
INTRODUCTION
Microtia is a congenital developmental malformation of the external ear that ranges in severity from mild differences in auricular shape and size to complete absence of the external ear with atresia or stenosis of the auditory canal [1,2]. Prevalence rates of this uncommon anomaly have been reported to vary from 0.83 to 4.34 per 10,000 [3]. It can occur unilaterally (79%-93%) and bilaterally [2–4], and approximately 60% of unilateral microtia are seen on the right side [5,6]. Microtia is divided into four grades, according to the Marx classification system, widely used to describe the degree of microtia. In grade I microtia, all of the outer ear structures are normal, but the pinna is small. This can occur with or without aural atresia. In grade II, auricle is abnormal, but definable. Grade III displays only soft tissue rudiments, with no definable structures of the auricle. Grade IV is the extreme case in which the external ear and auditory canal are absent, a condition called anotia [7].
Microtia can occur as an isolated birth defect or as part of a spectrum of anomalies or a syndrome. The most common associated malformations are cleft lip and/or palate, limb reduction defects, renal anomalies, vertebral abnormalities, and cardiac defects [3,4,8]. Although differences in the development of the external/middle ear and inner ear during embryogenesis mean that external and middle ear malformations can occur without inner ear malformations and vice versa, malformations in microtia can affect the external ear, middle ear, and/or inner ear in combination [3,4,8]. Swartz and Faerber reported inner ear malformation frequencies of 11% to 30% in individuals with outer and middle ear malformations [9].
In addition, many syndromic microtia patients present with inner ear malformations, including branchio-oculofacial, branchio-oto-renal/branchio-otic, CHARGE (coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities, ear anomalies), Nager (preaxial acrofacial dysostosis), Treacher Collins, Wildervanck (cervico-oculo-acoustic), LAMM (congenital deafness, inner ear agenesis, microtia, microdontia) and lacrimo-auriculodento-digital (LADD) syndromes, among others. The frequent malformations of the inner ear are common cavity, cochlear aplasia, Mondini defect, enlarged vestibular aqueduct, abnormal/absent semicircular canals, and bulbous IAC (internal auditory canal) [7]. Conductive hearing loss, sensorineural hearing loss, and mixed hearing loss can all be found in microtia patients [7]. Given these observations, we propose that malformations of the external/middle ear and inner ear may reflect the actions of certain genes in common.
To date, no studies have reported screens of deafness genes in microtia patients. The technology of targeted genomic capturing combined with next-generation sequencing (NGS) has proven to be effective and feasible and has been successfully used in several studies [10,11]. Here, we utilized this approach to screen for mutations in 307 targeted genes in 32 isolated unilateral microtia patients using a customized capture array that includes all known deafness genes, selected from human and animal experimental studies (Supplementary Table 1). We attempted to identify mutations or genes that may be associated with isolated unilateral microtia and the signaling pathways that may be responsible for this disease.
RESULTS
Variant identification
A total of 4899 exons comprising 2,362,343 bases (1,441,253 in target regions and 921,090 in flanking regions) were captured and sequenced in our study. After alignment to the reference human genome (NCBI37/hg19), 70.93% of the clean reads could be matched to the targeted regions. The average read depth for the targeted regions was 394.5-fold, and 97.68% of the targeted regions were covered by 20 or more reads, demonstrating the high quality of the sequencing. A detailed coverage analysis for each sample is provided in Supplementary Figure 1.
Identification of rare or novel mutations
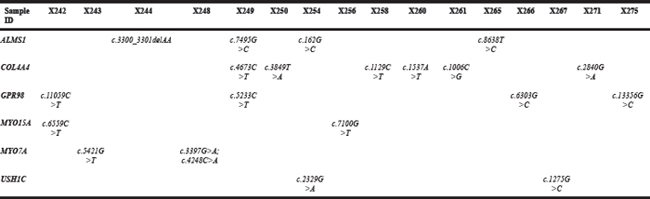
Using a rigorous filtering pipeline, we identified 42 very rare heterozygous mutations, including 22 novel mutations in 25 genes from 24 isolated unilateral microtia patients (Table 1). A breakdown of the 42 mutations is as follows: 36 missense mutations, 3 frameshift variants, 2 splice mutations and 1 nonsense mutation. Twenty-one of the 36 missense mutations were predicted to be pathogenic by the Condel program. The three frameshift variants were c.2081_2082insC (p.P695Tfs*71) in DIAPH1 (diaphanous-related formin 1), c.3300_3301delAA (p.K1103Afs*16) in ALMS1 (Alstrom syndrome 1), and c.6072delG (p.R2025Afs*6) in OTOG (otogelin). One mutation, c.4248C>A in MYO7A (myosin VIIA), was predicted to lead to a truncated protein owing to a premature stop codon. Mutations in ALMS1, COL4A4 (collagen type IV alpha 4 chain), GPR98 (G protein-coupled receptor 98), MYO15A, MYO7A, and USH1C (Usher syndrome 1C) genes occurred in two or more cases (Allele frequency ≥ 1.56%) (Table 2).
Table 1: Mutations identified in isolated unilateral microtia patients
Sample ID |
Gene Symbol |
Type of variation |
Mutation |
PhyloP in Vertebrates |
Condel Score |
Condel Prediction |
Novel |
|---|---|---|---|---|---|---|---|
X242 |
ESPN |
missense |
c.2171C>T, p.Pro724Leu |
1.116 |
0.836 |
deleterious |
Reported |
|
GPR98 |
missense |
c.11059C>T, p.Arg3687Cys |
3.779 |
0.69 |
deleterious |
Novel |
|
DIAPH1 |
frameshift |
c.2081_2082insC, p.Pro695Thrfs*71 |
. |
. |
. |
Reported |
|
MYO15A |
missense |
c.6559C>T, p.Arg2187Cys |
1.942 |
. |
. |
Reported |
X243 |
FGFR2 |
missense |
c.1388T>A, p.Val463Asp |
4.509 |
0.718 |
deleterious |
Reported |
|
MYO7A |
missense |
c.5421G>T, p.Glu1807Asp |
0.519 |
0.74 |
deleterious |
Novel |
X245 |
HOXA1 |
missense |
c.344A>G, p.Gln115Arg |
2.769 |
. |
. |
Novel |
X246 |
COL9A1 |
missense |
c.2636C>A, p.Pro879His |
3.371 |
0.857 |
deleterious |
Novel |
X248 |
MYO7A |
missense |
c.3397G>A, p.Gly1133Arg |
3.778 |
. |
. |
Novel |
|
MYO7A |
nonsense |
c.4248C>A, p.Tyr1416* |
2.927 |
. |
. |
Reported |
X250 |
COL4A4 |
missense |
c.3849T>A, p.Ser1283Arg |
-0.336 |
. |
. |
. Novel |
X251 |
DSPP |
missense |
c.2615A>C, p.Asp872Ala |
1.275 |
. |
. |
Reported |
X252 |
TRIOBP |
missense |
c.2777G>A, p.Arg926His |
2.956 |
0.786 |
deleterious |
Reported |
X254 |
ALMS1 |
missense |
c.162G>C, p.Glu54Asp |
0.429 |
. |
. |
. Novel |
|
USH1C |
missense |
c.2329G>A, p.Gly777Arg |
5.095 |
0.945 |
deleterious |
Reported |
|
MYO1A |
splice-5 |
c.1164+1delG |
5.634 |
. |
. |
Novel |
X256 |
MYO15A |
missense |
c.7100G>T, p.Gly2367Val |
0.015 |
. |
. |
Novel |
X260 |
COL4A4 |
missense |
c.1537A>T, p.Ser513Cys |
0.06 |
0.481 |
deleterious |
Novel |
X262 |
PCDH15 |
missense |
c.5213C>T, p.Pro1738Leu |
0.98 |
. |
. |
Novel |
X263 |
GPSM2 |
missense |
c.122G>A, p.Arg41His |
6.144 |
0.667 |
deleterious |
Reported |
X265 |
ALMS1 |
missense |
c.8638T>C, p.Cys2880Arg |
0.347 |
. |
. |
Reported |
X267 |
COL4A3 |
missense |
c.1103G>A, p.Arg368His |
0.727 |
0.74 |
deleterious |
Reported |
X267 |
USH1C |
missense |
c.1275G>C, p.Lys425Asn |
1.686 |
0.779 |
deleterious |
Reported |
X270 |
OTOG |
missense |
c.8129T>A, p.Val2710Glu |
3.867 |
0.704 |
deleterious |
Novel |
X274 |
WFS1 |
missense |
c.292G>A, p.Gly98Arg |
4.158 |
0.945 |
deleterious |
Novel |
X274 |
DIAPH3 |
missense |
c.772T>C, p.Tyr258His |
3.193 |
0.8 |
deleterious |
Novel |
X275 |
GPR98 |
missense |
c.13356G>C, p.Leu4452Phe |
0.082 |
0.75 |
deleterious |
Novel |
|
ESPN |
missense |
c.2171C>T, p.Pro724Leu |
1.116 |
0.836 |
deleterious |
Reported |
X244 |
ALMS1 |
frameshift |
c.3300_3301delAA, p.Lys1103Alafs*16 |
. |
. |
. |
Novel |
X249 |
BSND |
missense |
c.919C>T, p.Leu307Phe |
3.393 |
0.809 |
deleterious |
Reported |
|
ALMS1 |
missense |
c.7495G>C, p.Glu2499Gln |
0.375 |
. |
. |
Novel |
|
COL4A4 |
missense |
c.4673C>T, p.Ala1558Val |
2.594 |
. |
. |
Reported |
|
GPR98 |
missense |
c.5233C>T, p.Arg1745Cys |
2.647 |
0.945 |
deleterious |
Novel |
|
LOXHD1 |
missense |
c.4459G>A, p.Asp1487Asn |
5.879 |
0.849 |
deleterious |
Novel |
X258 |
COL4A4 |
missense |
c.1129C>T, p.Arg377Cys |
0.582 |
. |
. |
Reported |
|
OTOG |
frameshift |
c.6072delG, p.Arg2025Alafs*6 |
3.056 |
. |
. |
Novel |
X261 |
COL4A4 |
missense |
c.1006C>G, p.Leu336Val |
-0.162 |
. |
. |
Reported |
|
TPRN |
missense |
c.1927G>A, p.Val643Met |
0.363 |
0.676 |
deleterious |
Novel |
X266 |
*USH2A |
splice-3 |
c.8559-2A>G |
4.853 |
. |
. |
Novel |
|
GPR98 |
missense |
c.6303G>C, p.Lys2101Asn |
1.027 |
0.481 |
deleterious |
Reported |
|
PCDH15 |
missense |
c.2047C>T, p.Arg683Cys |
4.357 |
0.799 |
deleterious |
Reported |
X271 |
COL11A1 |
missense |
c.1289C>A, p.Pro430His |
5.849 |
. |
. |
Reported |
|
COL4A4 |
missense |
c.2840G>A, p.Arg947Gln |
-4.674 |
. |
. |
Reported |
*indicates the mutation has been reported in HGMD database
Table 2: Mutations identified in the same gene in two or more patients
In addition, two intronic variants were identified; one, a c.8559-2A>G mutation in USH2A, has been reported to be pathogenic in HGMD, and the other, a c.1164+1delG mutation in MYO1A, was predicted to most likely affect splicing by the Human Splicing Finder program (http://www.umd.be/HSF3/). An alignment of a selected region of the HOXA1 protein showed that amino acid 115 exhibited very high evolutionary conservation (Figure 1). A structural analysis of HOXA1 protein performed using the HOPE server suggested that the mutation (c. 344 A>G, p. Q115R), which introduces a positively charged arginine for the negatively charged glutamine in the wild-type protein, could cause repulsion of ligands or other residues with the same charge.
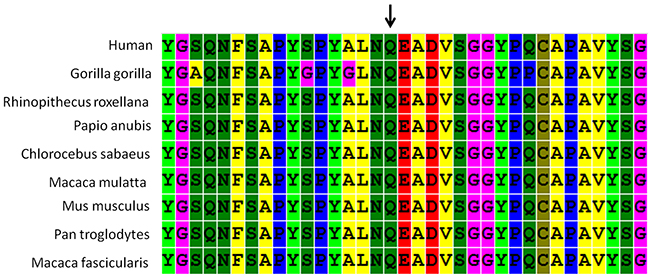
Figure 1: ConSeq conservation analysis of amino acid Q115 in the homeodomain of HOXA1. This analysis is based on an alignment of nine full-length HOXA1 proteins from different species. Different colors indicate different types of residues. The arrow shows the Q115 residue.
KEGG pathway enrichment analysis
To further determine the signaling pathways in which the 25 rare mutated genes found in microtia patients are involved, we performed a KEGG pathway enrichment analysis. This analysis revealed that five pathways are significantly associated with isolated unilateral microtia: the protein digestion and absorption pathway, the focal adhesion pathway, the extracellular matrix (ECM)-receptor interaction pathway, the amoebiasis pathway, and the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway (Figure 2). Among these pathways, two (the focal adhesion pathway and ECM-receptor interaction pathway) may play important roles in auricular development, because both have been reported to be associated with cartilage destruction and the main component of the auricle is cartilage [12]. Notably, the two pathways are connected to the downstream Wnt signaling pathway. Based on these considerations, we propose that both pathways are linked to microtia (http://www.genome.jp/kegg/pathway.html).
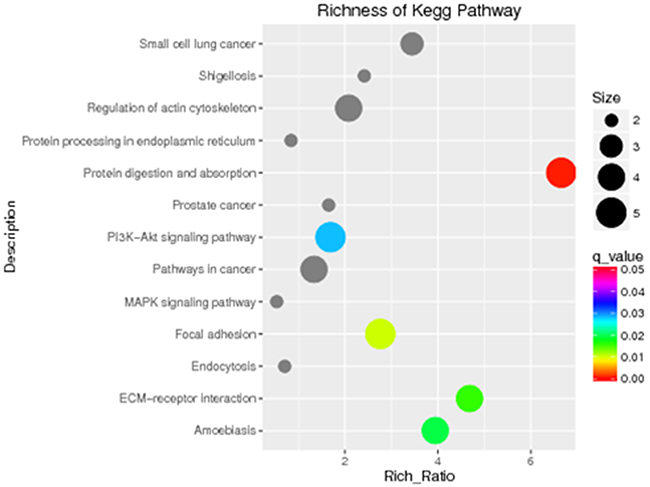
Figure 2: KEGG pathway analysis identified five signaling pathways that are significant in microtia (P < 0.05). KEGG pathway enrichments are displayed in a scatter diagram, where each point represents the enrichment level, the color corresponds to the q-value, and the size corresponds to the number of genes enriched for the given pathway. Red indicates the q-value; the smaller the value, the more significant the pathway enrichment.
Association analysis
To test whether low-frequency mutations exist that might be risk factors for microtia, we conducted an association analysis of 32 isolated unilateral microtia cases and 208 healthy controls from China. After correction for multiple testing (P = 2.51 × 10-4), MUC4 (mucin 4), MUC6, COL4A4, MYO7A, AKAP12 (A-kinase anchoring protein 12), COL11A1, DSPP (dentin sialophosphoprotein), ESPN (espin), GPR98, PCDH15 (protocadherin 15), BSN (bassoon), CACNA1D (calcium channel, voltage-dependent, L type, alpha 1D subunit), TPRN (taperin), and USH1C were identified as significantly associated with isolated unilateral microtia (Table 3, Supplementary Table 2). Of these genes, two were related to the pathways we identified above using KEGG pathway enrichment analysis. COL4A4 was related to the pathways for ECM-receptor interaction and focal adhesion, and COL11A1 was related to protein digestion and absorption (http://www.genecards.org/).
Table 3: Results of gene-based, low-frequency variant association tests (significance level P = 2.51 × 10-4)
Gene |
Markers |
P-value |
|---|---|---|
MUC4 |
141 |
3.68 × 10-17 |
MUC6 |
95 |
1.20 × 10-11 |
COL4A4 |
9 |
2.24 × 10-9 |
MYO7A |
8 |
2.02 × 10-7 |
AKAP12 |
6 |
1.82 × 10-6 |
COL11A1 |
7 |
1.82 × 10-6 |
DSPP |
3 |
1.82 × 10-6 |
ESPN |
5 |
1.82 × 10-6 |
GPR98 |
9 |
3.42 × 10-6 |
PCDH15 |
5 |
1.58 × 10-5 |
BSN |
7 |
5.97 × 10-5 |
CACNA1D |
4 |
0.000133 |
TPRN |
4 |
0.000133 |
USH1C |
4 |
0.000133 |
DISCUSSION
During early development of vertebrates, signaling between neural crest cells and other embryonic cell types (e.g., endothelia and craniofacial ectoderm) plays a critical role in driving facial outgrowth and morphogenesis, including that of the external and inner ear [13]. The external ear begins its development during the fifth week, when hillocks developed from the first and second pharyngeal arches progress over several months to form the auricle. The otic placode, also derived from second pharyngeal arch begins to form on inner ear structures in the third week of embryonic development and is completed after several weeks [13]. Hypoplasia of the first and second pharyngeal arches is the underlying cause of microtia, a congenital malformation of the external ear. Thus, we propose that external and inner ear development may be regulated by certain genes in common. To date, no studies have reported an investigation of the associations between external ear deformities and deafness genes.
In the current study, we screened all exons of 307 deafness-associated genes, identifying 42 rare or novel mutations of 25 genes in isolated unilateral microtia patients. All of these mutations add to the database of potential pathogenic mutations in isolated unilateral microtia cases. Among these muations, only one, c.8559-2A>G in USH2A, has been reported to be pathogenic; when combined with p.T3936P or p.R34fs as a compound heterozygous mutation, it results in Usher syndrome type II [15]. This information could be helpful in providing genetic counseling for patients with related conditions.
The wingless/INT (Wnts) signaling pathway, together with bone morphogenetic proteins (BMPs) and fibroblast growth factors (FGFs), have been reported to be involved in external ear development [16]. Here, through pathway enrichment analysis of the 25 genes, we identified additional five pathways—protein digestion and absorption pathway, focal adhesion pathway, ECM-receptor interaction pathway, amoebiasis pathway and PI3K-Akt signaling pathway—that may be related to microtia. Of these pathways, two (focal adhesion pathway and ECM-receptor interaction pathway) may especially play more important roles during auricular development. Because both have been reported to be associated with cartilage destruction, and the main component of the auricle is cartilage [12]. More importantly, both pathways are connected to downstream Wnt signaling pathway, and thus we propose that they linked to microtia.
In addition to exploring rare mutations that may cause microtia, we also employed associated studies to identify potential pathogenic genes among low-frequency variants. The contribution of rare and low-frequency variants to human traits has been largely unexplored [17,18]. Using the candidate gene approach for complex traits, several previous rare variant association studies (RAVS) have successfully identified novel associations [19,20]. In the current study, through the low-frequency variants association studies by SKAT method, we identified several genes significantly associated with isolated unilateral microtia, including MUC4, MUC6, COL4A4, MYO7A, AKAP12, COL11A1, DSPP, ESPN, GPR98, PCDH15, BSN, CACNA1D, TPRN, and USH1C. Of these genes, two were related to the pathways we identified using KEGG pathway enrichment analysis. COL4A4 was related to the pathways for ECM-receptor interaction and focal adhesion, and COL11A1 was related to protein digestion and absorption. Thus we suggest that the two genes, COL4A4 and COL11A1, may be strongly involved in the mechanism of microtia and are worth exploring in the context of the pathogenesis of microtia.
Additionally, a novel heterozygous mutation (c. 344A>G) in HOXA1 identified in one isolated unilateral microtia patient also attracted our interest. It resulted in substitution of an arginine for a glutamine at position 115 (Q115R), which is highly evolutionarily conserved. A structural analysis of HOXA1 protein performed using the HOPE server indicated that this mutation, which replaces a negatively charged residue in the wild-type with a positively charge amino acid, could cause repulsion of ligands or other residues with the same charge. Hox genes encode proteins that share a 60-amino-acid domain called the homeodomain. HoxA cluster genes play a fundamental role in building the sensorimotor circuitry in vertebrates. Mutations in either HOXA1 or HOXA2 have reported to result in external ear deformities, although the phenotypes caused by HOXA1 mutations are more severe than those caused by HOXA2 mutations. Diseases associated with HOXA1 mutations include Athabaskan Brainstem Dysgenesis Syndrome and Bosley-Salih-Alorainy syndrome [21,22].
A small number of studies have also focused on the HOXA2 gene as a genetic cause of isolated microtia. Identification of pathogenic mutations in HOXA2 was reported in three different isolated bilateral microtia families [23–25], prompting researchers to propose that HOXA2 may be among genes responsible for the pathogenesis of isolated microtia. However, studies that have focused on sporadic microtia patients have found no pathogenic mutations in HOXA2 [26,27]. Consistent with this, we failed to identify any mutations in HOXA2, suggesting that there may be distinct etiologies that result in isolated microtia.
In conclusion, some deafness genes may be associated with microtia, and certain genes may affect both external/middle and inner ear development. Pathway analysis showed that two pathways—the focal adhesion and ECM-receptor interaction pathways—were associated with microtia. The deafness genes panel has the potential to enable the discovery of pathogenic genes in microtia patients and can be reliably used in such patients.
MATERIALS AND METHODS
Recruitment of subjects
Thirty-two isolated unilateral microtia patients (22 males, 10 females; mean age, 9 years; range, 6–28 years) were recruited from Peking Union Medical College Hospital (PUMCH). They were all classified as grade III external ear deformities, according to the classification by the Marx classification system [7], and had atresia of the auditory canal. None of the patients exhibited other complicating deformities. Patients with syndromic microtia such as Treacher Collins syndrome, Miller syndrome, CHARGE syndrome or branchio-oto-renal (BOR) syndrome, were excluded from the study. All patients were asked to provide a detailed family history, and none had a family history of microtia.
Ethical approval for this study was obtained from the institutional review board of PUMCH. All subjects or their parents gave written, informed consent to participate in this study.
Targeted genomic capture and next-generation sequencing
Patients’ genomic DNA (gDNA) was extracted from blood samples using a Blood DNA kit (TIANGEN BIOTECH, Beijing, China), and 1 ug of purified gDNA was fragmented into 200–300-base-pair lengths using an ultrasonoscope. Libraries were prepared by performing end-repair, adenylation, and adapter ligation using a protocol provided by the manufacturer of the NGS system (HiSeq2000; Illumina).
A customized capture array (Roche-NimbleGen) containing 307 deafness genes was designed to capture all exons and flanking intron (±10 bp) sequences of the target genes (Supplementary Table 1). The same amount of each library was pooled and then hybridized to the customized capture array. Paired-end reads (PE90) were generated by sequencing using an Illumina HiSeq2000 platform, according to the manufacturer’s instructions [28].
Data filtering, mapping and variant detection
Illumina Pipeline (version 1.9.4) was used to perform image analysis, error estimation and base calling, yielding the primary data. After removing low-quality reads from the primary data using a local algorithm, data analysis and bioinformatics processing were performed based on the reference sequences of the NCBI37/hg19 assembly of the human genome using the Burrows-Wheeler alignment tool. Single nucleotide variations (SNVs) and small insertions and deletions (Indels) were detected using GATK (Genome Analysis Toolkit), and in-house scripts were used for annotation of variants.
Identification of rare or novel variants
To identify the most likely pathogenic mutations, we filtered out 1) synonymous and non-coding variants (with the exception of splicing site mutations that might create an ectopic splicing site); 2) variants with allele frequencies of 0.0005 or higher in the dbSNP, 1000 Genomes Project, ESP6500 or ExAC database; and 3) missense variants that were predicted to be neutral by the Condel program, which assesses the functional effects of non-synonymous variants using a CONsensus DELeteriousness (Condel) score that combines various tools (SIFT, Polyphen2) [29]. The strength of ectopic splicing sites created by two intronic variants was evaluated by the Human Splicing Finder program (http://www.umd.be/HSF/). The HOPE server (http://www.cmbi.ru.nl/hope/) was used to analyze and predict structural variations in mutant HOXA1 (homeobox A1) protein and the ConSeq server was used for determining the conservation of HOXA1 amino acid position 115. Additionally, all rare and novel mutations were checked against the Human Gene Mutation Database (HGMD) to determine whether any identified mutations had been reported to be pathogenic.
Principle of KEGG pathway enrichment
The number of potential pathogenic genes and all genes in each pathway were determined, and then the number of all potential pathogenic genes and all genes in all pathways were determined separately. Ultimately, a hypergeometric test was applied to determine the statistical significance of the enrichment pathway (P), calculated according to the following formula:
where N represents the gene count in all pathways, n represents the z potential pathogenic gene count in N, M represents the gene count in each pathway, and m represents the potential pathogenic gene count in each pathway. Pathways with a q-value < 0.05, determined from the p-value through multiple comparisons, were considered to be significantly enriched.
Association analysis of low-frequency variants
To test whether low-frequency mutations (MAF < 0.01 in the 1000 Genomes Project) that might be risk factors for microtia, we performed a gene-based test comparing the burden of low-frequency variants in cases and controls using a sequence kernel association test (SKAT) implemented in the SKAT software package. Only nonsynonymous variants were included in the analysis. Genotype data for the Han Chinese in Beijing (CHB) and Southern Han Chinese (CHS) populations were extracted from the 1000 Genomes Project. Finally, all 32 microtia cases and 208 normal controls from the 1000 Genome Project were genotyped for the selected variants. A Bonferonni correction was used to account for multiple testing (P = 2.51 × 10-4).
Abbreviations
NGS: Next generation sequencing
PTA: Pure-tone audiometry
AC: Air conducted
AF: Allele Frequency
KEGG: Kyoto Encyclopedia of Genes and Genomes
Author contributions
XWC conceived and designed the experiments. XMF and YBW ascertained the probands and collected the DNA samples. PW and YF analyzed the data and wrote the paper. XWC, YPL and SYZ revised it critically for important intellectual content. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
This work was supported by grant to Xiaowei Chen from the General Programs of National Natural Science Foundation of China (No. 81271053) and Shuyang Zhang from the National Key Research and Development Program of China (No. 2016YFC0901501). We are grateful to Prof. Dengge Ma for reviewing the English script of the paper at Beijing International Studies University.
CONFLICTS OF INTEREST
The authors report no conflicts of interest and have received no payment in preparation of this manuscript. The authors alone are responsible for the content and writing of this paper.
REFERENCES
1. Okajima H, Takeichi Y, Umeda K, Baba S. Clinical analysis of 592 patients with microtia. Acta Otolaryngol Suppl. 1996; 525:18-24.
2. Suutarla S, Rautio J, Ritvanen A, Ala-Mello S, Jero J, Klockars T. Microtia in Finland: comparison of characteristics in different populations. Int J Pediatr Otorhinolaryngol. 2007; 71:1211-1217.
3. Cox TC, Camci ED, Vora S, Luquetti DV, Turner EE. The genetics of auricular development and malformation: new findings in model systems driving future directions for microtia research. Eur J Med Genet. 2014; 57:394-401.
4. Llano-Rivas I, Gonzalez-del AA, Del CV, Reyes R, Carnevale A. Microtia: a clinical and genetic study at the National Institute of Pediatrics in Mexico City. Arch Med Res. 1999; 30:120-124.
5. Forrester MB, Merz RD. Descriptive epidemiology of anotia and microtia, Hawaii, 1986-2002. Congenit Anom (Kyoto). 2005; 45:119-124.
6. Shaw GM, Carmichael SL, Kaidarova Z, Harris JA. Epidemiologic characteristics of anotia and microtia in California, 1989-1997. Birth Defects Res A Clin Mol Teratol. 2004; 70:472-475.
7. Bartel-Friedrich S. Congenital auricular malformations: description of anomalies and syndromes. Facial Plast Surg. 2015; 31:567-580.
8. Sanchez O, Mendez JR, Gomez E, Guerra D. [Clinico-epidemiologic study of microtia]. [Article in Spanish]. Invest Clin. 1997; 38:203-217.
9. Swartz JD, Faerber EN. Congenital malformations of the external and middle ear: high-resolution CT findings of surgical import. AJR Am J Roentgenol. 1985; 144:501-506.
10. Prasad MK, Geoffroy V, Vicaire S, Jost B, Dumas M, Le Gras S, Switala M, Gasse B, Laugel-Haushalter V, Paschaki M, Leheup B, Droz D, Dalstein A, et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J Med Genet. 2016; 53:98-110.
11. Lim EC, Brett M, Lai AH, Lee SP, Tan ES, Jamuar SS, Ng IS, Tan EC. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum Genomics. 2015; 9:33.
12. Wang W, Liu Y, Hao J, Zheng S, Wen Y, Xiao X, He A, Fan Q, Zhang F, Liu R. Comparative analysis of gene expression profiles of hip articular cartilage between non-traumatic necrosis and osteoarthritis. Gene. 2016; 591:43-47.
13. Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005; 207:575-601.
14. Sai X, Ladher RK. Early steps in inner ear development: induction and morphogenesis of the otic placode. Front Pharmacol. 2015; 6:19.
15. Dai H, Zhang X, Zhao X, Deng T, Dong B, Wang J, Li Y. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008; 14:2067-2075.
16. Luquetti DV, Heike CL, Hing AV, Cunningham ML, Cox TC. Microtia: epidemiology and genetics. Am J Med Genet A. 2012; 158A:124-139.
17. Tada H, Kawashiri MA, Konno T, Yamagishi M, Hayashi K. Common and rare variant association study for plasma lipids and coronary artery disease. J Atheroscler Thromb. 2016; 23:241-256.
18. Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, Daly MJ, Neale BM, Sunyaev SR, Lander ES. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014; 111:E455-E464.
19. Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004; 305:869-872.
20. Johansen CT, Wang J, McIntyre AD, Martins RA, Ban MR, Lanktree MB, Huff MW, Peterfy M, Mehrabian M, Lusis AJ, Kathiresan S, Anand SS, Yusuf S, et al. Excess of rare variants in non-genome-wide association study candidate genes in patients with hypertriglyceridemia. Circ Cardiovasc Genet. 2012; 5:66-72.
21. Higley MJ, Walkiewicz TW, Miller JH, Curran JG, Towbin RB. Bilateral complete labyrinthine aplasia with bilateral internal carotid artery aplasia, developmental delay, and gaze abnormalities: a presumptive case of a rare HOXA1 mutation syndrome. AJNR Am J Neuroradiol. 2011; 32:E23-E25.
22. Bosley TM, Salih MA, Alorainy IA, Oystreck DT, Nester M, Abu-Amero KK, Tischfield MA, Engle EC. Clinical characterization of the HOXA1 syndrome BSAS variant. Neurology. 2007; 69:1245-1253.
23. Alasti F, Sadeghi A, Sanati MH, Farhadi M, Stollar E, Somers T, Van Camp G. A mutation in HOXA2 is responsible for autosomal-recessive microtia in an Iranian family. Am J Hum Genet. 2008; 82:982-991.
24. Brown KK, Viana LM, Helwig CC, Artunduaga MA, Quintanilla-Dieck L, Jarrin P, Osorno G, McDonough B, DePalma SR, Eavey RD, Seidman JG, Seidman CE. HOXA2 haploinsufficiency in dominant bilateral microtia and hearing loss. Hum Mutat. 2013; 34:1347-1351.
25. Piceci F, Morlino S, Castori M, Buffone E, De Luca A, Grammatico P, Guida V. Identification of a second HOXA2 nonsense mutation in a family with autosomal dominant non-syndromic microtia and distinctive ear morphology. Clin Genet. 2016.
26. Monks DC, Jahangir A, Shanske AL, Samanich J, Morrow BE, Babcock M. Mutational analysis of HOXA2 and SIX2 in a Bronx population with isolated microtia. Int J Pediatr Otorhinolaryngol. 2010; 74:878-882.
27. Hao S, Jin L, Li C, Wang H, Zheng F, Ma D, Zhang T. Mutational analysis of GSC, HOXA2 and PRKRA in 106 Chinese patients with microtia. Int J Pediatr Otorhinolaryngol. 2017; 93:78-82.
28. Xie S, Lan Z, Qu N, Wei X, Yu P, Zhu Q, Yang G, Wang J, Shi Q, Wang W, Yang L, Yi X. Detection of truncated dystrophin lacking the C-terminal domain in a Chinese pedigree by next-generation sequencing. Gene. 2012; 499:139-142.
29. Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011; 88:440-449.