
INTRODUCTION
The prognosis for most patients with gastric cancer (GC) is poor and has improved little over the past several decades [1]. Currently, GC prognosis is based on pathology (i.e., histological type, invasion and metastasis), radiological imaging (for staging) and other clinical factors (age and comorbidity). However, these traditional clinicopathological factors have significant limitations, and major efforts are therefore being made to develop molecular signature-based methods to complement the traditional histopathological methods for diagnosis, classification and prognosis in GC [2-5]. Unfortunately, the current molecular biomarkers of clinical GC (e.g., p27, cyclin E, E-cadherin, HER2, c-Myc and p53) lack the sensitivity and specificity required for screening an asymptomatic population for the purpose of early detection [6-8].
With the development of microarray technologies, it is not surprising that the majority of molecular signatures for GC have been derived from gene expression microarray studies [9-13]. Several studies have identified genetic markers or gene expression profiles capable of distinguishing normal from malignant gastric growth, and these markers can aid in the prognosis and estimation of survival rates for GC patients. However, these signatures often contain large numbers of genes, which reduces their application in daily clinical practice. Alternatively, other mainstream high-throughput proteomic profiling techniques include gel-based methods, gel-free mass spectrometry (MS)-based methods and surface-enhanced laser desorption/ionization (SELDI) time-of-flight (TOF) MS, and the application of these techniques in tissue-based clinical studies has the potential to provide efficient biomarkers for GC [14-20]. However, the requirement of sophisticated devices greatly limits their broad application in routine clinical practice. Moreover, technological limitations due to patient-to-patient variability and loss of signal from low-abundance proteins have negatively impacted the field of disease proteomics focused on separation techniques coupled with MS-based protein identification.
Routine proteomic testing in patients at risk for GC is not practical or realistic on a large scale. Therefore, other simple and inexpensive tests will be necessary for such purposes. The use of capture reagents such as antibodies in affinity proteomics has emerged as a new tool capable of gathering information on the global level in a high-throughput format using multiple versions of affinity reagents (e.g., full-length antibodies, aptamers, affibody molecules, single-chain variable fragments of antibodies) and various capture formats (e.g., planar arrays, beads, antibodies in the array format) [21,22]. Moreover, antibody microarray technology has rapidly evolved from proof-of-concept to state-of-the-art technology capable of targeting complex, nonfractionated protein samples. Therefore, antibody arrays represent a new paradigm for biomarker proteomics that has the potential to accelerate biomarker discovery and validation compared to traditional methods of proteomics [23-29]. Antibody microarray-based technology, which can simultaneously detect the expression levels of multiple proteins and can combine the advantages of the specificity of ELISA, sensitivity of enhanced-chemiluminescence (ECL) and high-throughput capacity of microspot, represents a promising tool for the field of oncoproteomics. One of the most important applications of such technology is the comparison of proteome expression signatures in cancerous versus normal samples. Indeed, an ever-growing number of publications have documented the suitability of sandwich-based antibody arrays; these arrays are the most common of the antibody arrays used for protein detection and can characterize differential protein expression patterns using various sample types including serum, plasma, cell conditioned media, cell and tissue lysates, cerebrospinal fluid, urine, abscess fluid, sputum, breath condensates, saliva, tears, prostatic fluids, milk, colostrum, etc.
We hypothesized that antibody microarray analyses using whole-tumor samples as a starting material for protein profiling might provide a functional perspective to the view that GC emerges from active inflammatory cross-talk between tumor cells and the surrounding stroma. Thus, the interactions in the GC microenvironment should produce inflammation-associated proteomic profiles able to specifically identify the absence/presence of tissue malignancy [30-35]. To test the hypothesis that pathological processes causally linking inflammation with GC would produce disease-specific molecular changes during cancer development in the gastric mucosa, we herein applied the innovative, simple, flexible and cost-effective antibody-based protein array system. The aim of this study was to identify a unique “oncoproteomic signature” in a pilot study for biomarker discovery in patients with GC. We focused specifically on secreted signaling proteins including cytokines, angiogenic factors, growth factors, inflammatory factors and metalloproteinases, as these factors constitute the primary means of communication between cells in our body. We examined 120 cytokines, 43 angiogenic factors, 41 growth factors, 40 inflammatory factors and 10 metalloproteinases simultaneously in matched pairs of tumor/normal gastric tissues using commercially available Human Antibody Arrays (RayBiotech, Inc.). This system enabled the robust and accurate identification of more than 250 proteins in an inexpensive fashion. Importantly, the experiments were performed in a general laboratory setting without any specialized equipment or special training. Using this approach, we successfully identified a signature of 21 proteins for discriminating GC from noncancerous gastric mucosa with high sensitivity and specificity; we termed this signature INPROGAS (Inflammatory PROtein-driven GAstric cancer Signature).
RESULTS
Supervised proteomic analysis identified differentially expressed proteins in GC.
A supervised approach has the advantage of identifying proteins whose expression levels best correlate with clinical data. Therefore, we initially grouped a training set of paired samples (n=10) consisting of GC tissues and their corresponding adjacent, non-GC sections (Fig. 1A). To ensure the identification of a robust set of proteins differentially expressed in GC tissues, we used an analytic workflow with two stringent criteria. First, to minimize the individual differences among the patients’ samples, equal amounts of protein from each sample, either the GC or the adjacent tissue, were applied to several microarrays of antibodies strictly in parallel. Second, following processing with antibody-based microarrays of the pairs of samples according to their known “GC” and “non-GC” phenotypic status, the ratio of the GC sample/non-GC sample cell lysates for each of the > 250 antibodies on the arrays was calculated to determine a unique value for the specific protein. The inflammatory proteome was then screened for proteins that were significantly altered from the non-GC spot intensities in the paired GC sample (either upregulated or downregulated; Fig. 1B) from each patient. All of the proteins that were significantly expressed in a given GC tissue were arbitrarily placed into the following groups based on their intensities relative to the paired non-GC tissue: very high abundance (10-fold and over), high abundance (3-fold to < 10-fold), no-change (< ±3-fold), low abundance (-3-fold to < -10-fold) and very low abundance (-10-fold and under). With this 2-step strategy, we identified 21 differentially expressed proteins in at least one of the paired samples in the training set, including 19 upregulated (GRO, MMP-9, IL-8, MMP-8, TIMP-1, Acrp30, ICAM-1, NAP-2, Angiogenin, HGF, b-FGF, RANTES, ENA-78, uPAR, sTNF RII, TIMP-2, EGFR, MCP-1 and IL-1β) and 2 downregulated (MIP-1δ and IGFBP-2) proteins in GC tissues. These identifications were grouped in the heat map shown in Fig. 2.
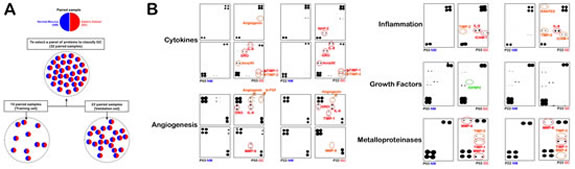
Figure 1:INPROGAS: Study outline and representative antibody-based array chips. A. Informed consent was obtained from all human subjects according to the ethics committee guidelines at the Hospital Dr. Josep Trueta, Girona (Spain). A total of 32 paired GC/non-GC samples were separated into a training set (n=10) and a validation set (n=22), as indicated. B. This figure shows antibody-based array chips encompassing 120 cytokines, 43 angiogenic factors, 41 growth factors, 40 inflammatory factors and 10 metalloproteinases in duplicates probed with whole lysates from paired GC and non-GC mucosae in patients #3 and #22 (NM: Normal mucosa; GC: Gastric carcinoma). The membranes were treated with antibody cocktails, developed by an ECL kit and exposed to an X-ray film as described in the “Materials and Methods”. The intensity of each signal was evaluated photometrically using integrator software and normalized to the background noise in each spot relative to the negative controls. The spot intensities of each protein in replicates were then merged and expressed as a mean value relative to the average signals of the positive controls (membrane-bound biotin-conjugated antibodies) on the array chip analyzed for each experimental (GC) and control (NM) paired group.
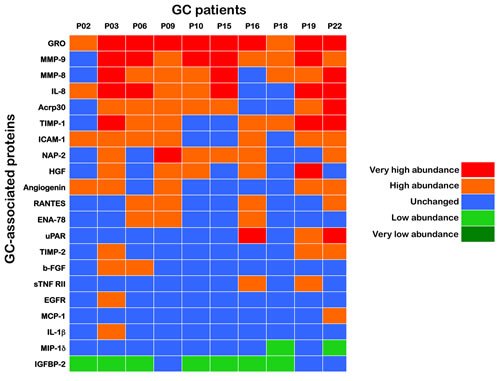
Figure 2:INPROGAS: A 21-protein signature that discriminates GC from noncancerous gastric mucosa. Upon calculation of fold-changes for expression in GC relative to the matched non-GC sample, all proteins that were significantly expressed in a given GC tissue were arbitrarily placed into several “expression groups” based on their intensities relative to the paired non-GC tissue. We analyzed normalized array measurements in the training set to discover differences in protein abundance between samples of GC and those of non-GC to generate a signature of the inflammatory proteome (INPROGAS). Patient data were arranged in columns, and the proteins are listed in rows. Red shades, very high abundance (10-fold and over); orange shades, high abundance (3-fold to < 10-fold); blue shades, no-change (< ±3-fold); light green, low abundance (-3-fold to < -10-fold); dark green, very low abundance (-10-fold and under).
INPROGAS: An INflammatory PROtein-driven GAstric cancer Signature.
When the 21 proteins with highly significant differences in expression between GC and non-GC tissues were assigned into categories based on Gene Ontology (GO) Consortium-defined molecular function, more than one-third (36%) of the proteins demonstrated cytokine/chemokine activity. When the proteins were classified based on GO Consortium-defined biological process, more than one-third (35%) of the proteins were involved in inflammatory/immune and chemotactic responses. Next, to reveal the key signaling pathways or networks related to the set of biomarkers identified in the training set, we imported the list of these 21 proteins into the IPA software (Fig. 3). According to the IPA knowledge base, 3 major signaling networks comprised of 35 nodes each were associated with this set of proteins (Fig. 3, top panels). Network 1 (cell morphology, cellular development, embryonic development) included 8 out of the 21 differentially expressed proteins (P-score=17) and mainly involved MMP- or tissue inhibitor of metalloproteinase (TIMP)-associated signaling. Network 2 (cell-to-cell signaling and interaction, cellular movement) included 7 out of the 21 differentially expressed proteins (P-score=15) and mainly involved MCP-1 (CCL2)- and CCL5-associated signaling. Network 3 (cellular growth and proliferation, cellular movement) included 5 out of the 21 differentially expressed proteins (P-score=10) and mainly involved interleukin (IL)-8- and IL-1-associated signaling. The pro-inflammatory action of the chemokines MCP-1 and IL-1 was central in a merged network combining the top 3 signaling networks with the highest IPA scores (Fig. 3, bottom panels). When we overlaid “diseases and disorders”, “molecular and cellular functions” and “physiological system” onto the merged core networks in IPA, 19 proteins were associated with the “inflammatory response” (p value = 6.98E-21), 20 proteins were associated with “cellular movement” (p value = 1.77E-23), and 18 proteins were associated with “immune cell trafficking” (p value = 1.77E-23), respectively. We termed this signature of 21 predictors that could discriminate GC from noncancerous gastric mucosa “INPROGAS” (INflammatory PROtein-driven GAstric cancer Signature; Fig. 4, top panel)
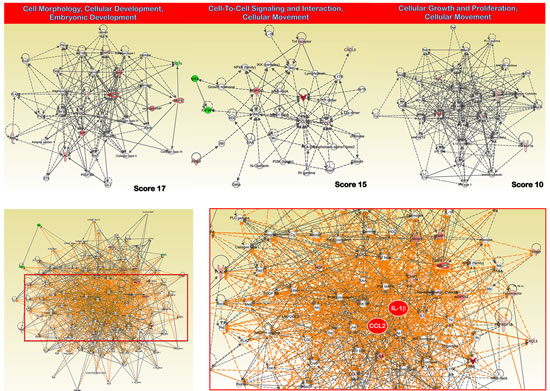
Figure 3:INPROGAS: a functional analysis. Network analysis of differentially expressed proteins included in INPROGAS. A dataset containing the differentially expressed biomarkers in GC tissues (called the focus molecules, n=21) was overlaid onto a global molecular network developed from information contained in the IPA Knowledge Base. Networks of these focus molecules were then algorithmically generated based on their connectivity. Top. The figure shows the networks with the 3 highest IPA scores (a composite measure indicating the statistical significance of the interconnection between the molecules depicted in the network). The focus molecules are colored according to the gene expression (fold change) value; red gene symbols indicate upregulation, and green gene symbols indicate downregulation. The nodes are displayed using various shapes that represent the functional class of the gene product. Edges with dashed lines indicate indirect interactions, while continuous lines represent direct interactions. Bottom. Merged network combining major signaling networks depicted in top panels associated with the proteins included in INPROGAS.
Performance of INPROGAS in the classification of unknown gastric samples.
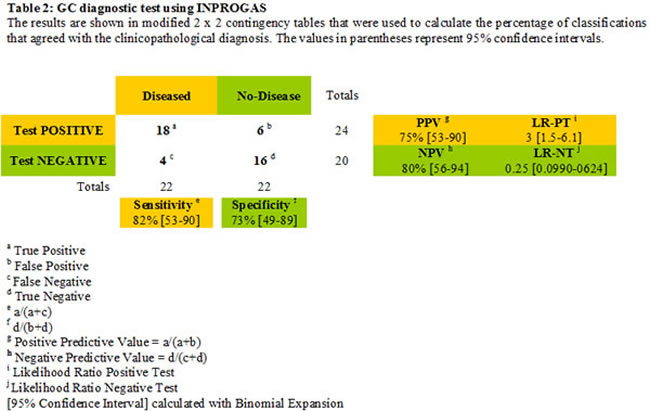
Unlike gene expression profiles, in which the numbers of features vastly outnumber the size of the sample cohort, the limited number of features included on a protein array notably reduces the risk of over fitting when correlating expression with diagnosis. Nevertheless, we sought to validate the performance of INPROGAS in an independent sample cohort and performed GC profiling on a new set of 22 pairs of samples. None of these samples were included in the training set. Thus, to assess the predictive performance of INPROGAS in the classification of unknown samples, we performed a prediction for “GC” or “non-GC” phenotype (a so-called “2-class” prediction) in a blinded set containing paired samples of GC and normal mucosae obtained from 22 patients. For each sample, INPROGAS was performed, and samples were classified into “GC” or “non-GC” groups according to the discovery of positive biomarkers (positivity for at least 80% of the INPROGAS biomarkers was considered a “GC” identification) (Fig. 4, bottom panel). Using this expanded independent validation cohort, INPROGAS classified gastric samples as “GC” or as “non-GC” with a sensitivity of 82% (95% CI 59-94) and a specificity of 73% (95% CI 49-89) (Table 2). Moreover, INPROGAS demonstrated a high positive predictive value (75%; 95% CI 53-90) and negative predictive value (80%; 95% CI 56-94) in the validation cohort.
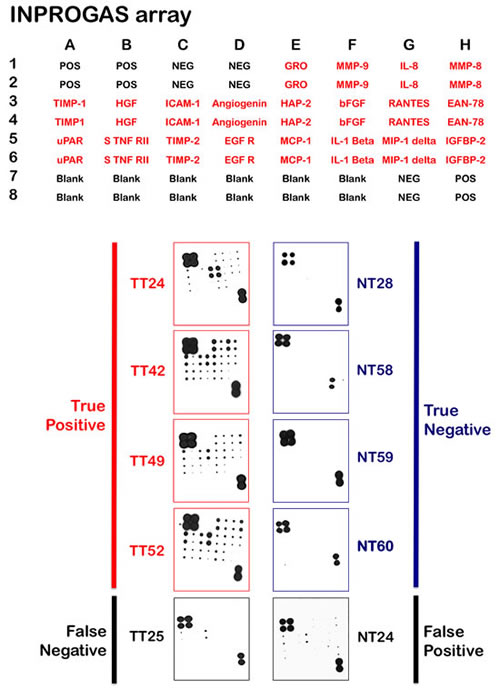
Figure 4:Classification and prediction of GC diagnosis using INPROGAS. A. This figure shows antibody-based array INPROGAS chips for assessing the performance of the in the classification of unknown samples. B. The INPROGAS predictors identified in the training set were used for GC and non-GC class prediction in a blinded test set including 22 paired samples. This figure shows representative antibody-based array chips encompassing the 20 INPROGAS predictors (Acrp30 was excluded from the INPROGAS chip due to technical issues) in duplicates probed with whole lysates from non-tumor tissue (NT) and tumoral tissue (TT). The figure shows several representative images of tissues catalogued as true positive, true negative, false negative, and false positive.
DISCUSSION
Rudolph Virchow first suggested a causal link between inflammation and cancer in 1863 when he demonstrated the presence of leucocytes in neoplastic tissue. Although Virchow’s original hypothesis was repeatedly revisited and there were ample data corroborating inflammation-mediated oncogenesis, inflammation was not recognized among the 6 bona fide hallmarks of cancer in a seminal contribution by Hanahan and Weinberg more than a decade ago [36]. Interestingly, recent years have seen a renaissance of the inflammation-cancer connection, leading to a generally accepted paradigm for inflammation in tumorigenesis, i.e., an inflammatory microenvironment is an essential component of all tumors, including GC [37-44]. We now acknowledge that aberrant proliferation is insufficient to cause cancer, which additionally requires a cancer-promoting microenvironment rich in factors that support cellular survival, growth and angiogenesis. In fact, many cancer-related cytokines, chemokines, metalloproteinases, growth factors and angiogenic factors are produced not only by the tumor cells themselves but also by activated stroma and immune cells associated with tumors. All of these factors that accumulate in situ during chronic inflammation not only exert profound effects on (transformed) epithelial, endothelial and mesenchymal cells but also recruit immune cells. These findings highlight the close parallels between tumor initiation and wound inflammation (i.e., “cancers as wounds that do not heal”).
The inflammatory mediators produced by immunocompetent cells and cancer cells can directly stimulate carcinogenesis (e.g., via the induction of genetic instability in the epithelium of the human stomach), cancer proliferation, angiogenesis, cell adhesion, migration and invasion. In addition, the tumor microenvironment contributes to the systemic anti-inflammatory state associated with cancer (i.e., inflammatory cytokines and chemokines leaking into the systemic circulation are likely to desensitize circulating leukocytes), actively promotes the tumor cells’ ability to subvert the host’s anti-tumorigenic innate and adaptive immune responses and indirectly leads to cancer progression [37-44]. Increasing evidence supports the hypothesis that the seventh hallmark of cancer, i.e., cancer-related inflammation, is a key component of GC formation and progression. Our study hypothesized that processes such as aberrant cell growth, cell invasion, alterations in immune system function and the inflammatory response actively generate an altered or unbalanced stoichiometry of numerous molecules (i.e., growth factors, pro-inflammatory molecules, cytokines, metalloproteinases and angiogenic/lymph angiogenic factors) in the tumor microenvironment compared to the “normal” microenvironment. We further speculated that these qualitative and/or quantitative changes could be characterized in the form of proteomic profiles that could differentiate GC from non-GC in the human stomach mucosae. The description of proteomic profiles specifically representative of the molecular interactions that occur in the GC tumor microenvironment may significantly increase the specificity and sensitivity provided by existing GC diagnostic markers, especially during early disease stages. Our current study confirmed that antibody-based microarray analyses of complex proteomes in gastric tissue-based studies are useful tools to define GC disease-associated protein signatures. Moreover, our proteomic profiling using whole-tumor samples supported the view that GC emerges from active cross-talk between tumor cells and the surrounding stroma, as many of the differentially regulated proteins were pro-inflammatory chemokines and MMPs.
A large number of the GC samples included in the training set revealed a role for the chemokine GRO/CXCL1, which plays a key role in inflammation, immunity, angiogenesis and cell movement [32, 45, 46]. In addition, MMP-9 and MMP-8, which act on pro-inflammatory cytokines, chemokines and other proteins to regulate varied aspects of inflammation and immunity [47, 49], showed a high prevalence among GC samples. Very high incidence rates were also observed for the pro-inflammatory factor IL-8, which has an established role in the chronic inflammation that underpins the development of a number of human cancers [50-52]. Moreover, TIMP-1, which has recently emerged as an important multifunctional protein capable of regulating inflammation, also reached a high prevalence among GC samples [53-55]. Paradoxically, Acrp30/adiponectin, an adipokine regulating glucose and lipid metabolism with well-known anti-inflammatory properties [56, 57], was found to be upregulated in a significant number of GC samples. Adiponectin has been shown to have antiproliferative effects on GC, and adiponectin expression is inversely correlated with clinical staging of the disease [58, 59]. Nevertheless, it should be noted that due to problems with combinatorial optimization during the design and production of the INPROGAS signature by RayBio Inc., the Acrp30/adiponectin marker was not included in custom microarrays. Therefore, the actual impact of Acrp30/adiponectin on GC diagnosis was not assessed in the current study. Similarly, one of the 21 predictors included in the INPROGAS signature was hepatocyte growth factor (HGF). HGF is a potent angiogenic factor that stimulates growth and motility of endothelial cells and has potent anti-inflammatory effects in multiple animal models of disease in various organs; in particular, HGF functions by suppressing NF-κB and downstream endothelial inflammation [60-62]. However, whether adiponectin and/or HGF act to protect the organism from systemic inflammation as part of the paradoxical local inflammation and systemic anti-inflammation during the development of GC remains to be answered in forthcoming studies. A high incidence rate was also found for intracellular adhesion molecule-1 (ICAM-1), a key contributor to vascular inflammation, as ICAM-1 ligation produces pro-inflammatory effects such as inflammatory leukocyte recruitment [63, 64]. In addition, angiogenin, a heparin-binding 14-kDa plasma protein that has been demonstrated to stimulate angiogenesis and is induced by pro-inflammatory cytokines to mediate local inflammation [65-67], was found to be significantly upregulated in GC samples. High incidence rates were also observed for the CXC chemokine neutrophil-activating peptide-2 (NAP-2), a chemoattractant that is rapidly generated within the vasculature early during inflammation and potently induces effector functions in neutrophils, such as chemotaxis and degranulation [68, 69]. The platelet-derived pro-inflammatory chemokines RANTES and ENA-78 were found to be significantly co-upregulated among GC samples, and significant incidence rates were also detected for the urokinase-type plasminogen activator receptor (uPAR). uPAR expression is elevated during inflammation and tissue remodeling and in many poor-prognosis human cancers [70-72]. The anti-angiogenesis and anti-inflammatory factor TIMP-2 was also significantly upregulated in GC samples [73].
In contrast, other biomarkers included in the INPROGAS signature displayed lower incidence rates, including the cytokine inhibitor soluble tumor necrosis factor receptor II (sTNFRII) and the epidermal growth factor receptor (EGFR). EGFR is a signaling hub for an increasing list of growth factors, cytokines and inflammatory mediators that connects the inflammatory reaction to tumor development [74, 75]. Basic fibroblast growth factor (bFGF) is known to potentiate leukocyte recruitment to sites of inflammation by enhancing endothelial adhesion molecule expression [76]. The pro-inflammatory cytokine IL-1β is a key component of the multiprotein inflammasome complexes [77, 78]. Monocyte chemoattractant protein (MCP)-1 is a key chemokine of the C-C type that recruits circulating monocytes to sites of inflammation [79, 80], and MIP-1δ (CCL15) is a member of the macrophage inflammatory protein (MIP) family of CC-type chemokines that are mainly produced by leukocytes after exposure to inflammatory cytokines [81]. MIP-1δ play a major role in the recruitment of immune cells to sites of injury or infection and was found to be downregulated in 20% of the training set samples. Of note, in 70% of the training set samples, the binding protein for insulin-like growth factor (IGFBP)-2 was underexpressed. IGFBP-2 is crucial for modulating the levels of the IGF-1R ligand IGF-2, and loss of this regulatory protein leads to an increased availability of IGF-2 and thus constitutive activation of IGF-1R [82, 83]. The IGF-1R signaling pathway is involved in the carcinogenesis of GC through inhibiting cell apoptosis [84]. In the complex inflammatory scenario captured by the INPROGAS signature, it is reasonable to suggest that the local inflammatory network in GC is determined according to the expressed inflammation-related proteins, inflammatory receptor expression patterns and relative concentrations of pro- and anti-inflammatory biomarkers. Thus, the net inflammatory environment likely fluctuates during various stages of GC development.
Early diagnosis is likely to improve the outcome and prognosis of most solid tumors, including biologically aggressive, chemotherapy-refractory GC. Moreover, there is an urgent need to develop new approaches to detect and measure biomarkers in tissues and/or the blood because these markers could lead not only to early detection of GC but also to improved targeted treatments for GC patients. In this regard, because the discovery and validation of GC-associated proteomic profiles could transcend the problems of tumor heterogeneity and population dynamics, there is an urgent need to develop and validate new, simple, flexible, effective and highly sensitive proteomic analysis techniques. New techniques will also allow for the simultaneous analysis of several biomarkers in a single assay in a low-cost format for proper carcinoma diagnosis and/or staging during routine hospital practice. Here, we demonstrated that low-cost proteomic analysis using antibody-based protein microarrays represents a useful new tool for the routine early detection, diagnosis and perhaps therapeutic intervention of GC in hospitalized patients. To elucidate the core molecular networks underlying INPROGAS, the 21 biomarkers were analyzed using the Ingenuity Knowledge Base, which consists of expert-curated molecular interactions. By merging the three INPROGAS-associated core networks, we identified MCP-1 and IL-1β as hub proteins related to the “inflammatory response”, “cellular movement” and “immune cell trafficking” biofunctions. The IPA Knowledge Database further suggested that the involvement of these signaling networks could be essential for cancer development in the human stomach. Because perturbations of these hub proteins have functional effects on GC, small molecule inhibitors of nodes in INPROGAS-related networks may lead to dynamic changes in protein expression. This possibility could open new avenues for the manipulation of cytokine expression and function in cancer immunotherapy for GC.
In summary, we measured the relative abundance of more than 200 signaling proteins in an initial set of samples from 10 matched pairs of tumor/normal gastric tissues and found significantly different expression patterns of 21 proteins, which we have termed the INPROGAS (Inflammatory PROtein-driven GAstric cancer Signature). When INPROGAS was tested in 22 independent samples, gastric mucosae were classified as “tumor” or “normal” with a sensitivity of 82% and a specificity of 72%. Although further tests are needed before this approach can be used in patients, the identification of a disease-specific biomarker panel early during gastric mucosa cancer development could facilitate more effective interventions against GC. Because the candidate biomarkers identified through antibody microarray-based oncoproteomics of the tumor-host GC microenvironment were found to mostly belong to the secreted class of proteins present in tissue and body fluids (i.e., the secretome), our current findings may provide a great basis for non-invasive, blood-based identification of GC biomarkers for screening purposes. Thus, imbalances in the network of communication between cells in disease states may not only serve as a diagnostic indicator but could potentially reveal mechanistic insight into cancer development in the human stomach.
METHODS
Patients and tissue samples
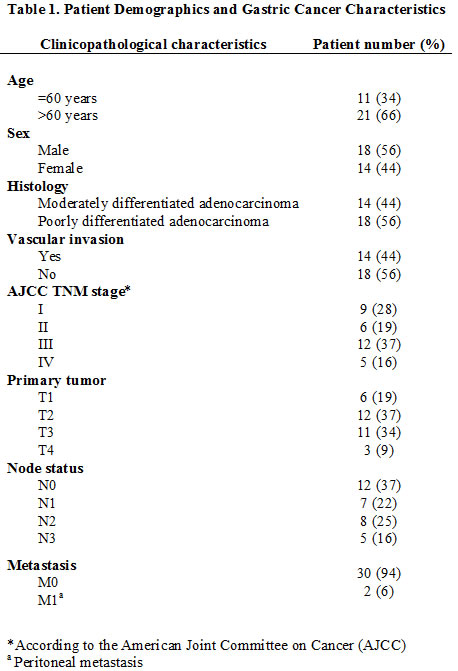
This was a prospective, controlled, single blind analysis study. Pairs of GC and adjacent noncancerous mucosa were obtained after informed consent was received from patients (n=32) who underwent D2 gastrectomy (i.e., radical gastrectomy with level 2 extended lymphadenectomy) between January 2009 and July 2011 at the Hospital Universitari de Girona Dr. Josep Trueta in Girona, Catalonia, Spain. The study was reviewed and approved by the institutional review board and ethics committee. The prospective subject cohort consisted of matched pairs of tumor/normal gastric tissues from GC patients who fulfilled the following criteria: a) histological diagnosis of GC; b) any tumor node metastasis (TNM) stage; c) gastric resection with curative/radical intention; d) no chemotherapy or radiotherapy treatment prior to surgery; and e) signed informed consent. The clinicopathological data of the patients are summarized in Table 1.
To ensure the purity of the GC tissues, the specimens were excised from the cancerous cores. Thus, tumor samples of 3 x 3 x 5 mm3 were taken from areas without gross necrosis. Adjacent nontumor mucosa samples of 3 x 3 x 5 mm3 were taken from the same patient at a location 5 cm away from the tumor margin and were defined as the controls for each GC patient. The representative tumors and adjacent normal tissues of these patients were washed with physiological saline and subsequently frozen within 30 minutes of removal in a liquid nitrogen tank after immediate pathological examination. The senior pathologists routinely conducted the diagnosis for GC based upon Hematoxylin and Eosin (HE) staining. The TNM stage of the tumor was assigned according to the American Joint Committee on Cancer.
Summary of experimental design
Thirty-two paired tumor and adjacent normal tissue samples were extracted, all of which were used to assess the expression level of more than 200 proteins with 5 different proteomic microarrays (see below). These 32 paired samples were divided into a training set (n=10), which was used to select the protein panel to distinguish between normal and GC tumor tissues of GC, and a validation set (n=22), which was used to confirm the ability of candidate proteins selected in the training set to classify blinded gastric samples as “GC” or “non-GC”.
Proteomic chip-based analysis of protein expression in gastric mucosa
We employed protein arrays based on recently developed antibodies by RayBiotech (Norcross, GA, USA), which are capable of rapidly and specifically detecting the expression levels of numerous cytokines, growth factors, soluble receptors of growth factors, angiogenic factors, metalloproteinases and other proteins using small amounts of experimental samples in a single experiment. This technology is designed around the “sandwich immunoassay” principle. A panel of antibodies (capture antibodies) is immobilized at specific locations scored on the surface of a solid membrane, and incubation of the membrane arrays with biological samples results in the capture of soluble proteins by their corresponding antibodies. The bound proteins are detected by incubation with a cocktail of biotinylated antibodies, and corresponding signals are then visualized using enhanced chemiluminescent (ECL) techniques, colorimetry or infrared fluorescence. The following microarrays were used for each of the samples from patients enrolled in the study (i.e., tumor tissue lysates versus adjacent normal tissue lysates): RayBio® Human Cytokine Antibody Array C Series 1000 (Array VI + VII, which detects the expression of 120 cytokines in 2 membranes); RayBio® Human Angiogenesis Antibody Array C Series 1000 (Array 1 + Array 2, which detects the expression of 43 angiogenic factors in 2 membranes); RayBio® Human Matrix Metalloproteinases (MMPs) Antibody Array 1 (which detects the expression of 10 MMPs in a single membrane); RayBio® Human Growth Factor Antibody Array 1 (which detects the expression of 41 growth factors in a single membrane); and RayBio® Human Inflammation Antibody Array 3 (which detects the expression of 40 inflammatory factors in a single membrane). The information provided by the 5 proteomic microarrays was monitored in a quantitative manner (see below), and the expression levels of over 200 proteins were analyzed in each sample.
The protein test using antibody-based microarrays was performed according to the manufacturer’s instructions. Briefly, prior to the start of the analysis, the membranes were blocked with 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS; 0.01 M Tris HCl, pH 7.6, 0.15 M NaCl) for 1 h. After blocking to reduce the amount of non-specific binding, membranes were incubated with 750 µg total protein/tissue sample or 1 ml of serum for 2 h. After extensively washing the membranes with 0.1% Tween-20 in TBS (v/v) (i.e., 3 times for 5 minutes each) and TBS (2 times for 5 minutes each) to remove non-bound material, the membranes were incubated with a cocktail of biotin-labeled antibodies directed against the immobilized proteins by capture antibodies. Following antibody incubation, the membranes were washed as described above and subsequently incubated with horseradish peroxidase (HRP)-conjugated streptavidin (2.5 pg/ml) for 1 h at room temperature. Excess HRP-streptavidin was removed by washing in 0.1% TBS/Tween 20 and TBS. Finally, specific expression signals were detected using the ECL system.
The results obtained in each of the microarrays were evaluated using Analysis Tool Software, a data analysis program specifically designed for RayBio® Antibody Arrays. This analytical tool allows the user to perform the following tasks: a) locate the signal intensities (expression levels) in the antibody array map; b) provide a list of differentially expressed proteins (i.e., candidate markers); c) calculate mean signal intensities; d) analyze and subtract background data (i.e., noise); e) standardize data from different samples; and f) obtain comparative patterns of expression levels between different samples. This program operates based on Microsoft (MS) Excel computer software.
Ingenuity analysis. Signaling networks were constructed using Ingenuity Pathway Analysis (Ingenuity® Systems, Redwood City, CA, USA). Data sets containing identifiers of proteins that were significantly up- or downregulated were uploaded into the application. The ‘focus genes’ were then overlaid on the global molecular network developed from information in the Ingenuity Pathway Knowledge Base. Networks of these ‘focus genes’ (nodes) were algorithmically generated based on the principle that highly connected gene networks are the most biologically meaningful networks. All edges were supported by at least one reference from the literature stored in the Ingenuity Pathway Knowledge Base (the IPA interaction database is manually curated by scientists and updated quarterly). Briefly, the user-input or ‘focus genes’ list was compared to the ‘global molecular network’ (GMN) database consisting of thousands of genes and interactions. The focus genes were sorted based on highest to lowest connectivity within the GMN, and networks of approximately 35 genes were generated starting with the most connected focus gene. IPA assigns a p-value for a network of size n and an input focus gene list of size f by calculating the probability of finding f or more focus genes in a randomly selected set of n genes from the GMN. The intensity of the node color indicated the degree of expression (green scale for downregulated nodes; red scale for upregulated nodes). The nodes were displayed using various shapes, each of which represented a functional class of the gene products. The score indicated the likelihood of the genes in a network being found together due to random chance. Using a 99% confidence interval, scores of ≥ 3 were deemed significant.
ACKNOWLEDGEMENTS
This work was financially supported by the Ministerio de Ciencia e Innovación (SAF2009-11579 and SAF2012-38914 Plan Nacional de I+D+I, MICINN, Spain). This work was also supported in part by a Pilot Research Grant to Manuel Puig-Costa and Javier A. Menendez by the Girona Biomedical Research Institute (IDIBGI).
CONTRIBUTIONS
M.P-C., A.C-C., J.R., R.G., B.M-C., J. J. and J.A.M. contributed to experimental design. E.C-P., C.O-F., S.C., S.F., F.L-P., E.P-A., B.C-F., E.C., R.O. and E.L-B. were responsible for performing experiments. M.P-C., J.J., and J.A.M. contributed to data analysis. J.A.M. wrote the paper.
COMPETING FINANCIAL INTERESTS
The authors of this manuscript have no competing financial interests to declare.
REFERENCES
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008; 58:71-96.
2. Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T. p27 expression and gastric carcinoma. Nat Med. 1997; 3:593.
3. Akama Y, Yasui W, Yokozaki H, Kuniyasu H, Kitahara K, Ishikawa T, Tahara E. Frequent amplification of the cyclin E gene in human gastric cancer. Jpn J Cancer Res. 1995; 86: 617-621.
4. Graziano F, Mandolesi A, Ruzzo A, Bearzi I, Testa E, Arduini F, Silva R, Muretto P, Mari D, Berardi R, Scartozzi M, Lai V, Cascinu S, Magnani M. Predictive and prognostic role of E-cadherin protein expression in patients with advanced gastric carcinomas treated with palliative chemotherapy. Tumour Biol. 2004; 25:106-110.
5. Sanz-Ortega J, Steinburg SM, Moro E, Saez M, Lopez JA, Sierra E, Sanz-Esponera J, Merino MJ. Comparative study of tumor angio- genesis and immunohistochemistry for p53, c-erbB2, c-myc and EGFr as prognostic factors in gastric cancer. Histol Histopathol. 2000; 15:455-462.
6. Zheng L, Wang L, Ajani J, Xie K. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004; 7:61-77.
7. Jang BG, Kim WH. Molecular pathology of gastric carcinoma. Pathobiology. 2011; 78:302-310.
8. Cappellani A, Zanghi A, Di Vita M, Zanet E, Veroux P, Cacopardo B, Cavallaro A, Piccolo G, Lo Menzo E, Murabito P, Berretta M. Clinical and biological markers in gastric cancer: update and perspectives. Front Biosci (Schol Ed). 2010; 2:403-412.
9. Chen CN, Lin JJ, Chen JJ, Lee PH, Yang CY, Kuo ML, Chang KJ, Hsieh FJ. Gene expression profile predicts patient survival of gastric cancer after surgical resection. J Clin Oncol. 2005; 23:7286-7295.
10. Takeno A, Takemasa I, Doki Y, Yamasaki M, Miyata H, Takiguchi S, Fujiwara Y, Matsubara K, Monden M. Integrative approach for differ- entially overexpressed genes in gastric cancer by combining large- scale gene expression profiling and network analysis. Br J Cancer. 2008; 99:1307-1315.
11. Nagarajan N, Bertrand D, Hillmer AM, Zang ZJ, Yao F, Jacques PE, Teo AS, Cutcutache I, Zhang Z, Lee WH, Sia YY, Gao S, Ariyaratne PN, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012; 13:R115.
12. Kim HK, Kim J, Korolevich S, Choi IJ, Kim CH, Munroe DJ, Green JE. Distinctions in gastric cancer gene expression signatures derived from laser capture microdissection versus histologic macrodissection. BMC Med Genomics. 2011; 4:48.
13. Cho JY, Lim JY, Cheong JH, Park YY, Yoon SL, Kim SM, Kim SB, Kim H, Hong SW, Park YN, Noh SH, Park ES, Chu IS, et al. Gene expression signature-based prognostic risk score in gastric cancer. Clin Cancer Res. 2011; 17:1850-1857.
14. Kuramitsu Y, Nakamura K. Proteomic analysis of cancer tissues: shedding light on carcinogenesis and possible biomarkers. Proteomics. 2006; 6:5650-5661.
15. Ebert MP, Röcken C. Molecular screening of gastric cancer by proteome analysis. Eur J Gastroenterol Hepatol. 2006; 18:847-853.
16. Lin LL, Huang HC, Juan HF. Discovery of biomarkers for gastric cancer: a proteomics approach. J Proteomics. 2012; 75:3081-3097.
17. Wu W, Chung MC. The gastric fluid proteome as a potential source of gastric cancer biomarkers. J Proteomics. 2013; 90:3-13.
18. Sousa JF, Ham AJ, Whitwell C, Nam KT, Lee HJ, Yang HK, Kim WH, Zhang B, Li M, LaFleur B, Liebler DC, Goldenring JR. Proteomic profiling of paraffin-embedded samples identifies metaplasia-specific and early-stage gastric cancer biomarkers. Am J Pathol. 2012; 181:1560-1572.
19. Meding S, Nitsche U, Balluff B, Elsner M, Rauser S, Schöne C, Nipp M, Maak M, Feith M, Ebert MP, Friess H, Langer R, Höfler H, et al. Tumor classification of six common cancer types based on proteomic profiling by MALDI imaging. J Proteome Res. 2012; 11:1996-2003.
20. Kim HK, Reyzer ML, Choi IJ, Kim CG, Kim HS, Oshima A, Chertov O, Colantonio S, Fisher RJ, Allen JL, Caprioli RM, Green JE. Gastric cancer-specific protein profile identified using endoscopic biopsy samples via MALDI mass spectrometry. J Proteome Res. 2010; 9:4123-4130.
21. Stoevesandt O, Taussig MJ. Affinity proteomics: the role of specific binding reagents in human proteome analysis. Expert Rev Proteomics. 2012; 9:401-414.
22. Medvedev A, Kopylov A, Buneeva O, Zgoda V, Archakov A. Affinity-based proteomic profiling: problems and achievements. Proteomics. 2012; 12:621-637.
23. Borrebaeck CA, Wingren C. Design of high-density antibody microarrays for disease proteomics: key technological issues. J Proteomics. 2009; 72:928-935.
24. Wingren C, Borrebaeck CA. Antibody-based microarrays. Methods Mol Biol. 2009; 509:57-84.
25. Voshol H, Ehrat M, Traenkle J, Bertrand E, van Oostrum J. Antibody-based proteomics: analysis of signaling networks using reverse protein arrays. FEBS J. 2009; 276:6871-6879.
26. Brennan DJ, O’Connor DP, Rexhepaj E, Ponten F, Gallagher WM. Antibody-based proteomics: fast-tracking molecular diagnostics in oncology. Nat Rev Cancer. 2010; 10:605-617.
27. Sanchez-Carbayo M. Antibody array-based technologies for cancer protein profiling and functional proteomic analyses using serum and tissue specimens. Tumour Biol. 2010; 31:103-112.
28. Sanchez-Carbayo M. Antibody microarrays as tools for biomarker discovery. Methods Mol Biol. 2011; 785:159-182.
29. Vazquez-Martin A, Colomer R, Menendez JA. Protein array technology to detect HER2 (erbB-2)-induced ‘cytokine signature’ in breast cancer. Eur J Cancer. 2007; 43:1117-1124.
30. Milne AN, Carneiro F, O’Morain C, Offerhaus GJ. Nature meets nurture: molecular genetics of gastric cancer. Hum Genet. 2009; 126:615-628.
31. Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012; 143:550-563.
32. Verbeke H, Geboes K, Van Damme J, Struyf S. The role of CXC chemokines in the transition of chronic inflammation to esophageal and gastric cancer. Biochim Biophys Acta. 2012; 1825:117-129.
33. Tsujimoto H, Ono S, Ichikura T, Matsumoto Y, Yamamoto J, Hase K. Roles of inflammatory cytokines in the progression of gastric cancer: friends or foes? Gastric Cancer. 2010; 13:212-221.
34. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo C, Zaninotto G, Rugge M. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur J Cancer Prev. 2008; 17:195-200.
35. Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007; 117:60-9.
36. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
37. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009; 30:1073-1081.
38. Mantovani A. Molecular pathways linking inflammation and cancer. Curr Mol Med. 2010; 10:369-373.
39. Solinas G, Marchesi F, Garlanda C, Mantovani A, Allavena P. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev. 2010; 29:243-248.
40. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010; 11:889-896.
41. Allavena P, Germano G, Marchesi F, Mantovani A. Chemokines in cancer related inflammation. Exp Cell Res. 2011; 317:664-673.
42. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
43. Galdiero MR, Garlanda C, Jaillon S, Marone G, Mantovani A. Tumor associated macrophages and neutrophils in tumor progression. J Cell Physiol. 2013; 228:1404-1412.
44. Mantovani A, Germano G, Marchesi F, Locatelli M, Biswas SK. Cancer-promoting tumor-associated macrophages: new vistas and open questions. Eur J Immunol. 2011; 41:2522-2525.
45. Eck M, Schmausser B, Scheller K, Brändlein S, Müller-Hermelink HK.Pleiotropic effects of CXC chemokines in gastric carcinoma: differences in CXCL8 and CXCL1 expression between diffuse and intestinal types of gastric carcinoma. Clin Exp Immunol. 2003; 134:508-515.
46. Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008; 267:226-244.
47. Dufour A, Overall CM. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci. 2013; 34:233-242.
48. Herszényi L, Lakatos G, Hritz I, Varga MZ, Cierny G, Tulassay Z. The role of inflammation and proteinases in tumor progression. Dig Dis. 2012; 30:249-54.
49. Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG. Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol. 2012; 181:1895-1899.
50. Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol. 2013; 14:e218-e228.
51. Vendramini-Costa DB, Carvalho JE. Molecular link mechanisms between inflammation and cancer. Curr Pharm Des. 2012; 18:3831-52.
52. McLean MH, El-Omar EM. Genetics of inflammation in the gastrointestinal tract and how it can cause cancer. Recent Results Cancer Res. 2011; 185:173-183.
53. Rhee JS, Diaz R, Korets L, Hodgson JG, Coussens LM. TIMP-1 alters susceptibility to carcinogenesis. Cancer Res. 2004; 64:952-961.
54. Zhang M, Zhu GY, Gao HY, Zhao SP, Xue Y. Expression of tissue levels of matrix metalloproteinases and tissue inhibitors of metalloproteinases in gastric adenocarcinoma. J Surg Oncol. 2011; 103:243-247.
55. Grunnet M, Mau-Sørensen M, Brünner N. Tissue inhibitor of metalloproteinase 1 (TIMP-1) as a biomarker in gastric cancer: a review. Scand J Gastroenterol. 2013; 48:899-905.
56. Turer AT, Scherer PE. Adiponectin: mechanistic insights and clinical implications. Diabetologia. 2012; 55:2319-2326.
57. Buechler C, Wanninger J, Neumeier M. Adiponectin, a key adipokine in obesity related liver diseases. World J Gastroenterol. 2011; 17:2801-2811.
58. Ishikawa M, Kitayama J, Yamauchi T, Kadowaki T, Maki T, Miyato H, Yamashita H, Nagawa H. Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci. 2007; 98:1120-1127.
59. Seker M, Bilici A, Sonmez B, Ustaalioğlu BB, Gumus M, Gozu H, Sargin M, Orcun A, Gezen C, Eser M, Bildik N, Salepci T. The association of serum adiponectin levels with histopathological variables in gastric cancer patients. Med Oncol. 2010; 27:1319-1323.
60. Sakon M, Kita Y, Yoshida T, Umeshita K, Gotoh M, Kanai T, Kawasaki T, Kambayashi J, Monden M. Plasma hepatocyte growth factor levels are increased in systemic inflammatory response syndrome. Surg Today. 1996; 26:236-41.
61. Beppu K, Uchiyama A, Morisaki T, Nakamura K, Noshiro H, Matsumoto K, Nakamura T, Tanaka M, Katano M. Elevation of serum hepatocyte growth factor concentration in patients with gastric cancer is mediated by production from tumor tissue. Anticancer Res. 2000; 20:1263-1267.
62. Fujita K, Ewing CM, Sokoll LJ, Elliott DJ, Cunningham M, De Marzo AM, Isaacs WB, Pavlovich CP. Cytokine profiling of prostatic fluid from cancerous prostate glands identifies cytokines associated with extent of tumor and inflammation. Prostate. 2008; 68:872-882.
63. Kobayashi H, Boelte KC, Lin PC. Endothelial cell adhesion molecules and cancer progression. Curr Med Chem. 2007; 14:377-386.
64. Jung WC, Jang YJ, Kim JH, Park SS, Park SH, Kim SJ, Mok YJ, Kim CS. Expression of intercellular adhesion molecule-1 and e-selectin in gastric cancer and their clinical significance. J Gastric Cancer. 2012; 12:140-148.
65. Shimoyama S, Kaminishi M. Increased angiogenin expression in gastric cancer correlated with cancer progression. J Cancer Res Clin Oncol. 2000; 126:468-474.
66. Lioté F, Champy R, Moenner M, Boval-Boizard B, Badet J. Elevated angiogenin levels in synovial fluid from patients with inflammatory arthritis and secretion of angiogenin by cultured synovial fibroblasts. Clin Exp Immunol. 2003; 132:163-168.
67. Chen Y, Zhang S, Chen YP, Lin JY. Increased expression of angiogenin in gastric carcinoma in correlation with tumor angiogenesis and proliferation. World J Gastroenterol. 2006; 12:5135-5139.
68. Schwartzkopff F, Brandt E, Petersen F, Flad HD, Bock L, Ludwig A. The CXC chemokine NAP-2 mediates differential heterologous desensitization of neutrophil effector functions elicited by platelet-activating factor. J Interferon Cytokine Res. 2002; 22:257-267.
69. Smith C, Damås JK, Otterdal K, Øie E, Sandberg WJ, Yndestad A, Waehre T, Scholz H, Endresen K, Olofsson PS, Halvorsen B, Gullestad L, Frøland SS, Hansson GK, Aukrust P. Increased levels of neutrophil-activating peptide-2 in acute coronary syndromes: possible role of platelet-mediated vascular inflammation. J Am Coll Cardiol. 2006; 48:1591-1599.
70. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010; 11:23-36.
71. Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003; 22: 205-222.
72. Blasi F, Sidenius N. The urokinase receptor: focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010; 584:1923-1930.
73. Stetler-Stevenson WG. The tumor microenvironment: regulation by MMP-independent effects of tissue inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 2008; 27:57-66.
74. Berasain C, Perugorria MJ, Latasa MU, Castillo J, Goñi S, Santamaría M, Prieto J, Avila MA. The epidermal growth factor receptor: a link between inflammation and liver cancer. Exp Biol Med (Maywood). 2009; 234:713-725.
75. Pastore S, Mascia F, Mariani V, Girolomoni G. The epidermal growth factor receptor system in skin repair and inflammation. J Invest Dermatol. 2008; 128:1365-1374.
76. Zittermann SI, Issekutz AC. Endothelial growth factors VEGF and bFGF differentially enhance monocyte and neutrophil recruitment to inflammation. J Leukoc Biol. 2006; 80:247-257.
77. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013; 13:397-411.
78. Yin Y, Pastrana JL, Li X, Huang X, Mallilankaraman K, Choi ET, Madesh M, Wang H, Yang XF. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Front Biosci (Landmark Ed). 2013; 18:638-649.
79. Coll B, Alonso-Villaverde C, Joven J. Monocyte chemoattractant protein-1 and atherosclerosis: is there room for an additional biomarker? Clin Chim Acta. 2007; 383:21-9.
80. Kuroda T, Kitadai Y, Tanaka S, Yang X, Mukaida N, Yoshihara M, Chayama K. Monocyte chemoattractant protein-1 transfection induces angiogenesis and tumorigenesis of gastric carcinoma in nude mice via macrophage recruitment. Clin Cancer Res. 2005; 11:7629-7636.
81. Maurer M, von Stebut E. Macrophage inflammatory protein-1. Int J Biochem Cell Biol. 2004; 36:1882-1886.
82. Nordqvist AC, Peyrard M, Pettersson H, Mathiesen T, Collins VP, Dumanski JP, Schalling M. A high ratio of insulin-like growth factor II/insulin-like growth factor binding protein 2 messenger RNA as a marker for anaplasia in meningiomas. Cancer Res. 1997; 57:2611-2614.
83. Kang Z, Yu Y, Zhu YJ, Davis S, Walker R, Meltzer PS, Helman LJ, Cao L. Downregulation of IGFBP2 is associated with resistance to IGF1R therapy in rhabdomyosarcoma. Oncogene. 2013 Dec 2. doi: 10.1038/onc.2013.509
84. Adachi Y, Yamamoto H, Ohashi H, Endo T, Carbone DP, Imai K, Shinomura Y. A candidate targeting molecule of insulin-like growth factor-I receptor for gastrointestinal cancers. World J Gastroenterol. 2010;16:5779-5789.