
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a pathological condition histologically categorized into nonalcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH) [1]. NAFLD can progress to cirrhosis and end-stage liver diseases such as hepatocellular carcinoma [2, 3].
Accumulation of unfolded proteins in the endoplasmic reticulum (ER) causes ER stress, which triggers an adaptive response called the unfolded protein response (UPR) to restore ER homeostasis [4]. The UPR pathway is also required to maintain hepatic lipid metabolism [5]. The UPR is coordinated primarily by three ER transmembrane stress transducers, protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol requiring enzyme 1 (IRE1).
Prolonged ER stress leads to PERK signaling-mediated upregulation of C/EBP homologous protein (CHOP), a pro-apoptotic transcription factor [6, 7]. One mechanism by which CHOP induces apoptosis is via inhibition of B-cell lymphoma-2 (Bcl-2) expression and induction of bcl-2-like protein 11 (Bim) expression [8, 9]. ATF6 is a membrane-bound transcription factor, but it is cleaved to release its cytoplasmic domain in response to ER stress. Cleaved ATF6 transcriptionally activates X-box–binding protein 1 (XBP1). XBP1 mRNA is spliced by IRE1 during ER stress to produce the transcription factor, XBP1s [4]. These ER stress-related transcription factors are involved in activation of various ER chaperones and folding-related proteins that directly execute protein quality control [10, 11].
Glucose-regulated protein 78 (GRP78, also known as BiP) and glucose-regulated protein 94 (GRP94) are molecular chaperones regulating protein quality control and degradation. GRP78 also plays a pivotal role in activation of the UPR, in which GRP78 is released from PERK, IRE1 and ATF6 and activates them. The lectin calnexin is a transmembrane ER chaperone involved in folding of newly synthesized glycoproteins [12].
Protein disulfide isomerase (PDI) is a member of PDI superfamily that is involved in oxidative protein folding [13]. ER protein 72 (ERp72) and ER protein 44 (ERp44) are also oxidoreductases in the ER belonging to the PDI family [14]. ER oxireductin 1 (Ero1)-Lα is an oxidase activated during UPR [15, 16].
The intrinsic pathway to apoptosis predominantly leads to cytochrome c release from the mitochondria into the cytosol. The intrinsic pathway is strictly controlled by anti-apoptotic (Bcl-2 and myeloid cell leukemia-1 (Mcl-1)) and pro-apoptotic (such as Bim and bcl-2 homologous antagonist/killer (Bak)) Bcl-2 family proteases. The extrinsic pathway to apoptosis can bypass the mitochondrial step. The intrinsic and extrinsic pathways activate caspase-3 protease, which is central to execution of apoptosis [17]. Poly (ADP-ribose) polymerase (PARP) is a cellular substrate of caspases. Cleavage of PARP is considered to be an apoptosis marker [18].
Autophagy is an intracellular pathway responsible for turnover of long-lived proteins [19]. Beclin-1 regulates autophagy, forming a multiprotein complex that initiates autophagosome formation [20]. Autophagy protein 16L1 (Atg16L1) mediates conjugation between autophagy protein 5 (Atg5) and autophagy protein 12 (Atg12) and delivers this complex to autophagosomes. Atg5–Atg12 conjugates convert the cytoplasmic form of microtubule-associated proteins 1A/1B light chain 3A (LC3A/B-I) to the membrane-bound form, referred to as LC3A/B-II. The conversion of LC3A/B-I to LC3A/B-II is a pivotal process for maturation of autophagosomes, enabling their fusion with lysosomes and autophagosome cargo degradation [21]. The protein p62 is a selective substrate for autophagy [19, 22].
Recent evidence suggests the involvement of ER stress and the UPR in development of many chronic liver diseases such as NAFLD [23–28]. Inadequate response to ER stress may cause fat accumulation, insulin resistance, inflammation, autophagy and apoptosis, all of which are critical to pathogenesis of NAFLD [29, 30]. In our study, expression of various proteins associated with ER stress, autophagy and apoptosis was analyzed in NAFL and NASH tissues to elucidate the roles of those proteins in pathogenesis of the critical metabolic disorder.
RESULTS
Enhanced expression of transcription factors associated with ER stress in NASH tissues
Expression of three ER stress-responsive transcription factors was analyzed in NASH, NAFL and normal liver tissues by western blotting (Figure 1). Levels of cleaved ATF6, XBP1s and CHOP were higher in NASH than in normal tissues (P < 0.05), suggesting that there was activation of these main UPR transducers in NASH. Levels of cleaved ATF6 appeared to be very low in normal and NAFL tissues. It appeared that CHOP levels in NAFL tissues were between those in NASH and normal tissues. The two NAFL tissues displayed highly variable levels of XBP1s and CHOP.
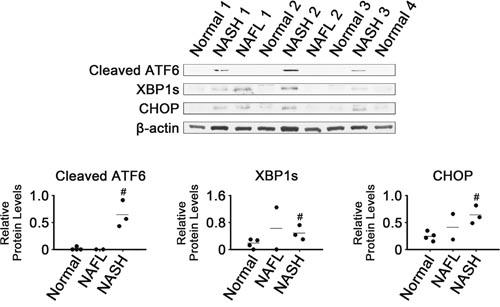
Figure 1: Expression of ER stress-associated transcription factors in NASH, NAFL and normal liver tissues. β-Actin was used as an internal control. Horizontal lines represent means of densitometry signals from the western blot analyses for all tissue groups. #, significant differences in signals between NASH and normal liver tissues (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
Decreased expression of ER chaperones in NASH tissues
In contrast to the enhanced expression of ER stress-associated transcription factors, levels of some ER chaperones were decreased in NASH tissues (Figure 2). Levels of GRP78 and GRP94 were much lower in NASH than in normal tissues (P < 0.05; Figure 2A). As found in the whole tissues, the NASH microsomes had lower levels of GRP78 and GRP94 than did normal microsomes (P < 0.05; Figure 2B).
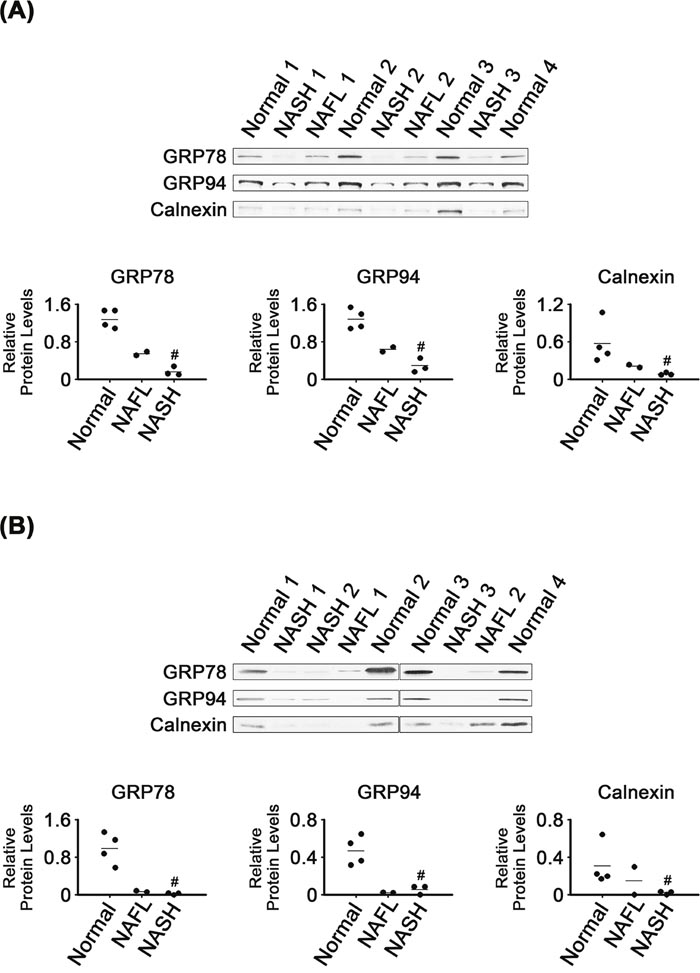
Figure 2: Expression of ER chaperones in NASH, NAFL and normal liver tissues (A) and in their microsomes (B). β-Actin was used as an internal control for the whole tissue samples, as described for Figure 1. Horizontal lines represent means of densitometry signals from the western blot analyses for the sample groups. #, significant differences in signals between NASH and normal liver tissues or microsomes (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
It appeared that levels of GRP78 and GRP94 in the NAFL tissues were between those in NASH and normal tissues. The microsomal levels of the two chaperones in NAFL tissues were as low as in NASH microsomes.
The NASH tissues and their microsomes also had lower calnexin levels, as compared with normal tissues and their microsomes, respectively (P < 0.05; Figure 2A and 2B). Calnexin levels in the NAFL tissues appeared to be between those in normal and NASH tissues. Levels of calnexin in NAFL microsomes also tended to be between those in normal and NASH microsomes.
Dysregulated expression of protein foldases in NASH tissues
Levels of some enzymes related to protein folding were lower in NASH than in normal tissues. There were lower levels of ER foldases such as PDI, ERp44 and ERp72 in NASH tissues and their microsomes, as compared with in normal tissues and their microsomes, respectively (P < 0.05; Figure 3). It appeared that levels of the three foldases in NAFL tissues and microsomes were between those in normal and NASH samples. However, Ero1-Lα levels were not different among the groups, in either tissues or their microsomes.
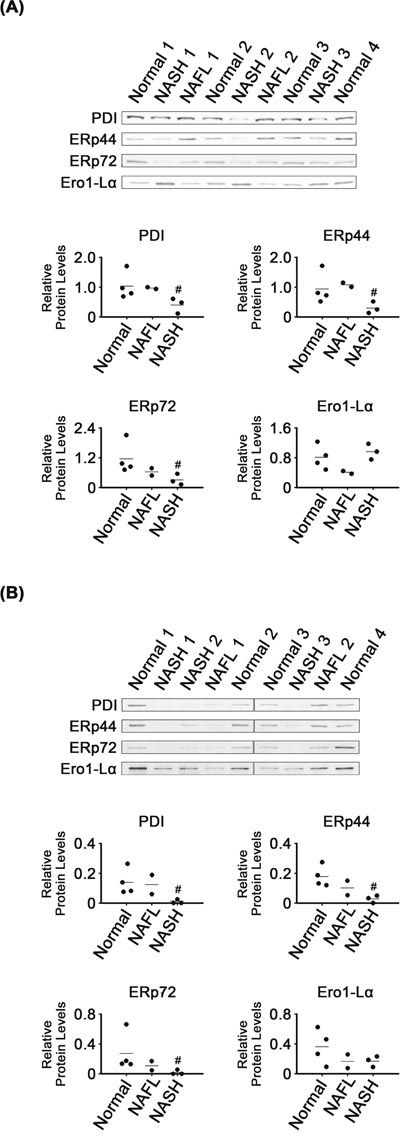
Figure 3: Expression of ER-related foldases in NASH, NAFL and normal liver tissues (A) and in their microsomes (B). β-Actin was used as an internal control for the whole tissue samples, as described for Figure 1. Horizontal lines represent means of densitometry signals from the western blot analyses. #, significant differences in signals between NASH and normal liver tissues or microsomes (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
Reduced expression of apoptosis markers in NASH tissues
Cytosolic cytochrome c levels were lower in NASH than in normal tissues (P < 0.05; Figure 4A), whereas there were no clear differences in cytochrome c levels in mitochondria from these two tissue groups. There were also no significant differences in cleaved caspase-3 levels in normal and NASH tissues (Figure 4B). However, cleaved PARP levels were lower in NASH than in normal tissues (P < 0.05). It appeared that cleaved PARP levels in NAFL tissues were as high as in the normal tissues.
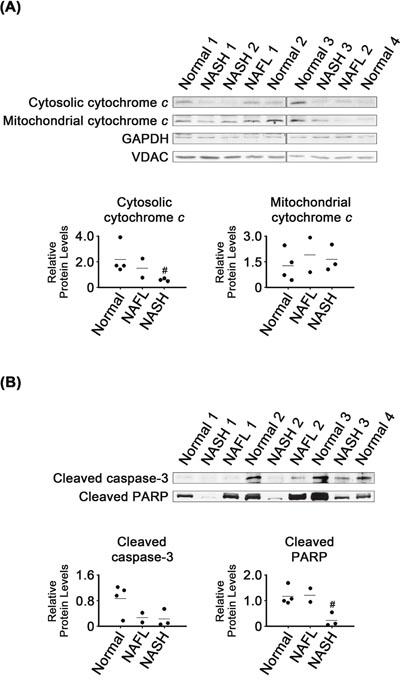
Figure 4: Expression of apoptosis marker proteins in NASH, NAFL and normal liver tissues (A and B). Glyceraldeyde-3-phosphate dehydrogease (GAPDH) and voltage-dependent anion-selective channel (VDAC) were used as internal controls for cytosolic and mitochondrial samples, respectively (A). β-Actin was used as an internal control for the whole tissue samples (B), as described for Figure 1. Horizontal lines represent means of densitometry signals from western blot analyses. #, significant differences in signals between NASH and normal liver tissues (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
Dysregulated expression of Bcl-2 family proteins in NASH tissues
Bcl-2 levels were much higher in NASH than in normal liver tissues (P < 0.05; Figure 5A). Bcl-2 levels in NAFL were as low as in the normal tissues. NASH mitochondria also had higher Bcl-2 levels than those of normal (P < 0.05) and NAFL tissues (Figure 5B). Bim levels were lower in NASH tissues and their mitochondria than in the corresponding normal samples (P < 0.05). It appeared that expression of the two CHOP-regulated Bcl-2 family proteins was regulated, in NASH tissues, in a manner promoting cell survival.
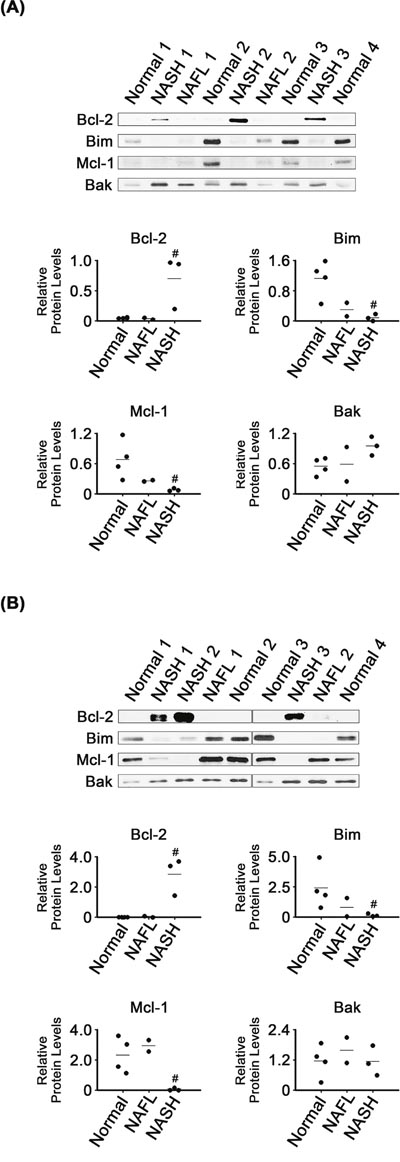
Figure 5: Expression of Bcl-2 family proteins in NASH, NAFL and normal liver tissues (A) and in their mitochondria (B). β-Actin and VDAC were used as internal controls for the whole tissue and mitochondria samples, as described for Figures 1 and 4, respectively. Horizontal lines represent means of densitometry signals from the western blot analyses. #, significant differences in signals between NASH and normal liver tissues (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
In contrast to the enhanced expression of Bcl-2, that of Mcl-1, another anti-apoptotic protein, was decreased in NASH tissues and their mitochondria, as compared with in corresponding normal samples (P < 0.05). However, it was unclear whether Bak levels were different in NASH and normal tissues or mitochondria.
Dysregulated expression of autophagy-related proteins
Expression of autophagy-related proteins was also analyzed (Figure 6). The NASH tissues had higher levels of Atg16L1 and LC3A/B-II than the normal tissues (P < 0.05). It appeared that LC3A/B-II levels in NAFL tissues were between those in NASH and normal tissues. However, there were no clear differences in levels of Beclin1, Atg5-Atg12 conjugate or LC3A/B-I among all tissue groups analyzed. In addition, it appeared that levels of p62 were not different among all tissue groups.
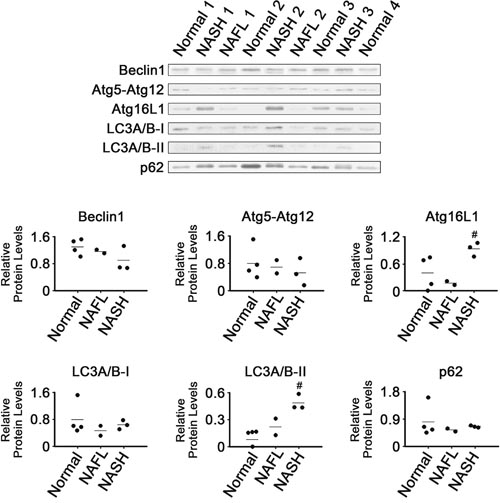
Figure 6: Expression of proteins related to autophagy in NASH, NAFL and normal liver tissues. β-Actin was used as internal control, as described for Figure 1. Horizontal lines represent means of densitometry signals from western blot analyses. #, significant differences in signals between NASH and normal liver tissues (P < 0.05). Data for NAFL tissues were not used for statistical comparisons because of limited sample number (n = 2).
DISCUSSION
XBP1s and cleaved ATF6 are among the major UPR transducers [31]. Thus, the enhanced expression of XBP1s and cleaved ATF6 in the NASH tissues (Figure 1) suggests that ER stress could be sensed within inflammatory tissues, resulting in UPR activation to restore cellular homeostasis. Induction of XBP1s and cleaved ATF6 was also observed in mouse models of NASH [32, 33].
Considering that GRP78 expression was increased in mouse NASH tissues [32, 34] as well as in hepatoma cells treated with palmitate [35], our observation of decreased GRP78 in NASH tissues (Figure 2) was unexpected. It was reported that the expression level of GRP78 mRNA was lower in human NAFL and NASH tissues than in normal liver tissues [27]. Notably, GRP78 downregulation occurred in tissues where expression of XBP1s and cleaved ATF6, two transcriptional regulators for GRP78, were induced (Figure 1) [36–38]. Decreased expression of GRP78 was reported in liver tissues of obese db/db mice, in which expression of cleaved ATF6 was enhanced [35].
Inhibition of GRP78 expression may cause fat accumulation in livers of mice. These mice exhibited increased GRP94 levels, PDI, CHOP, XBP1s and cleaved ATF6 [39]. GRP78 overexpression in the livers of obese ob/ob mice decreased hepatic TG and cholesterol content as well as hepatic expression of XBP1s and ATF6 [40]. Based on these findings, we assumed that decreased GRP78 levels (Figure 2) contributed to the disturbances related to NAFLD and induction of ER stress in liver tissues.
GRP94 depletion did not induce ER stress in the mouse liver, but led to high plasma low-density lipoprotein cholesterol levels [41] as well as to hyperproliferation of mouse liver progenitor cells [42]. In light of these findings, it is possible that GRP94 downregulation in NASH tissues (Figure 2) is involved in dysfunctional lipid metabolism as well as NASH-related proliferative diseases.
Like those of GRP78, GRP94 and calnexin, levels of the ER-associated protein foldases, PDI, ERp44 and ERp72, were also decreased in NASH tissues (Figure 3). Downregulation of the three foldases was unexpected, based on reports that XBP1s could regulate activation of ERp44 and PDI and that ATF6 could, similarly, activate ERp72 [5, 36, 43].
It was intriguing that levels of most of the ER-related chaperones and foldases analyzed in our study were decreased in NASH tissues (Figures 2 and 3), despite activation of the UPR transducers (Figure 1). Because chaperones and foldases are considered to directly execute protein quality control in the ER, it is possible that capacity for protein quality control was broadly compromised in the ER of NASH tissues.
When ER stress cannot be reversed, the UPR can trigger different pathways leading to cell death, such as apoptosis [44]. However, in our study, levels of two apoptosis markers, cytosolic cytochrome c and cleaved PARP, were decreased in NASH tissues (Figure 4), suggesting that apoptotic processes were less active than in normal liver. Apoptosis inhibition does not transform cells. However, when it is combined with activation of growth stimulatory signals, cancers can develop [45].
Activated CHOP can cause changes in gene expression favoring apoptosis, including increased Bim and decreased Bcl-2 expression [9, 46]. In contrast to those previously reported observations, CHOP induction (Figure 1) coincided with that of Bcl-2 as well as with inhibition of Bim expression in the NASH tissues (Figure 5). Based on their activities in apoptotic processes, expression of Bcl-2 and Bim was changed in a manner that would inhibit apoptosis in the NASH tissues. However, expression of another anti-apoptotic regulator, Mcl-1, was changed in a manner that would promote apoptosis. Thus, poorly controlled regulation of Bcl-2 family protein expression may contribute to dysregulation of apoptotic processes in NASH tissues.
Autophagy is a pathway mediating cell survival, although it can also promote cell death under certain conditions. Decreased autophagic function was reported to promote the initial development of NAFLD [47]. The observation that levels of p62 were unchanged suggests that the autophagy process was not activated in the NASH tissues (Figure 6), even though two major autophagy regulators, Atg16L1 and LC3, were induced.
NAFL may represent an intermediate state leading to NASH, the most extreme form of NAFLD. Five to twenty percent of patients with NAFL progress to NASH [48]. Levels of most of the proteins analyzed in NAFL tissues were either similar to those in one of the other two types, NASH and normal, or were somewhere in between (Figures 1–6). That is, NAFL tissues showed no unique expression patterns of any of the proteins analyzed, as compared with NASH and normal liver tissues.
Taken together, our findings suggest that many proteins related to UPR, apoptosis and autophagy were dysregulated in the NASH tissues. Some of the proteins were dysregulated in NASH tissues in a manner consistent with inhibition of UPR and apoptosis processes. Inhibition of UPR and apoptosis can cause prolonged accumulation of cellular stresses that may, in turn, result in cell transformation. This is interesting to consider because NASH is one of the risk factors for hepatocellular carcinoma. Future studies are warranted to determine tissue environmental factors and signaling pathways that regulate expression of proteins related to UPR and apoptosis in NAFLD tissues.
MATERIALS AND METHODS
Chemicals
All chemicals used in this study were of reagent grade or higher and were purchased from Sigma-Aldrich (St. Louis, MO, USA), unless otherwise specified.
Liver tissues
Frozen human liver tissues were obtained from the National Institutes of Health-funded Liver Tissue Cell Distribution System at the University of Minnesota, Virginia Commonwealth University and the University of Pittsburgh [49]. Patient IDs for the liver tissues used are listed in Supplementary Table 1. Clinical, histopathological, and donor information for the tissues was described previously [49].
Tissue subcellular fractionation
Subcellular extraction of the liver tissues was performed as described by Cox and Emili [50]. Briefly, tissues were homogenized using a tight-fitting Teflon pestle in ice-cold lysis buffer containing 250 mM sucrose, 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2, 1 mM dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was centrifuged at 6000 x g for 15 min at 4°C and the pellet resuspended to obtain mitochondrial proteins solubilized in extraction buffer containing 20 mM Tris-HCl (pH 7.8), 0.4 M NaCl, 15% glycerol, 1 mM DTT, 1 mM PMSF and 1.5% Triton-X-100. The supernatant was centrifuged at 100,000 x g for 1 h at 4°C, and the pellet resuspended to extract microsomal proteins into the extraction buffer. Protein concentrations were determined with a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA).
Western blot analysis
Sample preparation and western blotting were performed as previously described [51]. Anti-Atg5-Atg12 complex (#4180), -Atg16L1 (#8089), -Bak (#12105), -Beclin1 (#3495), -Bim (#2933), -Calnexin (#2679), -CHOP (#2895), -cleaved PARP (#9541), -cytochrome c (#4280), -Ero1-Lα (#3264), -ERp44 (#3798), -ERp72 (#5033), -GRP94 (#2104), -LC3A/B-I/II (#12741), -Mcl-1 (#5453), -PDI (#3501) and -VDAC (#4661) antibodies were from Cell Signaling Technology (Beverly, MA, USA). Anti-β-actin (ab8226), -Bcl-2 (ab692), -cleaved caspase-3 (ab2302), -GAPDH (ab9485), -GRP78 (ab21685) and -p62 (ab56416) antibodies were from Abcam (Cambridge, MA, USA). Anti-XBP1s (sc-7160) antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-cleaved ATF6 (NBP1-40256) antibody was from Novus Biologicals (Littleton, CO). Horseradish peroxidase-conjugated goat anti-mouse and anti-rabbit immunoglobulin G secondary antibodies were from GenDEPOT (Barker, TX, USA). The blots were developed using a chemiluminescent detection kit (Ab Frontier, Seoul, Republic of Korea). Densitometric quantification of western blot bands was performed using Image J software, version 1.49 (http://rsb.info.nih.gov/ij/index.html).
Statistics
The nonparametric Mann-Whitney U test was performed to compare specific protein abundances in NASH and normal liver tissue groups (SAS 9.13 statistical program, SAS Institute, Cary, NC, USA). A P-value of < 0.05 was considered significant.
Abbreviations
ATF6, activating transcription factor 6; Atg5, autophagy protein 5; Atg12, autophagy protein 12; Atg16L1, autophagy protein 16L1; Bak, bcl-2 homologous antagonist/killer; Bcl-2, B-cell lymphoma-2; Bim, bcl-2-like protein 11; CHOP, C/EBP homologous protein; DTT, dithiothreitol; ER, endoplasmic reticulum; Ero1, ER oxireductin 1; ERp44, ER protein 44; ERp72, ER protein 72; GAPDH, glyceraldeyde-3-phosphate dehydrogease; GRP78, glucose-regulated protein 78; GRP94, glucose-regulated protein 94; IRE1, inositol requiring enzyme 1; LC3A/B, microtubule-associated proteins 1A/1B light chain 3A; Mcl-1, myeloid cell leukemia-1; NAFL, nonalcoholic fatty liver; NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; PARP, poly (ADP-ribose) polymerase; PDI, protein disulfide isomerase; PERK, protein kinase PMSF, phenylmethylsulfonyl fluoride; RNA-like ER kinase; UPR, unfolded protein response; VDAC, voltage-dependent anion-selective channel; XBP1, X-box–binding protein 1.
Author contributions
SL and DYR designed the experiments. SL and SK performed the experiments. SL, SH, CNJ, and DYR analyzed the experimental data. SL, CNJ, and DYR wrote the manuscript.
ACKNOWLEDGMENTS
The Liver Tissue Cell Distribution System was supported by the National Institutes of Health [Contract N01-DK-7-0004/HHSN267200700004C]. We appreciate the assistance from the KOBIC Research Support Program.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FINANCIAL SUPPORT
This research was supported by Research Institute for Veterinary Science, Seoul National University and by the National Institutes of Health [Grants ES006694 and HD062489].
REFERENCES
1. Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012; 55:2005-2023.
2. Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010; 51:1972-1978.
3. Younossi ZM, Otgonsuren M, Henry L, Venkatesan C, Mishra A, Erario M, Hunt S. Association of nonalcoholic fatty liver disease (NAFLD) with hepatocellular carcinoma (HCC) in the United States from 2004 to 2009. Hepatology. 2015; 62:1723-1730.
4. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007; 8:519-529.
5. Wang S, Chen Z, Lam V, Han J, Hassler J, Finck BN, Davidson NO, Kaufman RJ. IRE1α-XBP1s induces PDI expression to increase MTP activity for hepatic VLDL assembly and lipid homeostasis. Cell Metab. 2012; 16:473-486.
6. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011; 13:184-190.
7. van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, Marciniak SJ, Goodall JC, Green AR, et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014; 510:268-272.
8. McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001; 21:1249-1259.
9. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm- Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007; 129:1337-1349.
10. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003; 23:7448-7459.
11. Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007; 87:1377-1408.
12. Coe H, Bedard K, Groenendyk J, Jung J, Michalak M. Endoplasmic reticulum stress in the absence of calnexin. Cell Stress Chaperones. 2008; 13:497-507.
13. Rutkevich LA, Williams DB. Participation of lectin chaperones and thiol oxidoreductases in protein folding within the endoplasmic reticulum. Curr Opin Cell Biol. 2011; 23:157-166.
14. Määttänen P, Gehring K, Bergeron JJ, Thomas DY. Protein quality control in the ER: the recognition of misfolded proteins. Semin Cell Dev Biol. 2010; 21:500-511.
15. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003; 11:619-633.
16. Sevier CS, Kaiser CA. Ero1 and redox homeostasis in the endoplasmic reticulum. Biochim Biophys Acta. 2008; 1783:549-556.
17. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9:47-59.
18. Tewari M, Quan LT, O'Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995; 81:801-809.
19. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010; 12:213-223.
20. Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008; 19:5360-5372.
21. Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008; 19:2092-2100.
22. Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007; 131:1149-1163.
23. Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, Sanyal AJ. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008; 134:568-576.
24. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010; 140:900-917.
25. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. 2012; 56:900-907.
26. Pagliassotti MJ. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu Rev Nutr. 2012; 32:17-33.
27. Lake AD, Novak P, Hardwick RN, Flores-Keown B, Zhao F, Klimecki WT, Cherrington NJ. The adaptive endoplasmic reticulum stress response to lipotoxicity in progressive human nonalcoholic fatty liver disease. Toxicol Sci. 2014; 137:26-35.
28. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014; 5:112.
29. Cao SS, Kaufman RJ. Targeting endoplasmic reticulum stress in metabolic disease. Expert Opin Ther Targets. 2013; 17:437-448.
30. Hou NS, Gutschmidt A, Choi DY, Pather K, Shi X, Watts JL, Hoppe T, Taubert S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc Natl Acad Sci U S A. 2014; 111:E2271-E2280.
31. Jiang S, Yan C, Fang QC, Shao ML, Zhang YL, Liu Y, Deng YP, Shan B, Liu JQ, Li HT, Yang L, Zhou J, Dai Z, et al. Fibroblast growth factor 21 is regulated by the IRE1α-XBP1 branch of the unfolded protein response and counteracts endoplasmic reticulum stress-induced hepatic steatosis. J Biol Chem. 2014; 289:29751-29765.
32. Rinella ME, Siddiqui MS, Gardikiotes K, Gottstein J, Elias M, Green RM. Dysregulation of the unfolded protein response in db/db mice with diet-induced steatohepatitis. Hepatology. 2011; 54:1600-1609.
33. Jung YA, Choi YK, Jung GS, Seo HY, Kim HS, Jang BK, Kim JG, Lee IK, Kim MK, Park KG. Sitagliptin attenuates methionine/choline-deficient diet-induced steatohepatitis. Diabetes Res Clin Pract. 2014; 105:47-57.
34. González-Rodríguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J, Lo Iacono O, Corazzari M, Fimia GM, Piacentini M, Muntané J, Boscá L, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014; 5:e1179.
35. Yamagishi N, Ueda T, Mori A, Saito Y, Hatayama T. Decreased expression of endoplasmic reticulum chaperone GRP78 in liver of diabetic mice. Biochem Biophys Res Commun. 2012; 417:364-370.
36. Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998; 273:33741-33749.
37. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003; 23:7448-7459.
38. Ashraf NU, Sheikh TA. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic Res. 2015; 49:1405-1418.
39. Ji C, Kaplowitz N, Lau MY, Kao E, Petrovic LM, Lee AS. Liver-specific loss of glucose-regulated protein 78 perturbs the unfolded protein response and exacerbates a spectrum of liver diseases in mice. Hepatology. 2011; 54:229-239.
40. Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferré P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009; 119:1201-1215.
41. Poirier S, Mamarbachi M, Chen WT, Lee AS, Mayer G. GRP94 regulates circulating cholesterol levels through blockade of PCSK9-induced LDLR degradation. Cell Rep. 2015; 13:2064-2071.
42. Chen WT, Tseng CC, Pfaffenbach K, Kanel G, Luo B, Stiles BL, Lee AS. Liver-specific knockout of GRP94 in mice disrupts cell adhesion, activates liver progenitor cells, and accelerates liver tumorigenesis. Hepatology. 2014; 59:947-957.
43. Sha H, Yang L, Liu M, Xia S, Liu Y, Liu F, Kersten S, Qi L. Adipocyte spliced form of X-box-binding protein 1 promotes adiponectin multimerization and systemic glucose homeostasis. Diabetes. 2014; 63:867-879.
44. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013; 1833:3460-3470.
45. Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis. 2005; 26:263-270.
46. Yamaguchi H, Wang HG. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem. 2004; 279:45495-45502.
47. Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011; 5:159-166.
48. Weiß J, Rau M, Geier A. Non-alcoholic fatty liver disease: epidemiology, clinical course, investigation, and treatment. Dtsch Arztebl Int. 2014; 111:447-452.
49. Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, Cherrington NJ. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009; 37:2087-2094.
50. Cox B, Emili A. Tissue subcellular fractionation and protein extraction for use in mass-spectrometry-based proteomics. Nat Protoc. 2006; 1:1872-1878.
51. Kim S, Lee SH, Lee S, Park JD, Ryu DY. Arsenite-induced changes in hepatic protein abundance in cynomolgus monkeys (Macaca fascicularis). Proteomics. 2014; 14:1833-1843.