,along with SPC24, are shown in SPC24-knockdown and control cells. (B) Migration of the knockdown and control cells was evaluated by Transwell migration assay. (C) Quantification of the cellular migration shown in (B). (Mean ± S.D., n = 3).
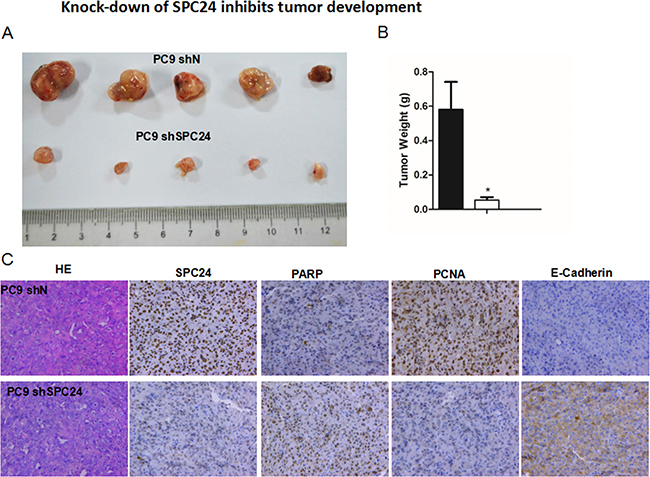
Figure 4: Knocking down SPC24 inhibits tumor growth in vivo. (A) Stable SPC24- knockdown (shSPC24) cell line of PC9 was generated by shRNA, and injected subcutaneously into 6-week old immunocompromised nude mice. At time of sacrifice, tumors were collected and weighed. (B) The weights of tumors are recorded as Mean ± S.D. (n = 3) *p < 0.05 (C) IHC staining of SPC24, PARP, PCNA, and E-cadherin was performed for SPC24-knockdown (shSPC24) or control (shN) tumors.Hematoxylin and eosin (HE) staining of the tumor tissues is also shown.
Knocking down SPC24 inhibits tumor growth in vivo
The marked in vitro phenotypes observed for SPC24 knockdown in the NSCLC cell lines prompted us to further evaluate its role in tumorigenic progression in vivo. We generated stable SPC24-knockdown cell line of PC9 (PC9 shSPC24) and injected the shSPC24 or control (shN) cells into 6-week old immunocompromised nude mice, and recorded in vivo tumor growth for either shSPC24 or shN cells. At time of sacrifice, the average weight of shSPC24 tumors were significantly smaller than the shN tumors (p < 0.05), suggesting SPC24 promotes tumorigenicity of lung cancer cells in vivo (Figure 4). IHC staining further showed that PARP (poly-ADP-ribose polymerase) and E-cadherin had been upregulated, yet PCNA (proliferating cell nuclear antigen) level downregulated, in the shSPC24 tumors, suggesting decreased SPC24 may have resulted in DNA damage-induced hyperactivation of PARP [19], compromised cell proliferation [20], and decreased metastasis in vivo. Taken together, our data indicate that SPC24 promotes NSCLC tumorigenicity both in vitro and in vivo.
SPC24 over-expression is observed in lung cancer patients who are smokers
Since smoking is the leading cause of small cell and non-small cell lung cancer, and contributes to more than 80 percent or more of lung cancer deaths, we further investigated the expression patterns of SPC24 among the lung cancer patients who were smokers. Noticeably, we observed SPC24 was significantly over-expressed in patients who were smokers in all four Oncomine expression datasets (p < 0.05, Figure 5) [15, 18, 21, 22]. Considering SPC24 may promotes tumorigenesis of lung adenocarcinoma, this result suggests a possibility that SPC24 may mediate smoking-induced tumorigenesis in lung cancer (see Discussion).
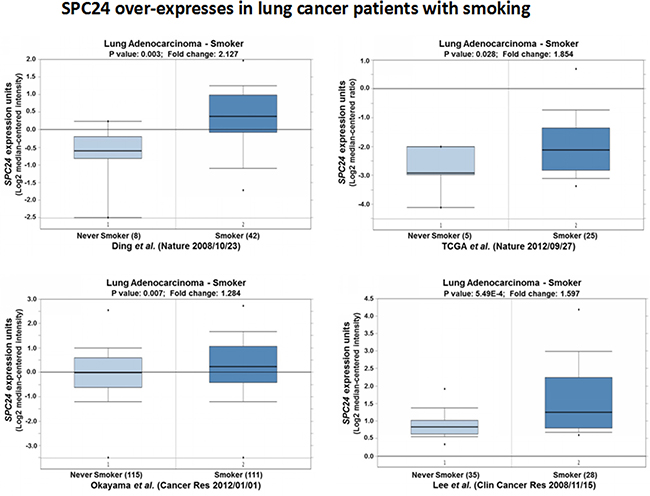
Figure 5: SPC24 over-expression is observed in lung tumors of whom the patients are smokers. Oncomine boxed plots of SPC24 levels in lung adenocarcinomas from smokers vs. non-smokers. Four Oncomine expression analyses are shown.
SPC24 over-expression is associated with lung cancer patients with recurrence or short survivals
So far, both the in vitro and in vivo data suggest SPC24 promotes tumorigenesis of lung cancer. Furthermore, we also observed significantly high levels of SPC24 in lung adenocarcinoma, especially the advanced stage, tumors. These results indicate SPC24 is a promising prognostic biomarker for lung cancer. We went forward to decide if SPC24 could serve as a potential biomarker for predicting the prognosis of patients who had either recurrent lung cancer or short survivals. The Oncomine expression analysis revealed that SPC24 was over-expressed in tumors from patients who had recurrent cancer after one year (p < 0.05, Figure 6) [22]. On the other hand, another Oncomine expression analysis performed on a more complete dataset showed that SPC24 was consistently over-expressed in tumors from patients who regained the cancer after one, three, or five years, compared with those who had not suffered recurrence. Most importantly, the later the patients had the recurrent cancer, the higher SPC24 levels were observed, and the trend became statistically significant along the timeline (Figure 6) [15].
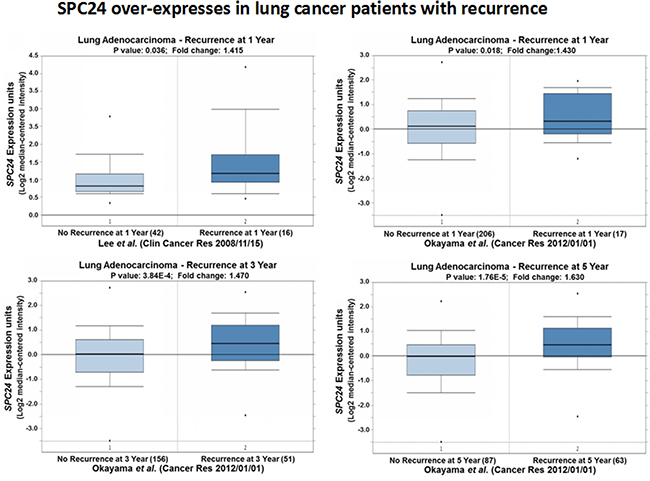
Figure 6: SPC24 over-expression is associated with lung tumors from patients with recurrence. Oncomine boxed plots of SPC24 levels in lung adenocarcinomas from patients with or without recurrent cancer. Four Oncomine expression analyses are shown.
Further Oncomine expression analyses on the lung cancer patients with short survivals recapitulated the over-expression pattern of SPC24 in the lung tumors: among the patients who died after one year, SPC24 levels were the highest compared with those who died three or five years later (Figure 7) [14, 15]. This result, along with the one obtained for recurrent tumors, suggests that SPC24 can serve as an efficient prognostic marker for lung cancer in the clinic.
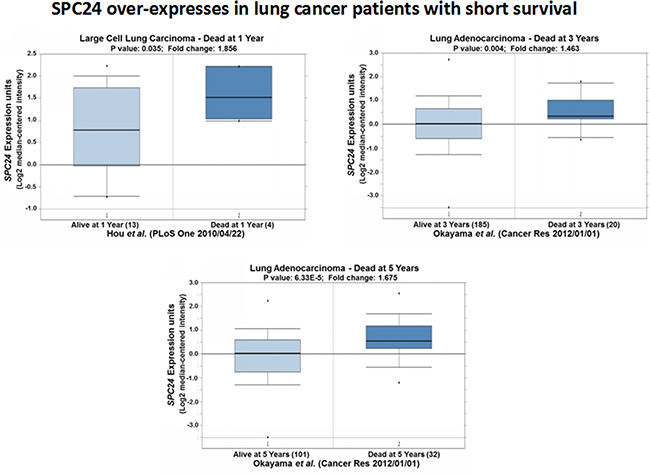
Figure 7: SPC24 over-expression is associated with lung tumors from patients with short survival. Oncomine boxed plots of SPC24 levels in lung adenocarcinomas from patients who died at 1, 3, or 5 years after diagnosis, compared with those who were still alive at the same time point.
DISCUSSION
In the current study, to the best of our knowledge, we for the first time provide evidence to support the oncogenic role of SPC24 in lung cancer development and progression. SPC24 is over-expressed in human lung adenocarcinoma tumors. Knocking down SPC24 in NSCLC cells significantly suppressed cellular proliferation and promoted apoptosis. SPC24-knockdown cells also displayed significantly compromised invasion in Transwell migration assay. In accordance with this phenotype, up-regulation of E-cadherin and down-regulation of N-cadherin and vimentin were observed in siSPC24 cells, suggesting that SPC24 may positively regulate EMT and metastasis in NSCLC. In vivo, SPC24-knockdown cells formed much smaller tumors compared to the unmodified control cells in the mouse xenograft model of NSCLC. Most importantly, multiple Oncomine expression analyses of SPC24 confirmed that high level of SPC24 is associated with lung tumors from smokers, recurrent patients, and patients with shorter survivals.
At the interface of the nuclear spindle microtubules and kinetochores, spindle machineries essentially control and ensure faithful chromosomal segregation. Disruption of spindle checkpoint proteins leads to chromosomal missegregation and alteration in chromosome numbers in the daughter cells, eventually causing massive genetic instability and aneuploidy, a hallmark of cancer [23]. In this study, we show direct evidence that down-regulation of SPC24, a key component linking inner and outer kinetochore, can reduce proliferation of lung cancer cells. The decreased viability is concomitant with increased apoptosis in SPC24-knockdown cells, suggesting SPC24 is essential for the survival of the cancer cells (Figure 2). Consistent with the in vitro phenotypes, the ability of the SPC24-knockdown cells to form tumors is significantly reduced, further attesting the importance of maintaining the integrity of the spindle machineries to promote tumorigenicity of lung cancer cells. IHC analysis on the xenograft tumors also revealed a decreased level of PCNA, a marker of cell proliferation, suggesting the viability of the SPC24-knockdown cells had been compromised in vivo. Oncomine expression analysis show that SPC24 is over-expressed in lung adenocarcinoma tumors, providing another layer of evidence that SPC24 positively regulates lung cancer development.
Supporting the notion that SPC24 may positively regulate metastasis of lung cancer, SPC24-knockdown cells display markedly decreased migration and invasion.EMT markers, such as E-cadherin, is up-regulated, along with down-regulation of N-cadherin and vimentin (Figure 3), suggesting that SPC24 may promote EMT, a hallmark of metastasis [24]. As a side note, down-regulation of E-cadherin has been implicated in invasive NSCLC in the clinic [25]. Thus, based on the results from the in vitro proliferation and invasion assays and the in vivo tumorigenicity assay, we conclude that SPC24 is a novel oncogene of lung cancer.
Consistent with this conclusion, we show that up-regulation of SPC24 is generally associated with poor prognosis of lung cancer patients. Up-regulation of SPC24 is associated with advanced lung adenocarcinoma tumors, and tumors from smokers. More importantly, patients with recurrent cancer or short survivals have higher levels of SPC24. Therefore, SPC24 is a potential prognostic marker of NSCLC. Previous work has suggested that standalone markers, such as p53, p21, Ki-67, PCNA, KRAS, and cyclin D1, cannot serve as efficient prognostic markers for NSCLC due to NSCLC heterogeneity [26–28]. Instead, combinatorial use of these markers will perform better for prognostic prediction, and thus is more clinically meaningful [26, 27, 29].Our current work suggests that SPC24 can serve as a potentially efficient marker for staging the lung tumors and predicting the clinical prognosis of the patients. As a result, it is anticipated that SPC24 would complement the currently available markers for prognostic prediction of NSCLC in the clinic.
In summary, our study reveals a new link between chromosomal missegregation-induced genetic instability and tumorigenesis of lung cancer. In light of the discovery, future studies should be directed to (1) interrogate the mechanism by which SPC24 stimulates development of lung cancer; (2) reveal how SPC24 promotes EMT and metastasis; (3) validate SPC24 as a prognostic marker of lung cancer in clinical cohorts; and (4) test if other components of the spindle checkpoint machinery will confer similar functions as what has been observed for SPC24. From the clinical perspective, confirmation of SPC24 as a prognostic biomarker can benefit the diagnosis, management, and targeted therapy of lung cancer.
MATERIALS AND METHODS
Animals
Female mice were purchased from Shanghai Laboratory Animal Co. Ltd. (SLAC, Shanghai, China), and maintained regularly with readily accessible food and water. All animal procedures were approved by the Institutional Bioethics Committee and complied with the regulations on animal welfare.
Cell culture
Human non-small cell lung adenocarcinoma cell lines PC9 and A549 were obtained from ATCC. Both cell lines were cultured in RPMI 1640 medium supplemented with 2 mM L-glutamine, 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 mg/ml streptomycin. The cell cultures were maintained at 37°C under a humidified atmosphere consisting of 95% air and 5% CO2.
RNA interference of SPC24
SPC24 small interference RNAs (siRNAs) were selected based on the program on http://jura.wi.mit.edu/bioc/siRNAext/ and synthesized at Shanghai GenePharma Co. The siRNA sequences are 5′-GAGCCU UCUCAAUGCGAAGTT-3′ and 5′-CUUCGCAUUGA GAAGGCUCTT-3′, and the following scrambled siRNA was used as the control: 5′-GAGUUAAAGUCAAAGUG ACTT-3′ and 5′-GUCACUUUGACUUUAACUCTT-3′. To exclude the off-target sequences, BLAST search was performed against the human genome database and the above sequence was confirmed to be SPC24-specific.
Proliferation assay
Cell proliferation assay was performed according to the manufacturer’s manual (Cell Counting Kit-8, Dojindo Molecular Technologies, Inc., USA), based on reduction of a tetrazolium salt into a water-soluble formazan dye in a living cell. Both targeted-knockdown (siSPC24) or knockdown control (siN) cells (1 × 105 cells/well) were cultured for 0, 24, 72, 96 h, the absorbance at 450 nm at each time point was assayed.
Analysis of apoptosis
Analysis of apoptosis is based on Annexin V-FITC-Propidium iodide (PI) staining method. Briefly, cells were harvested and washed by cold PBS at a density of 1 × 106 cells/mL. A mixture of PI (final concentration 100 μg/mL) and Annexin V-Alexa Fluor488 conjugate was then incubated with the cells at room temperature for 15 min. Flow cytometry was used to analyze apoptosis with the parameters of 494/518 nm set for Annexin V dectection and 535/617 nm for PI.
Cellular migration assay
A transwell insert (Falcon 354480; BD Biosciences) was used for cellular migration assay. Briefly, SPC24-knockdown and scramble iRNA controls cells were harvested and re-suspended with serum-free DMEM medium at a concentration of 1 × 105 cells/mL. Cell suspension (100 μL) was added to the upper chamber. The lower chamber was loaded with 600 μL of DMEM medium supplemented with 20% FBS. In control experiments, medium containing 1% FBS was added to the lower chamber. The unit was incubated at 37°C for 20 hr. Cells migrating through the membrane to the bottom chamber were then fixed by 4% paraformaldehyde and stained with 0.5% crystal violet for 20 min. Six microscopic fields (magnification, × 100) were randomly photographed for cell counting. Each experiment was repeated three times for the calculation of the means and standard deviations.
Mouse xenograft tumor model
Briefly, SPC24-knockdown and control cells were harvested and suspended at a concentration of 5 × 105 cells/100 μL in cold PBS. Freshly prepared cells were injected into the nude mice subcutaneously. Mice were examined twice a week for 10 weeks where tumor size and weight loss were assessed and recorded. Mice were euthanized by CO2 asphyxiation when they lost more than 20% of their body weight before injection. Tumors were then removed at the time of sacrifice, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) or immunohistochemistry (IHC).
Western blot
For Western blot, nuclear extracts were separated on 12% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes. Membranes were blocked in 5% non-fat milk in Tris-buffered saline (plus 0.1% Tween-20). SPC24 (Cat. ab169786), E-cadherin (Cat. ab1416), and β-actin (Cat. ab8226) antibodies were purchased from Abcam. Blots were detected by enhanced chemiluminescence and exposed on X-ray films.
Immunohistochemistry (IHC)
Continuous sections of tumor or normal tissues were prepared from paraffin-embedded blocks. After deparaffinization in xylene, the sections were rehydrated in a graded ethanol series. Antigen retrieval was performed by microwaving for 3 min in citrate-buffered solution (pH6.0). Endogenous peroxidase activity was quenched by incubation in 3% hydrogen peroxide for 20 min. Blocking was done by pre-incubation with 10% goat serum at room temperature for 30 min. Sections were incubated with antibodies indicated in this study (i.e. SPC24 [rabbit Ab 1:200 dilution Abcam]), PARP, PCNA, E-Cadherin) for overnight in a humidified container at 4°C. The following day, after PBS washes, incubation with the secondary antibody conjugated with horseradish peroxidase was performed for 1 hr at room temperature. Sections were finally stained with 3, 3- diaminobenzidine tetrahydrochloride (DAB) and counterstained with hematoxylin. A negative control was obtained by replacing the primary antibody with normal rabbit serum.
Statistical analysis
Student’s t tests were performed to analyze in vitro data. For mouse studies, two-tailed t test was used to compare tumor weights between control and SPC24-knockdown groups. P values < 0.05 were considered significant.
Abbreviations
immunohistochemistry: IHC; non-small cell lung cancer: NSCLC ; Epidermal growth factor receptor: EGFR; echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase: EML4-ALK; tyrosine-protein kinase Met: MET; nuclear division cycle 80: Ndc80; kinetochore associated 2: KNTC2; fetal bovine serum: FBS; small interference RNAs: siRNAs; hematoxylin and eosin: H&E; polyvinylidene difluoride; PVDF; 3, 3- diaminobenzidine tetrahydrochloride: DAB.
Authors’ contributions
Juan Zhou, Yang Yu and Yunfeng Pei designed the research; Yang Yu, Chunping Cao and Chen Ding performed the experiments; Duping Wang, Li Sun and Guoping Niu contributed new reagents and analytic tools; Yang Yu and Yunfeng Pei analyzed the data; Juan Zhou and Yang Yu wrote the manuscript.
ACKNOWLEDGMENTS
Not available.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This study was not supported by grant funding source.
REFERENCES
1. Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006; 56:106–30.
2. Spira A, Ettinger DS. Multidisciplinary management of lung cancer. N Engl J Med. 2004; 350:379–92.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129–39.
4. Liu J, Yu G, Zhao Y, Zhao D, Wang Y, Wang L, Liu J, Li L, Zeng Y, Dang Y, Wang C, Gao G, Long W, et al. REGgamma modulates p53 activity by regulating its cellular localization. J Cell Sci. 2010; 123:4076–84.
5. Bronte G, Rizzo S, La Paglia L, Adamo V, Siragusa S, Ficorella C, Santini D, Bazan V, Colucci G, Gebbia N, Russo A. Driver mutations and differential sensitivity to targeted therapies: a new approach to the treatment of lung adenocarcinoma. Cancer Treat Rev. 2010; 36:S21–9.
6. DeLuca JG, Musacchio A. Structural organization of the kinetochore-microtubule interface. Curr Opin Cell Biol. 2012; 24:48–56.
7. Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008; 9:33–46.
8. Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004; 23:2016–27.
9. Wigge PA, Kilmartin JV. The Ndc80p complex from Saccharomyces cerevisiae contains conserved centromere components and has a function in chromosome segregation. J Cell Biol. 2001; 152:349–60.
10. Kaneko N, Miura K, Gu Z, Karasawa H, Ohnuma S, Sasaki H, Tsukamoto N, Yokoyama S, Yamamura A, Nagase H, Shibata C, Sasaki I, Horii A. siRNA-mediated knockdown against CDCA1 and KNTC2, both frequently overexpressed in colorectal and gastric cancers, suppresses cell proliferation and induces apoptosis. Biochem Biophys Res Commun. 2009; 390:1235–40.
11. Zhu P, Jin J, Liao Y, Li J, Yu XZ, Liao W, He S. A novel prognostic biomarker SPC24 up-regulated in hepatocellular carcinoma. Oncotarget. 2015; 6:41383–97. doi: 10.18632/oncotarget.5510.
12. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004; 6:1–6.
13. Liu J, Cho SN, Akkanti B, Jin N, Mao J, Long W, Chen T, Zhang Y, Tang X, Wistub II, Creighton CJ, Kheradmand F, DeMayo FJ. ErbB2 Pathway Activation upon Smad4 Loss Promotes Lung Tumor Growth and Metastasis. Cell Rep. 2015 Mar 3. doi: 10.1016/j.celrep.2015.02.014. [Epub ahead of print].
14. Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, Grosveld F, Philipsen S. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS One. 2010; 5:e10312.
15. Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012; 72:100–11.
16. Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M, Siegmund KD, Koss MN, Hagen JA, Lam WL, Lam S, Gazdar AF, Laird-Offringa IA. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012; 22:1197–211.
17. Wei DC, Yeh YC, Hung JJ, Chou TY, Wu YC, Lu PJ, Cheng HC, Hsu YL, Kuo YL, Chen KY, Lai JM. Overexpression of T-LAK cell-originated protein kinase predicts poor prognosis in patients with stage I lung adenocarcinoma. Cancer Sci. 2012; 103:731–8.
18. Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–25.
19. Bey EA, Bentle MS, Reinicke KE, Dong Y, Yang CR, Girard L, Minna JD, Bornmann WG, Gao J, Boothman DA. An NQO1- and PARP-1-mediated cell death pathway induced in non-small-cell lung cancer cells by beta-lapachone. Proc Natl Acad Sci USA. 2007; 104:11832–7.
20. Wang SC. PCNA: a silent housekeeper or a potential therapeutic target? Trends Pharmacol Sci. 2014; 35:178–86.
21. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455:1069–75.
22. Lee ES, Son DS, Kim SH, Lee J, Jo J, Han J, Kim H, Lee HJ, Choi HY, Jung Y, Park M, Lim YS, Kim K, et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin Cancer Res. 2008; 14:7397–404.
23. Lee H. How chromosome mis-segregation leads to cancer: lessons from BubR1 mouse models. Mol Cells. 2014; 37:713–8.
24. Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. 2000; 36:1607–20.
25. Kase S, Sugio K, Yamazaki K, Okamoto T, Yano T, Sugimachi K. Expression of E-cadherin and beta-catenin in human non-small cell lung cancer and the clinical significance. Clin Cancer Res. 2000; 6:4789–96.
26. Groeger AM, Caputi M, Esposito V, Baldi A, Rossiello R, Santini D, Mancini A, Kaiser HE, Baldi F. Expression of p21 in non small cell lung cancer relationship with PCNA. Anticancer Res. 2000; 20:3301–5.
27. Nguyen VN, Mirejovsky P, Mirejovsky T, Melinova L, Mandys V. Expression of cyclin D1, Ki-67 and PCNA in non-small cell lung cancer: prognostic significance and comparison with p53 and bcl-2. Acta Histochem. 2000; 102:323–38.
28. Zhu CQ, Tsao MS. Prognostic markers in lung cancer: is it ready for prime time? Transl Lung Cancer Res. 2014; 3:149–58.
29. Coate LE, John T, Tsao MS, Shepherd FA. Molecular predictive and prognostic markers in non-small-cell lung cancer. Lancet Oncol. 2009; 10:1001–10.