
INTRODUCTION
Ovarian cancer (OC) is the most lethal gynecologic malignancy in the Western world, with an estimated 22,280 new cases and 14,240 deaths in the U.S. for 2016 [1]. Although most patients initially respond to surgical debulking and chemotherapy, the majority will experience recurrent disease that has become drug resistant [1, 2]. Oncology drugs lead other therapeutic areas in clinical development [3], but only 7% of drugs that enter Phase I clinical trials successfully attain marketing approval [4]. These statistics suggest that there is room for improvement in our approach to steering oncology drugs from the bench to the bedside.
AURKA was first discovered in Drosophila melanogaster as a gene that regulates mitotic spindle function [5] and has since been the focus of numerous studies documenting its essential roles in mitotic entry, centrosome function and bipolar spindle assembly [6]. AURKA is thought to play an important role in ovarian tumor biology, because activation of this kinase either by genomic amplification or increased expression is a common feature of ovarian carcinoma cell lines and primary tumors [7-13]. Further, high levels of active AURKA in ovarian tumors are also associated with supernumerary centrosomes and overall decreased survival [14].
Two small molecule inhibitors specific for AURKA, alisertib (Takeda Pharmaceuticals) and MK-5108 (Merck Research Laboratories), have been evaluated in clinical trials for the treatment of OC. Preliminary Phase I results for MK-5108 revealed activity as a single agent, as well as, in combination with docetaxel for the treatment of advanced solid tumors [15]. Alisertib (MLN8237) exhibits exquisite specificity for AURKA with >200-fold greater selectivity for AURKA than AURKB [16]. Using an orthotopic mouse model, we reported that alisertib specifically inhibited AURKA activity in vivo and exerts ovarian tumor growth inhibition (TGI) as a single agent [17]. Further, alisertib and paclitaxel combination therapy TGI was even more potent than that observed for monotherapy [17]. Alisertib showed modest effects for platinum-resistant and -refractory OC [18] when used as a single agent, and is currently in Phase II clinical trials in combination with paclitaxel [19, 20]. Early results from a Phase I/II alisertib and paclitaxel trial for ovarian and breast cancer reveal partial response in eight patients and stable disease for three patients [19]. The fact that alisertib, as a single agent or as part of a combination therapy regimen showed clinical activity in a subset of patients, underscores the need to improve our understanding of AURKA-regulated pathways that mediate tumor progression, including novel non-mitotic functions [17, 21-26].
While the role of AURKA in regulating mitosis has been extensively studied, little is known about the function of this kinase in mediating DNA repair and the DNA damage response (DDR). AURKA regulation of genomic instability has been linked to interactions with the caretakers of global chromosomal stability, BRCA1 and BRCA2. In the context of BRCA2, Yang et al [27] reported a functional interaction between AURKA and BRCA2 in sporadic disease and showed that AURKA inhibition of BRCA2 expression in vitro perturbs the DDR promoting cell cycle progression and genomic instability [27]. Analyses of 223 high-grade serous carcinomas uncovered an inverse correlation between AURKA and BRCA2 protein expression, with high AURKA to BRCA2 expression ratios predicting poor survival [27].
An inverse relationship between AURKA/B and BRCA1/2 has also been reported in vitro where silencing of AURKA/B by shRNA resulted in elevated expression of BRCA1/2 [28]. Further, downregulation of AURKA/B inhibited aberrant cytokinesis and diminished cell multinuclearity and chromosome tetraploidy, while a knockdown of BRCA1/2 expression had the opposite effect. Consistent with these observations, shRNA-mediated silencing of AURKA inhibited growth, while silencing of BRCA1/2, increased growth of tumor xenografts in mice [28].
AURKA may mediate chromosomal instability in tumor cells by regulating error-prone NHEJ DNA repair. In vitro studies in breast cancer cells revealed that AURKA overexpression diminished recruitment of RAD51 to sites of DSBs, which disrupted repair of DNA damage through the high-fidelity homologous recombination (HR)-dependent mechanism, thereby favoring the NHEJ pathway [25]. Moreover, loss of RAD51 recruitment to sites of DSBs required PLK1 inhibition of CHK1 activity [25].
Error-prone NHEJ results in chromosomal translocations and rearrangements [29, 30], leading to genomic instability. NHEJ is initiated when Ku80-Ku70 binds to DNA ends and recruits DNA-PKcs. DNA ends are then processed by several proteins, including Artemis, the polynucleotide kinase, and members of the polymerase X family [31-35], before ends are finally joined by ligase IV, which is part of a complex containing XRCC4 and Cernunos/Xlf [36-38].
Poly(ADP-ribose) polymerase 1 (PARP1) is a nuclear enzyme, which plays a critical role in DNA repair, including NHEJ. PARP1 (hereafter referred to as PARP) binds to damaged DNA and, when activated, produces poly(ADP-ribose) [pADPr] chains that binds covalently to chromatin proteins and to PARP itself, altering protein function [39-43]. A number of PARP inhibitors (PARPis) [e.g., rucaparib, niraparib, veliparib and talozaparib] are currently in clinical trials for the treatment of OC, and promising results led the Food and Drug Administration to approve olaparib (Lynparza) and to designate rucaparib as a Breakthrough Therapy [44]. PARPis were designed to target BRCA-mutated tumors since HR-based DNA repair is disrupted in these tumors [44]. Approximately 50% of high-grade serous ovarian carcinomas exhibit changes in genes (not limited to BRCA1/2) predicted to result in HR deficiency and these tumors are described as possessing ‘BRCAness’ [13, 45-48]. Patel et al [49] proposed a model in which PARPi is cytotoxic to ovarian carcinoma cells, because PARP inhibition stimulates NHEJ, thereby resulting in lethal genomic instability. Notably, PARPi stimulated error-prone NHEJ by activating DNA-PKcs only in HR-deficient and not in HR-proficient cells [49].
The current study tests the hypothesis that AURKA regulates the DDR and DNA repair pathways in ovarian carcinoma cells. Inhibition of AURKA activity significantly diminished cell growth, activated DNA-PKcs and decreased PARP expression and activity. Consistent with these observations, alisertib treatment also stimulated error-prone NHEJ DNA repair. Moreover, AURKA inhibition also decreased the expression of BRCA1 or BRCA2 and increased pH2AXS139 levels, suggesting that impaired HR pathway function and induction of DNA double-strand breaks (DSBs), respectively. These studies also promoted evaluation of combining alisertib and rucaparib to potentially extend the number of ovarian cancer patients with HR competent tumors who might benefit from a PARP inhibitor. Collectively, our findings reveal new functions for AURKA in the regulation of DNA repair, which shed new light on the complexity of AURKA-regulated pathways involved in the DDR.
RESULTS
Overexpression of AURKA has been shown to disrupt HR, thereby stimulating NHEJ[25], yet our knowledge of AURKA regulation of error-prone NHEJ repair remains incomplete. Given the success of PARPis for the treatment of OC, particularly HR-deficient cancers, we explored the potential connection of AURKA activation on cell growth, the DDR and DNA repair in PARPi-sensitive (HR-deficient) and PARPi-resistant (HR-proficient) ovarian carcinoma cells.
AURKA inhibits PARPi-sensitive and -resistant ovarian carcinoma cell growth and clonal survival
To understand the effects of AURKA inhibition, in the context of HR-proficiency and HR-deficiency, we employed a large panel of established ovarian carcinoma cell lines, including PEO1, SKOV3ip2, PEO4, OVCA429, MDAH, A2780, OVCAR5 and OVCAR10. We first treated this panel of cell lines with the PARPi, rucaparib, and evaluated cell viability to determine the comparative sensitivity as an indication of HR status for each cell line. Results from this analysis showed a range of sensitivities among the eight ovarian cell lines tested (Supplementary Table 1), with PEO1, a BRCA2-mutated and HR-deficient cell line [50], being the most sensitive (IC50 = 0.324 µmol/L); PEO4 and SKOV3ip2 (which have no deleterious BRCA1/2 mutations) [50, 51] were resistant to rucaparib; OVCA429 (which has no deleterious BRCA1/2 mutations) [51] exhibited modest sensitivity to rucaparib (IC50 = 5.5 µmol/L). Primary normal human ovarian surface epithelial (HOSE) cells were also treated with rucaparib and exhibited resistance to PARPi (Supplementary Table 1).
We next examined the effect of alisertib treatment for seven days on ovarian carcinoma cell growth using PEO1, OVCA429, PEO4 and SKOV3ip2 cells (Figure 1A & Table 1), which exhibit high sensitivity, moderate sensitivity and resistance to rucaparib, respectively. We also treated HOSE cells with alisertib to determine the effects of this AURKA-specific small molecule inhibitor on normal cells. Alisertib potently inhibited the growth of PARPi-sensitive and PARPi-resistant ovarian carcinoma cells but had very little effect on the growth of normal HOSE cells (Figure 1A & Table 1). Similarly, treatment with nanomolar concentrations of alisertib dramatically decreased clonal survival in both PEO1 and SKOV3ip2 (Figure 1B) cells in a dose-dependent manner.
We previously reported alisertib had modest effects on ovarian carcinoma cell growth after a three-day treatment period [17], but our current findings suggest that, similar to PARPi, longer exposure to this small molecule inhibitor is required to see more dramatic effects on cell growth.
Table 1: Ovarian carcinoma cell line IC50 values for alisertib
Cell Line |
Mean IC50 ± SE (nM) |
PEO1 |
32.9 ± 2.69 |
OVCA429 |
17.7 ± 5.18 |
PEO4 |
47.8 ± 11.5 |
SKOV3ip2 |
31.1 ± 3.52 |
HOSE |
NA |
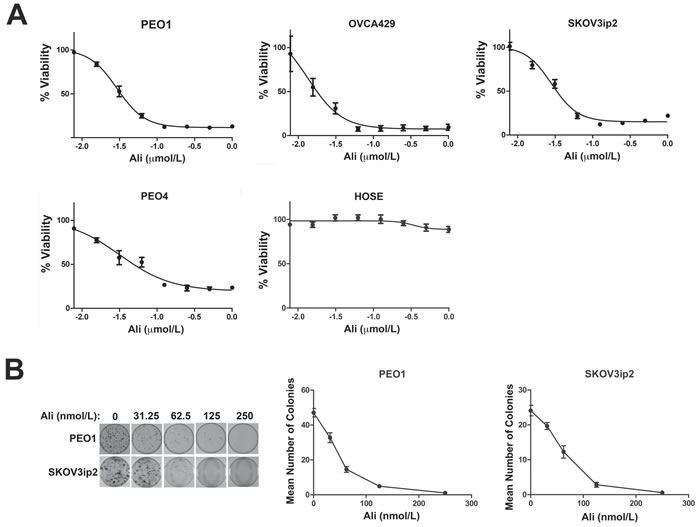
Figure 1: Inhibition of AURKA activity diminishes the growth and clonogenic survival of ovarian carcinoma cells. A. PEO1, PEO4, OVCA429 and SKOV3ip2, in addition to normal HOSE cells, were treated for seven days with increasing concentrations of alisertib (0.00781, 0.0156, 0.0313, 0.0625, 0.125, 0.250, 0.50, and 1.0 µmol/L) and then viability was assayed using Cell Titer Blue. Graphs that depict the mean percentage of viable cells ± SEM for each concentration of alisertib from three independent experiments are shown. The effect of AURKA inhibition on clonogenic survival of ovarian carcinoma cells was also analyzed by treatment with increasing concentrations of alisertib (0.0313, 0.0625, 0.125, and 0.250 µmol/L). B. Cells were stained with crystal violet and graphs depicting the mean number of PEO1 and SKOV3ip2 colonies ± SEM for each alisertib concentration from three independent experiments are shown. Cells were treated with vehicle (DMSO) as a control.
AURKA inhibition alters the expression of DDR proteins
To determine the effects of inhibiting AURKA activity on the DDR in ovarian carcinoma cells, we first analyzed the effect of alisertib treatment on the expression of important HR and NHEJ pathway signaling molecules, in PARPi-sensitive (PEO1) and -resistant (PEO4 and SKOV3ip2) cell lines. Alisertib treatment at nanomolar concentrations that are not expected to yield off-target effects [52], decreased the expression of PARP in a dose-dependent manner in all cell lines assayed, PEO1, PEO4 and SKOV3ip2 (Figure 2A). Alisertib treatment also decreased the expression of the HR pathway proteins, BRCA1 or BRCA2, in PEO1 and PEO4 cells or SKOV3ip2 (Figure 2A) cells, respectively.
To evaluate the potential effects of AURKA inhibition on the NHEJ pathway, we analyzed the phosphorylation of DNA-PKcs at Thr2609, which is required for DSB repair by NHEJ [53]. Alisertib treatment increased pDNA-PKcsT2609 levels in PARPi-sensitive (PEO1) and -resistant (SKOV3ip2 and PEO4) ovarian carcinoma cells (Figure 2A). Pharmacological inhibition of AURKA activity also increased pH2AXS139 levels in all cell lines assayed (Figure 2A). Collectively, these observations suggest that sustained inhibition of AURKA activity induces DNA damage, attenuates the HR pathway, and stimulates the NHEJ pathway.
To further confirm the specificity of the effect of AURKA inhibition on the NHEJ pathway, we transfected PEO1 and SKOV3ip2 cells with two independent siRNAs targeting AURKA and a non-targeted (NT) control. Since AURKA is essential for ovarian carcinoma cell survival, highly efficient AURKA knockdown is lethal to cells. Therefore, conditions resulting in partial knockdown of AURKA expression were employed, showing that depletion of AURKA expression increased phosphorylation of DNA-PKcs and H2AX in PEO1 and SKOV3ip2 cells (Figure 2B).
Collectively, these findings indicate that AURKA inhibition stimulates the NHEJ pathway by increasing DNA-PKcs activity and decreasing the expression of HR proteins, BRCA1/2. AURKA inhibition also elevated pH2AXS139 levels suggesting the induction of DNA DSBs.
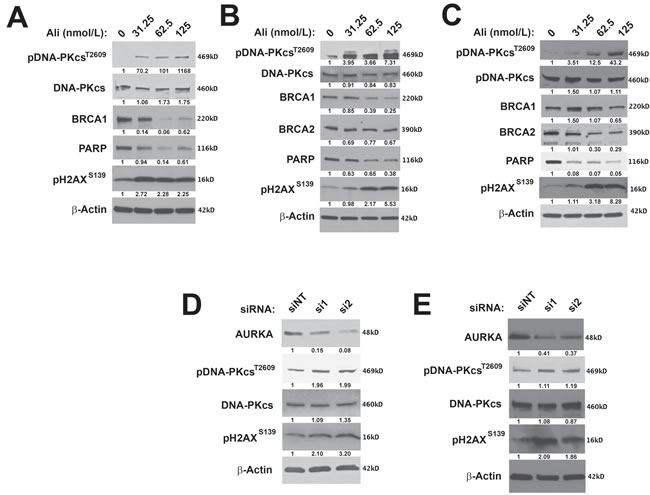
Figure 2: Inhibition of AURKA activity stimulates the NHEJ pathway and decreases the expression of HR proteins. A. PARPi-sensitive PEO1 and B. PARPi-resistant PEO4 and C. SKOV3ip2 cells were treated with vehicle (DMSO) or alisertib (31.25, 62.5, or 125 nmol/L) for 48 h and then total protein lysates were immunoblotted with antibodies against pDNA-PKcsT2609, total DNA-PKcs, BRCA1, BRCA2, PARP1, pH2AXS139, and β-Actin (loading control). Representative immunoblots are shown. To evaluate the effects of RNAi-mediated silencing of AURKA, D. PEO1 and E. SKOV3ip2 cells were transiently transfected with non-targeting (siNT) or AURKA-specific siRNAs (si1 and si2) for 48 h, and then total protein lysates were immunoblotted with antibodies against AURKA, pDNA-PKcsT2609, total DNA-PKcs, pH2AXS139, and β-actin (loading control). Representative immunoblots are shown with densitometry measurements normalized to both β-actin and experimental controls under each blot.
Inhibition of AURKA activity increases pH2AXS139 foci and error-prone NHEJ DNA repair
When DNA is damaged, the histone H2AX, is phosphorylated at S139 and recruits DNA repair proteins to sites of nascent DSBs (foci) within the nucleus [54]. To test whether AURKA inhibition induces DNA DSBs, we evaluated the effects of alisertib treatment on the formation of pH2AXS139 foci. Alisertib treatment induced a 3.7- and 2.5-fold increase in pH2AXS139 foci relative to vehicle treatment in PEO1 and in SKOV3ip2 (Figure 3A, P < 0.05) cells, respectively.
We next asked if inhibition of AURKA activity has a direct functional effect on error-prone NHEJ DNA repair. To answer this question, cells were treated with alisertib and then subjected to a validated functional assay for NHEJ using the Pem1-EGFP-Ad2 plasmid digested with either HindIII to generate DNA DSBs with compatible overhangs or I-SceI to generate DSBs with incompatible ends that require nucleolytic end processing before DNA repair (Figure 3B) [55]. Inhibition of AURKA activity in PARPi-sensitive PEO1 cells with 62.5 nmol/L alisertib stimulated a 1.8- and 2.5-fold increase in end-joining of the HindIII- and I-SceI-linearized plasmid, respectively (Figure 3C, P < 0.05). Although the effects of alisertib were less pronounced in PARPi-resistant SKOV3ip2 cells, there was still a significant increase in end-joining of the HindIII and I-SceI DNA substrates by 1.3- and 1.4-fold, respectively (Figure 3C, P < 0.05).
Taken together, these data suggest that AURKA inhibition induced DNA DSBs and elevated error-prone NHEJ repair of DNA DSBs with incompatible ends in both PARPi-sensitive and -resistant ovarian carcinoma cells.
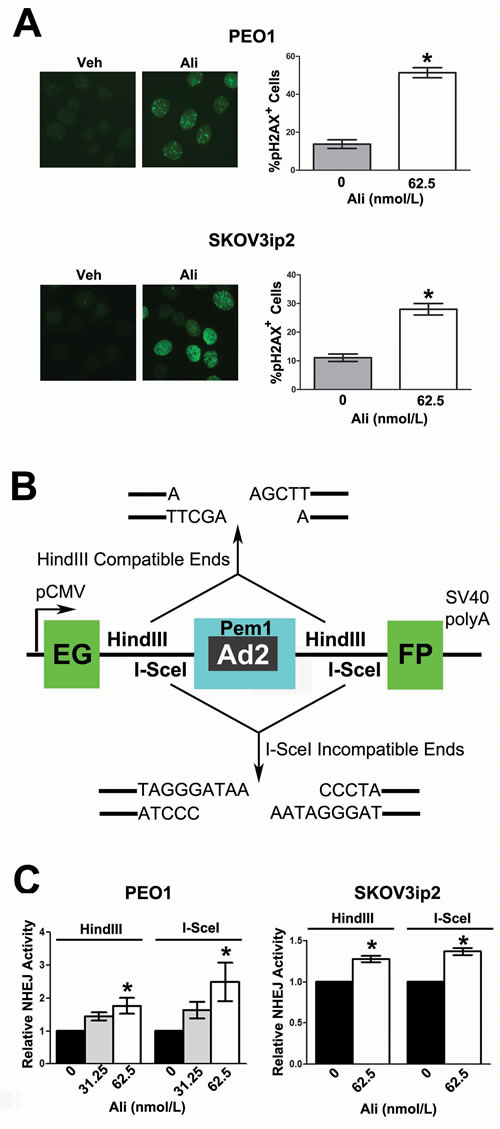
Figure 3: Inhibition of AURKA activity induces the formation of pH2AXS139 foci and stimulates error-prone NHEJ. A. PEO1 and SKOV3ip2 cells were treated with 62.5 nmol/L alisertib for 24 h before pH2AXS139 nuclear foci were enumerated. Graphs that depict the mean percentage of cells positive for ≥ 10 pH2AXS139 foci ± SEM (relative to vehicle-treated control) from three independent experiments are shown. Bars labeled with asterisks are statistically significant as analyzed by Wilcoxon matched-pairs signed rank test (*P < 0.05). To determine the functional effects of AURKA inhibition on NHEJ DNA repair, ovarian carcinoma cells were subjected to a validated NHEJ assay after alisertib treatment. A schematic of the B. pEGFP-Pem1-Ad2-GFP harboring the EGFP open-reading frame, which is interrupted by a Pem1 intron containing an adenoviral exon (Ad2) that is flanked on either side by HindIII and I-SceI restriction sites. Digestion with HindIII generates compatible DNA ends, while I-SceI digestion generates incompatible DNA ends. Cleavage with either HindIII or I-SceI creates a DNA DSB and removes the Ad2 exon; successful DNA repair results in recircularization of the plasmid, generating a functional EGFP. C. PARPi-sensitive PEO1 and PARPi-resistant SKOV3ip2 cells were transiently transfected with peEGFP-Pem1-Ad2 digested with either HindIII or I-SceI, in addition to an RFP plasmid (which served as a control for transfection efficiency). Cells were also treated with vehicle (DMSO) or alisertib (62.5 nmol/L) for 48 h, and then the percentage of EGFP+ and RFP+ cells was determined by flow cytometry. Graphs depicting the mean number of EGFP+ cells normalized to the number of RFP+ cells transfected with the HindIII- or I-SceI-digested DNA substrates ± SEM (relative to vehicle-treated control) from three independent experiments are shown. Bars labeled with asterisks are statistically significant as analyzed by one-way ANOVA followed by Tukey’s multiple comparison or Wilcoxon matched-pairs signed rank test (*P < 0.05).
AURKA regulates PARP activity
To determine if modulation of AURKA activity alters PARP activity, we examined the effects of inhibition of AURKA activity and enforced expression of AURKA on the expression of pADPr polymers. Inhibition of PARP activity with PARPi decreases pADPr levels in ovarian carcinoma cells [49]. Therefore, as a control, we treated PEO1 cells with increasing concentrations of the PARPi, rucaparib, and observed the expected reduction in pADPr levels (Figure 4A). Similarly, AURKA inhibition with low nanomolar concentrations of alisertib also diminished the expression of pADPr in PEO1 and SKOV3ip2 (Figure 4B) cells, indicating diminished PARP activity. Further, enforced expression of an AURKA-RFP construct resulted in elevated pADPr expression relative to RFP-transfected cells, suggesting that PARP activity is increased in PEO1 and SKOV3ip2 (Figure 4C) cells when AURKA is overexpressed. Although pADPr levels increase, PARP levels do not change as a result of enforced expression of AURKA in PEO1 and SKOV3ip2 (Figure 4C) cells. In addition, pDNA-PKcsT2609 and pH2AXS139 levels were not altered as a result of AURKA overexpression (Figure 4C), suggesting that DNA DSBs are not induced and the NHEJ pathway is not stimulated.
Taken together, these observations suggest that AURKA regulates PARP activity in PARPi-sensitive and -resistant ovarian carcinoma cells.
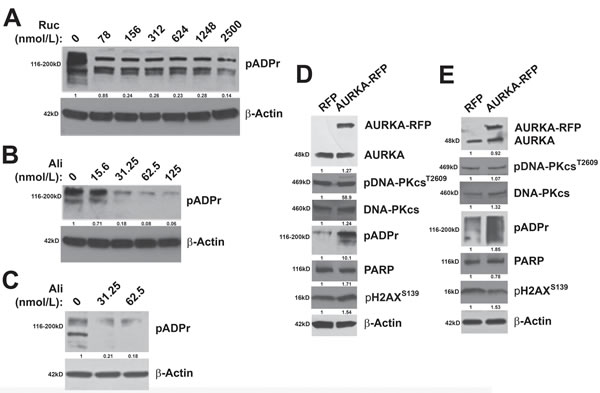
Figure 4: AURKA regulates PARP activity. BRCA2- and HR-deficient A. PEO1 cells were treated with the PARPi, rucaparib, as a control to demonstrate that pADPr levels decrease as a result of PARP activity inhibition. B. PEO1 and PARPi-resistant SKOV3ip2 cells were treated with vehicle (DMSO) or increasing concentrations of alisertib (31.25, and 62.5 nmol/L) for 48 h and total protein lysates were immunoblotted with antibodies against pADPr and β-Actin (loading control). Representative immunoblots are shown. To evaluate the effects of enforced AURKA expression, C. PEO1 and SKOV3ip2 cells were transiently transfected with an RFP or AURKA-RFP expression construct for 48 h and then total protein lysates were immunoblotted with antibodies against AURKA, pDNA-PKcsT2609, total DNA-PKcs, pADPr, PARP, pH2AXS139 and β-Actin (loading control). Representative immunoblots are shown with densitometry measurements normalized to both β-actin and experimental controls under each blot.
Alisertib treatment stimulates the NHEJ pathway and decreases PARP levels in vivo
Next, we sought to determine if AURKA regulation of the DDR and DNA repair pathways observed in vitro also occurs in vivo. To answer this question, we evaluated pAURKAT288, pDNA-PKcsT2609, Ku80, and PARP levels in SKOV3ip2 orthotopic xenografts. Briefly, SKOV3ip2 cells were implanted orthotopically by unilateral intrabursal (i.b.) injection into SCID mice (n = 5/arm) and tumors were allowed to develop for four weeks before treatment with vehicle or a low dose of alisertib, 10 mg/kg (four cycles of 5 days on/2 days off), to ensure that observations are not due to off-target effects. Mice were euthanized six hours after the final administration of alisertib and expression of PARP and signaling components in the NHEJ pathway were evaluated by immunohistochemistry (IHC). Detection of pAURKAT288 (H-score = 300 vs 200) showed that AURKA activity is decreased in alisertib-treated tumors relative to vehicle-treated tumors confirming drug activity (Figure 5). Notably, we observed elevated pDNA-PKcsT2609 levels (H-score = 100 vs 200, strong positive staining nuclei = 17 vs 43) and a modest increase in Ku80 expression (H-score = 200 vs 300) in alisertib-treated tumors compared to controls (Figure 5), suggesting stimulation of the NHEJ pathway. Conversely, we detected a decrease in PARP (H-score = 300 vs 200) levels in alisertib-treated tumors relative to vehicle-treated control tumors (Figure 5). Collectively, these observations suggest that AURKA inhibition decreases PARP expression and stimulates the NHEJ DNA repair pathway in PARPi-resistant ovarian tumor cells in vivo, consistent with our in vitro observations.
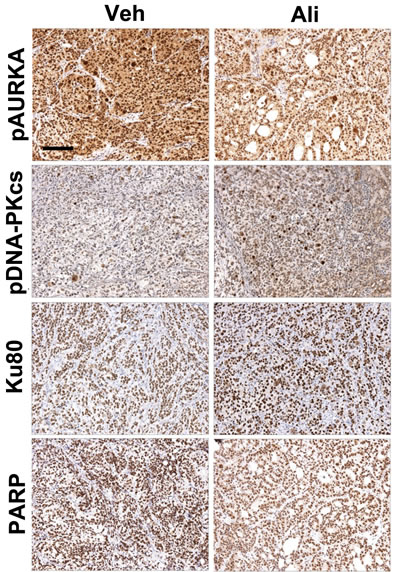
Figure 5: Inhibition of AURKA activity stimulates the NHEJ pathway and decreases PARP levels in vivo. SKOV3ip2 cells were orthotopically implanted into SCID mice and tumor cells were allowed to grow for one month before treatment with vehicle or alisertib (10 mg/kg) to evaluate the effects of AURKA inhibition on pAURKAT288, pDNA-PKcsT2609, Ku80, and PARP levels as assayed by IHC (n = 5 mice/arm). Representative brightfield images captured using the same exposure times are shown.
Alisertib stimulates BRCAness and synergy to PARP inhibition
While PARP inhibitors have been considered a breakthrough treatment in ovarian cancer, they are generally only recommended in BRCA mutant tumors. Finding clinical targets that can enhance “BRCAness”, like we have shown with AURKA inhibition, could open up a whole new cohort of patients that might benefit from PARP inhibitors. In a query of over 400 ovarian cancer tumors in TCGA [56], using the cBioPortal (www.cbioportal.org), we determined that copy number alterations of PARP1 and AURKA were often observed in the same tumors (Figure 6A) and observed a significant tendency towards co-occurrence in a mutual exclusivity analysis (Figure 6B, P= 0.043). In contrast, co-mRNA expression of both genes was not significantly correlated (data not shown). These genomic data suggest exploring a combination of alisertib and rucaprarib might have clinical benefit.
Synthetic lethality is the simultaneous alteration of two genes or proteins which causes cell death, while alteration of either gene/protein alone does not. Since we observed that AURKA inhibition leads to BRCAness by decreasing BRCA1/2 protein levels, we hypothesized that the combination of alisertib and rucaparib would be synergistic. First, using the functional assay for NHEJ shown in Figure 3, PEO1 cells treated with the combination of alisertib (31.25 nM) and rucaparib (313 nM) lead to a 1.3-fold increase in end-joining of the I-SceI-linearized plasmid relative to alisertib or rucaparib alone and a 2.9-fold increase versus vehicle (Figure 6C, P < 0.05). The enhancement in repair was also observed for the drug combination when using the HindIII assay, but the change was not deemed statistically significant. Next we evaluated the in vitro activities of the drug combination in both the BRCA wild-type (SKOV3IP) and BRCA defective (PEO1) cells using a combination matrix of rucaparib (2.5 µM, 1.25 µM, 625 nM, and 312 nM) and eight two-fold serial dilutions of alistertib (1 µM to 7.8 nM), with triplicate sampling. As shown in Figure 6D, the combined treatment of alisertib and rucaparib inhibited cell growth and was synergistic (CI < 1.0, red line) or strongly synergistic (CI < 0.3, green line) across both cell lines.
Taken together, these results suggest that induction of a BRCAness phenotype by alisertib, as a result of decreased BRCA protein expression and induction of NHEJ DNA repair, could help sensitize patient tumors without HR defects (e.g., wild-type for BRCA1/2) to PARPi. Importantly, this clinical approach could improve the efficacy and decrease the adverse side effects of both alisertib and PARPi, since the activity of the combination was more pronounced at lower dosages.
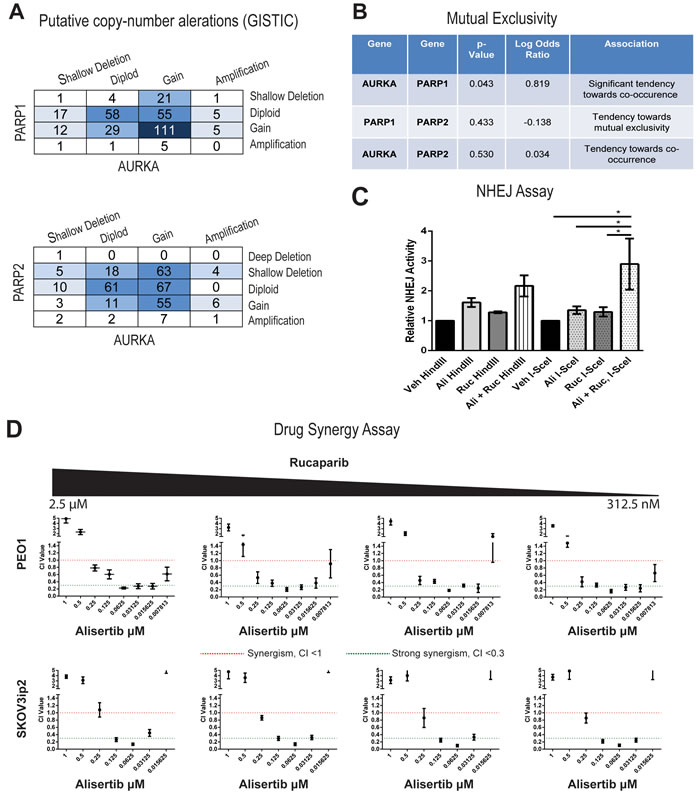
Figure 6: Drug synergy between alisertib and rucaparib in both PARPi sensitive and resistant cell lines. A. Copy number alterations are similar between AURKA and PARP1 as well as between AURKA and PARP2. B. Mutual exclusivity analysis suggest a significant association between AURKA and PARP1 and a trend of co-occurrence between AURKA and PARP2. C. Combinations of alisertib (31.25 nM) and rucaparib (313 nM) significantly enhance the NHEJ function in PEO1 cell lines compared to vehicle, alisertib alone (31.25 nM), or rucaparib alone (313 nM) (n = 5, *= P<0.05, two-way ANOVA). D. Combination index values for both PEO1 and SKOV3ip2 cells between rucaparib (2.5 µM, 1.25 µM, 625 nM, and 312 nM) and eight two-fold serial dilutions of alistertib (1 µM to 7.8 nM). CI values were considered either synergistic (CI< 1.0, red line) or strongly synergistic (CI< 0.3, green line). Each data point is represented of three independent experiments and plotted as mean ± SD.
DISCUSSION
Although it is known that AURKA regulation of mitosis and centrosome function plays an important role in OC biology [7-13], there remains a large gap in our knowledge of alternative, non-mitotic functions of this kinase in tumor cells. In the current study, we investigated the relatively undefined role of AURKA in mediating the DDR and DNA repair in ovarian carcinoma cells and discovered that AURKA regulates the activity of PARP and DNA-PKcs, a key signaling molecule in the error-prone NHEJ pathway.
Based on our findings, we propose a model in which inhibition of AURKA activity promotes NHEJ DNA repair through distinct mechanisms (Figure 7A). First, AURKA inhibition induces the formation of pH2AXS139 foci (i) in the nucleus, similar to the effects of PARPi on ovarian carcinoma cells [49]. AURKA inhibition also increases BRCAness by decreasing BRCA1/2 protein levels (ii) and stimulates the error-prone NHEJ pathway by diminishing PARP expression (iii) and increasing DNA-PKcs activity (iv). Since PARP and Ku directly compete for the binding of DNA ends [57], a decrease in PARP levels should favor Ku binding to DNA ends. All of these changes are predicted to favor the NHEJ repair pathway over the HR pathway [57].
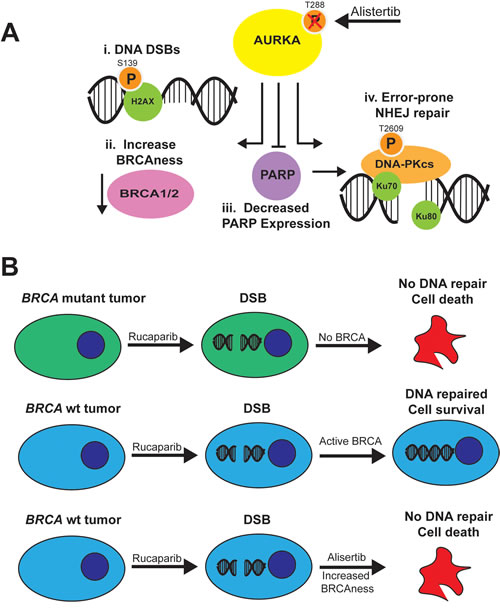
Figure 7: Model for effects of AURKA inhibition on NHEJ in ovarian carcinoma cells and combination with PARP inhibitors. A. Inhibition of AURKA activity stimulates error-prone NHEJ DNA repair of DNA DSBs with incompatible ends, which may result in genomic instability that is lethal to tumor cells. AURKA inhibition in ovarian carcinoma cells i) induces DNA DSBs as evidenced by an increase in gH2AX foci, ii) increases BRCAness as a result of decreased BRCA1 or BRCA2 levels, iii) decreases PARP expression and activity, which would relieve PARP suppression of Ku function, and iv) stimulates DNA-PKcs activity. The increase in BRCAness, decrease in PARP activity and stimulation of DNA-PKcs activity would favor the use of the more error-prone NHEJ pathway and combination therapy with PARP inhibitors. B. Rucaparib, a PARP inhibitor, is effective in BRCAmut tumors (top) but not BRCAwt tumors (middle) which repair DNA double strand breaks. Combination of rucaparib with alisertib, which induces a BRCAness phenotype, increases the function of PARPi in BRCAwt tumors (bottom).
Treatment with the PARPi, veliparib, resulted in distinct effects on HR-deficient versus HR-proficient ovarian carcinoma cells. Veliparib treatment increased pH2AXS139 and pDNA-PKcsT2609 levels in HR-deficient (PARPi-sensitive) but not HR-proficient (PARPi-resistant) ovarian carcinoma cells [49]. Also, PARPi stimulation of NHEJ repair resulted in genomic instability which was cytotoxic to HR-deficient but not -proficient cells. The same study demonstrated that RNAi-mediated silencing of PARP expression significantly reduced colony formation in HR-deficient cells, but had no effect on colony formation in HR-proficient cells [49].
In contrast to these findings for PARPi, we discovered that inhibition of AURKA activity with alisertib significantly diminished the growth and clonogenic survival of PARPi-sensitive and -resistant ovarian carcinoma cells. Alisertib treatment also decreased PARP expression and activity and activated DNA-PKcs, thereby stimulating error-prone NHEJ repair of DSBs with incompatible ends in both PARPi-sensitive and -resistant cells. To our knowledge, this study is the first to demonstrate that inhibition of AURKA activity increases error-prone NHEJ repair. These findings suggest that alisertib may be used in combination with PARPis to sensitize PARPi-resistant or otherwise weakly sensitive ovarian carcinoma cells to PARP inhibition, although we cannot rule out the possibility that alisertib treatment decreases ovarian carcinoma cell viability because other AURKA-regulated functions not related to DNA repair, are altered. Inhibition of AURKA activity also decreased the expression of BRCA1/2 in ovarian carcinoma cells. BRCA1 expression changes throughout the cell cycle, with highest expression observed during S phase, and high levels are maintained until G2/M before decreasing in early G1 [58, 59]. BRCA2 expression is lowest during G0 and early G1 and increases as cells enter S phase [60-63]; there are reports of roles for BRCA2 during G2/M [64, 66] and mitosis [66-71]. We do not know why BRCA1/2 expression decreased as a result of alisertib treatment, but this phenomenon might be able to be exploited to provide options for women whose tumors are wild-type for BRCA1/2 and related DNA repair genes and are not strong candidates for treatment with PARP inhibitors. One possible explanation is that AURKA activity may be required for the stability of BRCA1/2, since a number of studies report a physical and/or functional relationship between AURKA and BRCA1/2 [27, 28, 72, 73].
The modest effects of alisertib in clinical trials with platinum-resistant and -refractory patients may reflect the gap in our knowledge of the non-mitotic functions of AURKA. Elucidation of AURKA-regulated pathways that mediate the DDR and DNA repair may identify biomarkers for predicting response to molecular therapeutics which target these pathways and may also uncover novel targets for combination therapy strategies utilizing PARPi. In this paper, we showed that exploring the combination of AURKAi and PARPi showed singificant drug synergy and could open up the use of PARP inhibitors in non-BRCA mutant tumors (Figure 7B).
MATERIALS AND METHODS
Cell culture and transfection
The human ovarian carcinoma cell lines and culture medium used were: PEO1 A2780, MDAH, OVCAR5, OVCAR10 and PEO4 (kind gift of Dr. Michael White, University of Texas Southwestern) in RPMI supplemented with 10% FBS and insulin (0.25 units/mL); SKOV3ip2 [17] in McCoy’s 5A supplemented with 10% FBS, non-essential amino acids and 800 mg/mL G418; OVCA429 in MEM supplemented with 10% FBS, non-essential amino acids and sodium pyruvate. All media was supplemented with penicillin/streptomycin (100 mg/mL). The plasmid constructs, pcDNA3.1-AURKA-RFP and pcDNA3.1-RFP (generous gift of Dr. Erica Golemis, Fox Chase Cancer Center), were transiently transfected (4 µg DNA) using Lipofectamine 2000 (Thermo Scientific, Rockford, IL) or TransIT-LT1 (Mirus Bio, Madison, WI) according to the manufacturer’s instructions. The effect of RNAi-mediated AURKA knockdown was assayed in cells transfected twice (24 h apart) with 25 nmol/L (for SKOV3ip2) or 50 nmol/L (for PEO1) of AURKA-specific siRNA or the AllStars Negative Control siRNA (Qiagen, Valencia, CA; oligo sequence is proprietary) using Lipofectamine 2000. The sequences for AURKA-specific siRNA oligos utilized are: 5’-AAUAUUAGGAUGCCGAAGGTG-3’ (si1); 5’-UAAAUGGAGGUAGAGCACGTG-3’ (si2).
Drug treatment, cell viability and clonogenic assays
Cells were plated in 96-well black, clear bottom plates (Greiner Bio-One, Monroe, NC) and treated the following day with alisertib (Selleck Chemicals, Houston, TX). For each experiment, cells were treated in quadruplicate with vehicle (DMSO) or alisertib (0.00781, 0.0156, 0.0313, 0.0625, 0.125, 0.250, 0.50 and 1.0 µmol/L) for 7 days, and then viability was evaluated using the Cell Titer Blue assay (Promega, Madison, WI) by measuring fluorescent intensity on the Tecan M200 plate reader (Tecan US, Inc., Morrisville, NC) according to the manufacturer’s instructions. For PARPi treatment, cells were treated in quadruplicate with increasing concentrations of rucaparib (0.0781, 0.156, 0.313, 0.625, 1.25, 2.5, 5.0, and 10.0 mmol/L) [Selleck Chemicals] for 7 d. Nonlinear regression analysis was performed using GraphPad Prism version 5.01 (GraphPad Software, La Jolla, CA) to generate a log (inhibitor) versus drug response curve with variable slope and to calculate the IC50 ± SEM.
Clonogenic survival assays were performed by plating PEO1 (1000 cells/dish) and SKOV3ip2 (200 cells/dish) cells on 60 mm dishes and treating the next day with either vehicle (DMSO) or alisertib (0.0313, 0.0625, 0.125, and 0.250 µmol/L) for three days. Cells were then rinsed with fresh media and SKOV3ip2 and PEO1 cells were cultured in complete media for nine and ten days, respectively. Colonies (³ 50 cells) were fixed with 4% NBF for 10 min, stained with crystal violet for 30 min, and rinsed excessively with water. The plate was divided into four fields and the mean number of colonies per field ± SEM was calculated.
Immunoblotting
Cell monolayers were lysed in Mammalian Protein Extraction Reagent (MPER™, Thermo Scientific) supplemented with Halt™ Phosphatase Inhibitor Cocktail (Thermo Scientific) and Complete™ Mini Protease Inhibitor Cocktail (Roche Diagnostics, Indiannapolis, IN). Protein concentrations of cell lysates were determined using the BCA Protein Assay (Thermo Scientific) and proteins were resolved using 7.5%, 12%, or 4-20% gradient SDS-PAGE gels (Bio-Rad, Hercules, CA). Proteins were transferred to supported nitrocellulose membrane overnight at 25 V, and membranes were blocked in 5% non-fat dry milk or 5% BSA in TBST, incubated overnight at 4 C with primary antibody in block, followed by Immun-Star HRP-conjugated secondary antibody (Bio-Rad) at 1:10,000; signal was detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Antibodies directed against the following were used: p-DNA-PKcsT2609 (Thermo Scientific), DNA-PKcs (Santa Cruz Biotechnology, Dallas, TX); AURKA, BRCA1, PARP (Cell Signaling, Danvers, MA); BRCA2 (R & D Systems, Minneapolis, MN), pADPr or PAR (BD Biosciences, San Jose, CA), pH2AXS139 (Calbiochem), and β-Actin (Sigma-Aldrich). Pixel densities of blot images were calculated using Image-J software (NIH). Changes in protein levels were normalized to loading controls and expressed as fold change relative to treatment controls.
Immunofluorescent staining and microscopy
Cells were cultured on chamber slides and then treated with alisertib (62.5 nmol/L) for 24 h before incubating with extraction buffer (20 mmol/L HEPES, pH 7.5, 20 mmol/L NaCl, 5 mmol/L MgCl2, 0.5% Nonidet P-40), on ice for 20 min, then fixed in 4% NBF for 15 min, and permeabilized in 0.5% Triton-X100/PBS for 10 min at room temperature. Cells were blocked in 5% normal goat serum in TBS for 1 h at room temperature and then incubated with anti- pH2AXS139 diluted in block overnight at 4°C and then with goat-anti-mouse Dylight 488-conjugated secondary antibody for 1 h at room temperature. Immunofluorescent images were acquired using a Nikon Eclipse 80i microscope (Melville, NY) and digital camera with Metamorph software v. 7.7 (Molecular Devices, Sunnyvale, California) using identical exposure times. The number of nuclei with ≥ 10 pH2AXS139-positive foci was enumerated in five different fields for each treatment condition and the mean percentage of nuclei with ≥ 10 pH2AXS139 foci ± SE was calculated. Statistical analysis was performed with GraphPad Prism using Wilcoxon matched-pairs signed rank test; P < 0.05 was considered significant.
Nonhomologous end-joining DNA repair assay
Cells were treated with 5 µmol/L QVD-OPhe (Sigma-Aldrich), a broad spectrum caspase inhibitor, before being transfected twice (24 h apart) with 2 µg of pem1-Ad2-EGFP plasmid (generous gift of Dr. Eric A. Hendrickson, University of Minnesota Medical School) digested with either HindIII (Thermo Scientific) or I-SceI (New England Biolabs, Ipswich, MA), and 0.5 µg of pcDNA3.1-RFP plasmid using Lipofectamine 2000. Cells were then treated with either vehicle (DMSO) or 62.5 nmol/L alisertib for 48 h before analysis on the Becton Dickinson LSRII flow cytometer (Franklin Lakes, NJ) to detect GFP+ cells, which have successfully repaired DNA DSBs. The number of GFP+ cells was normalized to the number of RFP+ cells as a control for transfection efficiency, and statistical analysis was performed with GraphPad Prism using either one-way ANOVA followed by Tukey’s multiple comparison or by Wilcoxon matched-pairs signed rank test; P < 0.05 was considered significant.
RNA isolation and real-time PCR analyses
PEO1 cells were cultured in a 60 mm dish and treated with 62.5 nmol/L alisertib for 16 or 48 h before RNA isolation using the RNeasy Mini Kit (Qiagen). Genomic DNA was removed before cDNA was synthesized from 1 mg RNA using the Quantifect Reverse Transcription Kit (Qiagen), according to the manufacturer’s instructions. Real-time PCR was performed in quadruplicates to assay the expression of BRCA1, PARP1, and 53BP1 mRNA using Applied BiosystemsTM Taqman® Assays (ThermoFisher Scientific), according to the manufacturer’s instructions, and the Bio-Rad CFX96TM real-time detection system and accompanying software to analyze normalized relative mRNA expression and to perform statistical analyses. RPS13 was used as a control for normalizing gene expression P < 0.05 was considered significant.
Orthotopic xenografts and immunohistochemistry
The University of Kansas Medical Center Institutional Animal Care and Use Committee (IACUC) approved all procedures involving mice. Seven to fourteen week-old female C.B-17 severe combined immunodeficient (SCID) mice (Harlan Research Models, Indiannapolis, IN) were used for intrabursal (i.b.) injections as described [74]. Mice were given unilateral (left side) i.b. injections of SKOV3ip2-Luc-D3 cells (1 x 106), also referred to as SKOV3ip2 [17]. Alisertib (Selleck Chemicals) was suspended in 10% 2-hydroxypropyl-b-cyclodextrin (Sigma-Aldrich) with 1% sodium bicarbonate and 10 mg/kg administered orally to mice using a 5 day on/2 day off schedule. Mice were euthanized by CO2 inhalation, necropsied and examined for gross abnormalities. Paraffin-embedded tissue sections were used for immunohistochemical (IHC) staining with antibodies directed against pAURKAT288 (Bethyl Laboratories, Montgomery, TX); pDNA-PKcsT2609 (Thermo Scientific); and Ku80 and PARP (Cell Signaling). Brightfield images of IHC staining were acquired on a Nikon Eclipse 80i microscope and digital camera using identical exposure times. The level of expression of each antigen was calculated using a semi-quantitative approach for IHC staining, i.e., H-score: a summation of the percentage of area stained (0 to 100) at each intensity level multiplied by the weighted intensity (e.g., 1, 2, or 3; where 0 is no staining, 1 is weak staining, 2 is moderate staining and 3 is strong staining) [75]. For pDNA-PKcsT2609 the average number of positively staining nuclei per high powered field were compared in alisertib and vehicle treated tumors.
Drug combination assays
Drug combination studies were performed using the combination index (CI) method described by Chou and Talalay [76]. Cells from PEO1 or SKOV3ips cell lines were plated overnight and treated with rucaparib (2.5 µM, 1.25 µM, 625 nM, and 312 nM) and eight two-fold serial dilutions of alisertib (1 µM to 7.8 nM) in a combination matrix. Assays were performed as biological triplicate using triplicate wells within each experiment. Cell viability following 48 h of treatment from the serial dilutions was evaluated using CellTiter-Blue as previously described and the viability data were then analyzed using CalcuSyn (version 2.1, BioSoft, UK) to calculate the synergy between the two drugs at each molar ratio evaluated. Drug combinations which yielded CI values less than 1 were considered to be synergistic as previously reported [77, 78] and used in our laboratory [79, 80].
ACKNOWLEDGMENTS
The authors gratefully acknowledge Dr. Denise C. Connolly for critical review of this manuscript, as well as the support of the Biospecimen Repository Core Facility staff funded in part by the University of Kansas Cancer Center (P30 CA168524). We also acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the NIH/NIGMS COBRE grant (P30 GM103326).
CONFLICTS OF INTEREST
The authors have no conflicts of interest to disclose.
FUNDING
This work was supported by grants from the NCI R01 CA140323 and R01 CA214545, the Kansas Bioscience Authority Eminent Scholar Program, and the OVERRUN Ovarian Cancer Foundation (to AKG); the Marsha Rivkin Center for Ovarian Cancer Research Pilot Study Award (to T-VD). AKG is the Chancellors Distinguished Chair in Biomedical Sciences Endowed Professor.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Ganghadaran SG. Management of platinum resistant – refractory ovarian cancer: a short review. Journal of Cancer Research and Treatment. 2016; 4:32–36.
3. Lloyd I. Citeline Pharma R & D Annual Review. 2015. Pharma Intelligence, Informa. https://pharmaintelligence.informa.com/resources/product-content/citeline-pharma-rd-annual-review-2015
4. DiMasi JA, Feldman L, Seckler A, Wilson A. Trends in risks associated with new drug development: success rates for investigational drugs. Clin Pharmacol Ther. 2010; 87:272–77.
5. Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995; 81:95–105.
6. Nikonova AS, Astsaturov I, Serebriiskii IG, Dunbrack RL Jr, Golemis EA. Aurora A kinase (AURKA) in normal and pathological cell division. Cell Mol Life Sci. 2013; 70:661–87.
7. Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/Aurora-A in human ovarian cancer. Clin Cancer Res. 2003; 9:1420–26.
8. Hsu LC, Kapali M, DeLoia JA, Gallion HH. Centrosome abnormalities in ovarian cancer. Int J Cancer. 2005; 113:746–51.
9. Lassmann S, Shen Y, Jütting U, Wiehle P, Walch A, Gitsch G, Hasenburg A, Werner M. Predictive value of Aurora-A/STK15 expression for late stage epithelial ovarian cancer patients treated by adjuvant chemotherapy. Clin Cancer Res. 2007; 13:4083–91.
10. Tanner MM, Grenman S, Koul A, Johannsson O, Meltzer P, Pejovic T, Borg A, Isola JJ. Frequent amplification of chromosomal region 20q12-q13 in ovarian cancer. Clin Cancer Res. 2000; 6:1833–39.
11. Chung CM, Man C, Jin Y, Jin C, Guan XY, Wang Q, Wan TS, Cheung AL, Tsao SW. Amplification and overexpression of aurora kinase A (AURKA) in immortalized human ovarian epithelial (HOSE) cells. Mol Carcinog. 2005; 43:165–74.
12. Hu W, Kavanagh JJ, Deaver M, Johnston DA, Freedman RS, Verschraegen CF, Sen S. Frequent overexpression of STK15/Aurora-A/BTAK and chromosomal instability in tumorigenic cell cultures derived from human ovarian cancer. Oncol Res. 2005; 15:49–57.
13. Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, Godwin AK, Gross J, Hartmann L, et al. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474:609–15.
14. Landen CN Jr, Lin YG, Immaneni A, Deavers MT, Merritt WM, Spannuth WA, Bodurka DC, Gershenson DM, Brinkley WR, Sood AK. Overexpression of the centrosomal protein Aurora-A kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin Cancer Res. 2007; 13:4098–104.
15. Minton SE, LoRusso P, Lockhart AC, Saif M, Krishnamurthi SS, Pickett-Gies CA, Wang Y, Guan S, Roben E, Stein MN. A phase I study of MK-5108, an oral aurora A kinase inhibitor, in both monotherapy and in combination with docetaxel in patients with advanced solid tumors. J Clin Oncol. 2010; 28:e13026.
16. Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res. 2011; 17:7614–24.
17. Do TV, Xiao F, Bickel LE, Klein-Szanto AJ, Pathak HB, Hua X, Howe C, O’Brien SW, Maglaty M, Ecsedy JA, Litwin S, Golemis EA, Schilder RJ, et al. Aurora kinase A mediates epithelial ovarian cancer cell migration and adhesion. Oncogene. 2014; 33:539–49.
18. Matulonis UA, Sharma S, Ghamande S, Gordon MS, Del Prete SA, Ray-Coquard I, Kutarska E, Liu H, Fingert H, Zhou X, Danaee H, Schilder RJ. Phase II study of MLN8237 (alisertib), an investigational Aurora A kinase inhibitor, in patients with platinum-resistant or -refractory epithelial ovarian, fallopian tube, or primary peritoneal carcinoma. Gynecol Oncol. 2012; 127:63–69.
19. Falchook GS, Goff BA, Kurzrock R, Gray HJ, Martin LP, Coleman RL, Liu H, Zhou X, Benaim E, Schilder RJ. Phase I/II study of weekly paclitaxel with or without MLN8237 (alisertib), an investigational aurora A kinase inhibitor, in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer (OC), or breast cancer (BrC): Phase I results. J Clin Oncol. 2012; 30:5021.
20. Coleman RL, Roszak A, Behbakht K, Coquard RL, Matulonis U, Liu H, Schusterbauer C, Ullmann CD. Randomized phase 2 study of investigational, selective aurora A kinase inhibitor alisertib (MLN8237) with weekly paclitaxel vs paclitaxel alone. Ann Oncol. 2014; 25:iv305–26.
21. Kozyreva VK, Kiseleva AA, Ice RJ, Jones BC, Loskutov YV, Matalkah F, Smolkin MB, Marinak K, Livengood RH, Salkeni MA, Wen S, Hazard HW, Layne GP, et al. Combination of Eribulin and Aurora A Inhibitor MLN8237 Prevents Metastatic Colonization and Induces Cytotoxic Autophagy in Breast Cancer. Mol Cancer Ther. 2016; 15:1809–22.
22. Wang Y, Sun H, Wang Z, Liu M, Qi Z, Meng J, Sun J, Yang G. Aurora-A: a potential DNA repair modulator. Tumour Biol. 2014; 35:2831–36.
23. Wang K, Terai K, Peng W, Rouyanian A, Liu J, Roby KF, Wise AL, Ezzeldin M, Larson J, Woo RA, Lialyte K, Farassati F. The role of RalA in biology and therapy of ovarian cancer. Oncotarget. 2013.
24. Johnson EO, Chang KH, Ghosh S, Venkatesh C, Giger K, Low PS, Shah K. LIMK2 is a crucial regulator and effector of Aurora-A-kinase-mediated malignancy. J Cell Sci. 2012; 125:1204–16.
25. Sourisseau T, Maniotis D, McCarthy A, Tang C, Lord CJ, Ashworth A, Linardopoulos S. Aurora-A expressing tumour cells are deficient for homology-directed DNA double strand-break repair and sensitive to PARP inhibition. EMBO Mol Med. 2010; 2:130–42.
26. Pugacheva EN, Golemis EA. HEF1-aurora A interactions: points of dialog between the cell cycle and cell attachment signaling networks. Cell Cycle. 2006; 5:384–91.
27. Yang G, Chang B, Yang F, Guo X, Cai KQ, Xiao XS, Wang H, Sen S, Hung MC, Mills GB, Chang S, Multani AS, Mercado-Uribe I, Liu J. Aurora kinase A promotes ovarian tumorigenesis through dysregulation of the cell cycle and suppression of BRCA2. Clin Cancer Res. 2010; 16:3171–81.
28. Wang Y, Wang Z, Qi Z, Yin S, Zhang N, Liu Y, Liu M, Meng J, Zang R, Zhang Z, Yang G. The negative interplay between Aurora A/B and BRCA1/2 controls cancer cell growth and tumorigenesis via distinct regulation of cell cycle progression, cytokinesis, and tetraploidy. Mol Cancer. 2014; 13:94.
29. Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, Desmaze C, Sabatier L, Lopez BS. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004; 14:611–23.
30. Piganeau M, Ghezraoui H, De Cian A, Guittat L, Tomishima M, Perrouault L, René O, Katibah GE, Zhang L, Holmes MC, Doyon Y, Concordet JP, Giovannangeli C, et al. Cancer translocations in human cells induced by zinc finger and TALE nucleases. Genome Res. 2013; 23:1182–93.
31. Capp JP, Boudsocq F, Bertrand P, Laroche-Clary A, Pourquier P, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. The DNA polymerase lambda is required for the repair of non-compatible DNA double strand breaks by NHEJ in mammalian cells. Nucleic Acids Res. 2006; 34:2998–3007.
32. Capp JP, Boudsocq F, Besnard AG, Lopez BS, Cazaux C, Hoffmann JS, Canitrot Y. Involvement of DNA polymerase mu in the repair of a specific subset of DNA double-strand breaks in mammalian cells. Nucleic Acids Res. 2007; 35:3551–60.
33. Mahaney BL, Meek K, Lees-Miller SP. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009; 417:639–50.
34. Nick McElhinny SA, Ramsden DA. Sibling rivalry: competition between Pol X family members in V(D)J recombination and general double strand break repair. Immunol Rev. 2004; 200:156–64.
35. Moshous D, Callebaut I, de Chasseval R, Poinsignon C, Villey I, Fischer A, de Villartay JP. The V(D)J recombination/DNA repair factor artemis belongs to the metallo-beta-lactamase family and constitutes a critical developmental checkpoint of the lymphoid system. Ann N Y Acad Sci. 2003; 987:150–57.
36. Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006; 124:301–13.
37. Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche MC, Sanal O, Plebani A, Stéphan JL, Hufnagel M, le Deist F, Fischer A, Durandy A, de Villartay JP, Revy P. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006; 124:287–99.
38. Revy P, Malivert L, de Villartay JP. Cernunnos-XLF, a recently identified non-homologous end-joining factor required for the development of the immune system. Curr Opin Allergy Clin Immunol. 2006; 6:416–20.
39. de Murcia G, Ménissier de Murcia J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem Sci. 1994; 19:172–76.
40. Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012; 336:728–32.
41. Langelier MF, Pascal JM. PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr Opin Struct Biol. 2013; 23:134–43.
42. Hassler M, Ladurner AG. Towards a structural understanding of PARP1 activation and related signalling ADP-ribosyl-transferases. Curr Opin Struct Biol. 2012; 22:721–29.
43. Althaus FR, Richter C. ADP-ribosylation of proteins. Enzymology and biological significance. Mol Biol Biochem Biophys. 1987; 37:1–237.
44. Scott CL, Swisher EM, Kaufmann SH. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J Clin Oncol. 2015; 33:1397–406.
45. Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004; 4:814–19.
46. Walsh T, Lee MK, Casadei S, Thornton AM, Stray SM, Pennil C, Nord AS, Mandell JB, Swisher EM, King MC. Detection of inherited mutations for breast and ovarian cancer using genomic capture and massively parallel sequencing. Proc Natl Acad Sci USA. 2010; 107:12629–33.
47. Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011; 108:18032–37.
48. Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil C, Rendi M, Thornton A, Norquist BM, Casadei S, Nord A, Agnew KJ, Pritchard CC, Scroggins S, et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res. 2014; 20:764–75.
49. Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci USA. 2011; 108:3406–11.
50. Sakai W, Swisher EM, Jacquemont C, Chandramohan KV, Couch FJ, Langdon SP, Wurz K, Higgins J, Villegas E, Taniguchi T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009; 69:6381–86.
51. Stordal B, Timms K, Farrelly A, Gallagher D, Busschots S, Renaud M, Thery J, Williams D, Potter J, Tran T, Korpanty G, Cremona M, Carey M, et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol. 2013; 7:567–79.
52. Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ, Ray ET, Sells TB, Stringer B, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci USA. 2007; 104:4106–11.
53. Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002; 16:2333–38.
54. Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004; 64:2390–96.
55. Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010; 6:e1000855.
56. Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, Zhang Q, Koboldt DC, Xie M, Kandoth C, McMichael JF, Wyczalkowski MA, Larson DE, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014; 5:3156.
57. Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–82.
58. Vaughn JP, Davis PL, Jarboe MD, Huper G, Evans AC, Wiseman RW, Berchuck A, Iglehart JD, Futreal PA, Marks JR. BRCA1 expression is induced before DNA synthesis in both normal and tumor-derived breast cells. Cell Growth Differ. 1996; 7:711–15.
59. Ruffner H, Verma IM. BRCA1 is a cell cycle-regulated nuclear phosphoprotein. Proc Natl Acad Sci USA. 1997; 94:7138–43.
60. Bertwistle D, Swift S, Marston NJ, Jackson LE, Crossland S, Crompton MR, Marshall CJ, Ashworth A. Nuclear location and cell cycle regulation of the BRCA2 protein. Cancer Res. 1997; 57:5485–88.
61. Blackshear PE, Goldsworthy SM, Foley JF, McAllister KA, Bennett LM, Collins NK, Bunch DO, Brown P, Wiseman RW, Davis BJ. Brca1 and Brca2 expression patterns in mitotic and meiotic cells of mice. Oncogene. 1998; 16:61–68.
62. Rajan JV, Wang M, Marquis ST, Chodosh LA. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc Natl Acad Sci USA. 1996; 93:13078–83.
63. Su LK, Wang SC, Qi Y, Luo W, Hung MC, Lin SH. Characterization of BRCA2: temperature sensitivity of detection and cell-cycle regulated expression. Oncogene. 1998; 17:2377–81.
64. Lin HR, Ting NS, Qin J, Lee WH. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. J Biol Chem. 2003; 278:35979–87.
65. Lee M, Daniels MJ, Venkitaraman AR. Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression. Oncogene. 2004; 23:865–72.
66. Choi E, Park PG, Lee HO, Lee YK, Kang GH, Lee JW, Han W, Lee HC, Noh DY, Lekomtsev S, Lee H. BRCA2 fine-tunes the spindle assembly checkpoint through reinforcement of BubR1 acetylation. Dev Cell. 2012; 22:295–308.
67. Choi E, Choe H, Min J, Choi JY, Kim J, Lee H. BubR1 acetylation at prometaphase is required for modulating APC/C activity and timing of mitosis. EMBO J. 2009; 28:2077–89.
68. Park I, Lee HO, Choi E, Lee YK, Kwon MS, Min J, Park PG, Lee S, Kong YY, Gong G, Lee H. Loss of BubR1 acetylation causes defects in spindle assembly checkpoint signaling and promotes tumor formation. J Cell Biol. 2013; 202:295–309.
69. Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004; 306:876–79.
70. Lekomtsev S, Guizetti J, Pozniakovsky A, Gerlich DW, Petronczki M. Evidence that the tumor-suppressor protein BRCA2 does not regulate cytokinesis in human cells. J Cell Sci. 2010; 123:1395–400.
71. Mondal G, Rowley M, Guidugli L, Wu J, Pankratz VS, Couch FJ. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev Cell. 2012; 23:137–52.
72. Brodie KM, Henderson BR. Characterization of BRCA1 protein targeting, dynamics, and function at the centrosome: a role for the nuclear export signal, CRM1, and Aurora A kinase. J Biol Chem. 2012; 287:7701–16.
73. Zou J, Rezvani K, Wang H, Lee KS, Zhang D. BRCA1 downregulates the kinase activity of Polo-like kinase 1 in response to replication stress. Cell Cycle. 2013; 12:2255–65.
74. Connolly DC, Hensley HH. Xenograft and transgenic mouse models of epithelial ovarian cancer and non-invasive imaging modalities to monitor ovarian tumor growth in situ: applications in evaluating novel therapeutic agents. Current Protoc Pharmacol. 2009; 45:14.12.1-14.12.26.
75. Sethi G, Kwon Y, Burkhalter RJ, Pathak HB, Madan R, McHugh S, Atay S, Murthy S, Tawfik OW, Godwin AK. PTN signaling: components and mechanistic insights in human ovarian cancer. Mol Carcinog. 2015; 54:1772–85.
76. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27–55.
77. Chang TT, Chou TC. Rational approach to the clinical protocol design for drug combinations: a review. Acta Paediatr Taiwan. 2000; 41:294-302.
78. Chou TC. Preclinical versus clinical drug combination studies. Leuk Lymphoma. 2008; 49:2059–80.
79. Pessetto ZY, Ma Y, Hirst JJ, von Mehren M, Weir SJ, Godwin AK. Drug repurposing identifies a synergistic combination therapy with imatinib mesylate for gastrointestinal stromal tumor. Mol Cancer Ther. 2014; 13:2276–87.
80. Pathak HB, Zhou Y, Sethi G, Hirst J, Schilder RJ, Golemis EA, Godwin AK. A Synthetic Lethality Screen Using a Focused siRNA Library to Identify Sensitizers to Dasatinib Therapy for the Treatment of Epithelial Ovarian Cancer. PLoS One. 2015; 10:e0144126.