
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) comprise a variety of morphological and clinical phenotypes, resulting from miscellaneous, underlying molecular defects. Gene expression profiles and their immunohistochemical surrogate markers have been used to classify DLBCL into prognostic subgroups [1, 2], still, diversity in biology and clinical outcome exists within the individual categories. Only few genetic or epigenetic defects have been identified as prognostic markers. The data on TP53 mutation alone as a prognostic factor for survival in DLBCL have been inconsistent, however, a number of studies have shown that dual disruption of the p16/Rb and the ARF/p53 pathway, e.g. deletion of the INK4A/ARF locus, are strong negative prognostic factors for survival [32013;6]. Likewise, the combined translocations of oncogenic MYC and anti-apoptotic BCL-2 or BCL-6 (so-called “double hit” lymphoma) are associated with exceedingly poor prognosis [7, 8].
Most previous studies of tumor suppressor pathways in DLBCL have focused on the disruption of the coding sequences of genes. However, epigenetic alterations and aberrant expression of microRNAs (miRs) may be equally important for growth control. As for protein encoding genes, the transcription of miR genes may be inactivated by promoter hypermethylation [9, 10].
The members of the miR34 family (miR34A, miR34B, and miR34C) have been recognized as tumor suppressors, and implicated in a variety of cellular processes that control carcinogenesis including cell cycling, apoptosis, somatic cell reprogramming and metastasis [112013;14]. It has been shown that the p53-miR34 axis may be another link between the ARF/p53 pathway, the p16/Rb pathway and MYC regulated pathways. In a complex circuit, p53 promotes transcription of MIR34A and MIR34B/C , and the miR34s in turn act as mediators of p53 signaling [15, 16]. In addition, miR34s inhibit MYC and several proto-oncogens that counteract the p16/Rb tumor suppressor pathway [172013;19]. These observations place the miR34s at the center of cell cycle and apoptosis regulation, and loss of miR34 expression has been associated with poor response to therapy.
Several studies have implicated both miR34A [202013;22] and miR34B/C [15, 23, 24] in lymphoproliferative malignancies. In CLL, the p53-miR34A and p53-miR34B/C axes have been investigated in detail. Low expression levels of miR34A correlate to TP53 mutation or 17p deletion, and has negative prognostic impact on both treatment free survival [20] and survival of previously treated patients [22]. MiR34B and miR34C have been shown to act with miR15, miR16, p53 and ZAP70 [15]. In multiple myeloma the MIR34B/C cluster is downregulated by promoter methylation in a large proportion of cases at relapse [24]. In low-grade gastric MALT type lymphoma downregulation of miR34A is involved in the transformation to DLBCL, by deregulation of the oncogene FOXP1 [21]. However, in de novo DLBCLs, the miR34s have not previously been investigated, and in spite of their implication in several other lymphoproliferative malignancies, little is known about the role of the miR34s in normal B-cells.
Here, we investigated the expression of miR34A, miR34B and miR34C in normal and reactive B-cells. Given that MIR34A and MIR34B/C locate to regions of allelic loss in DLBCL (1p36.23 and 11q23.1, respectively) [4, 25, 26], and the importance of the miR34 targets in DLBCL pathogenesis, we also investigated a large panel of newly diagnosed cases of DLBCLs for MIR34A and MIR34B/C promoter methylation, TP53 mutational status, clinical presentation patterns, and outcome.
RESULTS
Several studies have shown the implication of the miR34s in CLL and multiple myeloma, and a single study implicated miR34A in the transformation of gastric MALT-type lymphomas to DLBCL [202013;24]. However, the role of the miR34s in de novo DLBCLs has not been analysed in detail, and little is known about the role of the miR34s in normal B-cells.
p53 has been shown to directly bind to and regulate the transcription of both miR34A and miR34B/C [16], and we speculated whether these molecules were involved in DLBCL lymphomagenesis in a mutually exclusive manner, as it was previously suggested in CLL [20]. DLBCL consists of mixtures of malignant B-cells and reactive cells, which complicates the measurement of miR34 expression. Therefore, we initially determined the expression of the individual miR34 family members in PBL-B and in reactive lymph nodes. Since these were differentially expressed, we next investigated miR34 regulation at the DNA level in DLBCL to avoid mis-interpretation of miR expression signals from infiltrating cells. Thus, we examined the components of the p53-miR34 axis including TP53 mutational status and hypermethylation of the MIR34A and MIR34B/C promoters in a panel of 150 newly diagnosed DLBCL and in 6 DLBCL cell lines, and compared these data to those obtained from PBL-B and reactive lymph nodes.
MiR34A, miR34B, and miR34C expression and regulation in non-cancerous B-cells
We began our investigations by examining the methylation and expression status of miR34A-5p, miR34A-3p, miR34B-5p, miR34B-3p, miR34C-5p and miR34C-3p in PBL-B from random donors and in reactive lymph nodes. DNA from normal PBL-B was unmethylated at both the MIR34A and MIR34B/C promoters. While miR34A-5p was expressed at low levels in normal PBL-B, no expression of miR34A-3p, miR34B-5p, miR34B-3p, miR34C-5p, and miR34C-3p could be detected in PBL-B (Ct values>35 cycles)). In reactive lymph nodes the expression of miR34A-5p was induced in average 70 fold (P<0.001), while no significant induction was observed for the rest of the investigated miRs, including the homologous miR34B-5p and miR34C-5p, indicating that the assay is highly specific to miR34A-5p (Figure 1A).
Since the reactive lymph nodes consist of a mixture of cells, we next stimulated PBL-B in vitro by LPS and IL-1α activation and observed significant induction of miR34A-5p (P= 0.017, t-test), but not of miR34B/C. In addition, normal CD3+ T cells showed no expression of miR34A, indicating that the response we observe in reactive lymph nodes most likely originate from activated B-cells (Figure 1A).
Given the low expression of miR34s in normal PBL-B, we investigated if miR34A, B and -C were downregulated by other epigenetic mechanisms. Chromatin immunoprecipitation (ChIP) at the MIR34A- and the MIR34B/C promoters showed a relative enrichment (>1,0 of input DNA) for the bivalent H3K27me3/H3K4me3 silencing mark at the MIR34B/C promoter (Figure 1B).
Taken together, these results indicate that in PBL-B both miR34A and miR34B/C are expressed at low levels. The miR34B/C cluster is downregulated by polycomb, and in contrast to miR34A, does not seem to be reactivated upon stimulation .
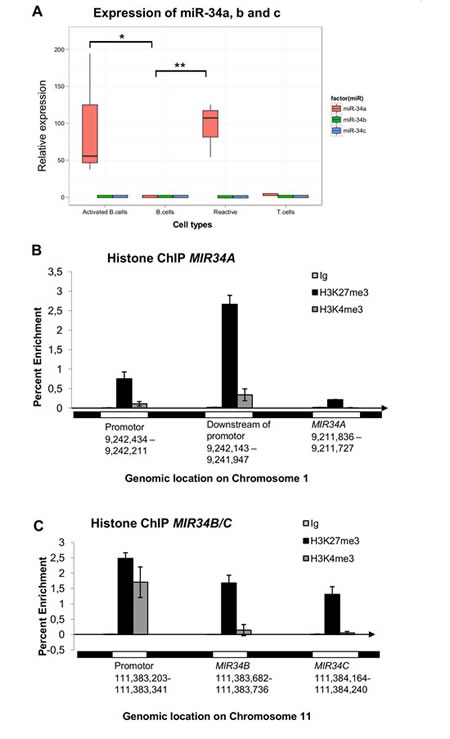
Figure 1: MiR34A, miR34B, and miR34C expression and regulation in normal B-cells, T-cells and reactive lymph nodes. (A) Normal peripheral blood CD19+ B-lymphocytes (PBL-B) have an unmethylated MIR34A promoter and show low expression of miR34A-5p, which, however, is significantly upregulated in reactive lymph nodes (P<0.001) and in vitro activated B-cells (P<0.017). MiR34A-5p is not expressed in normal CD3+ T-cells. The MIR34B/C promoter is unmethylated and miR34B and miR34C is not expressed in normal or in vitro activated B-cells, T-cells or reactive lymphnodes. (B) No enrichment of the H3K27me3 or H3K4me3 marks was observed at the TSS of MIR34A. (C) The downregulation of miR34B/C in PBL-B may be due to enrichment for the bivalent H3K27me3/H3K4me3 silencing mark at the MIR34B/C promoter.
MiR34A, -B, and -C expression is associated with promoter methylation in DLBCL cell lines and can occur in combination with TP53 mutations
We next investigated the methylation status of the MIR34A and MIR34B/C promoters in DLBCL-cell lines, and compared these data with TP53 mutational status. In all DLBCL cell lines the MIR34B/C promoter was methylated (Figure 2A) and miR34B/C not expressed (data not shown). By contrast, only the Toledo, DB1, HT, and U2932 cell lines showed a biphasic melting curve at the MIR34A promoter indicating that both methylated and unmethylated alleles were present, while the Farage and DOHH2 cell lines had a completely unmethylated MIR34A promoter (Figure 2B and Supplemental Figure 1). Bisulfite genomic sequencing of the HT-cell line showed that ~50 % of the MIR34A alleles were fully methylated and, ~50% were fully unmethylated , suggesting this could be caused by monoallelic methylation, or alternatively, be indicative of an unmethylated subclone (Figure 2C). While the completely unmethylated Farage and DOHH2 cells showed an expression level of miR34A-5p that was comparable to that of reactive lymph nodes, no expression of miR34A-5p was detected in the cell lines with both methylated and unmethylated alleles (~total 2 copies pr. cell as measured by quantification of a miR34A-5p mimic) (Figure 2D). These data indicate that the observed level of methylation at the MIR34A promoter is associated with blocked transcription of MIR34A.
Mutation analysis of exons 5-9 of the TP53 gene in the six DLBCL cell lines revealed a G to A transition causing the V216M hotspot mutation in the HT cell line (Figure 2E) and the C176Y mutation in U2932 was confirmed [27]. Surprisingly, both cell lines also showed both MIR34A and MIR34B/C promoter hypermethylation. Treatment of the TP53 mutant/MIR34A/B/C promoter methylated cell lines HT and U2932 with 5-aza-CdR showed that miR34A-5p (but none of the other miRs) are upregulated irrespective of the TP53 mutation (Figure 2F and Supplemental Figure 1 for U2932). Treatment with cytosine arabinoside did not induce miR34A-5p.
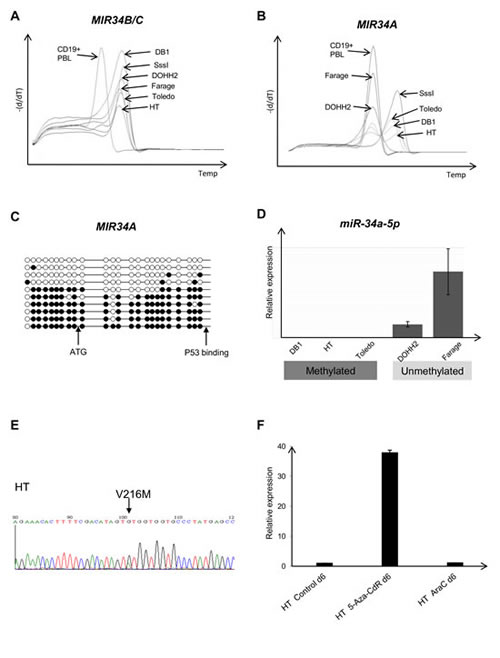
Figure 2: MIR34A, MIR34B/C and TP53 regulation in lymphoma cell lines. (A) MS-MCA showing that all cell lines are methylated at the MIR34B/C promoter in the DLBCL cell lines. (B) In the DLBCL cell lines Toledo, DB1 and HT, the MIR34A promoter shows a biphasic methylation pattern (methylated and unmethylated), while Farage and DOHH2 are completely unmethylated. (C) Bisulfite sequencing of the MIR34A promotor region showing methylation of ~50% of the MIR34A alleleles, methylated cytosines, unmethylated cytosines. (D) MIR34A promoter methylation correlates with expression. (E) Direct sequencing showing the concomitant TP53 mutation (V216M) in the MIR34A/B/C methylated cell line HT (F) Upregulation of miR34A in the MIR34A methylated and TP53 mutant cell line HT after treatment with the hypomethylating agent 5-aza-2’deoxycytidine (5-aza-CdR 0.5uM). No induction of miR34A was observed after treatment with cytosine arabinoside (araC 20nM).
Coordinated promoter methylation of MIR34A and MIR34B/C in primary DLBCL
To investigate whether these combined molecular alterations also occur in vivo, we next examined a panel of 150 primary DLBCL samples for promoter methylation of MIR34A and MIR34B/C by MS-MCA. MIR34A was methylated in 42 cases (28%) (Figure 3A), and MIR34B/C was methylated in 116 (78%) cases (Figure 3B). All except two of the cases with methylation of MIR34A also carried methylation of the MIR34B/C promoter (P=0.001, Fisher’s exact test), suggesting that the methylation of these miRs may be interdependent and occur in coordinated manner. The MIR34A methylated cases did not differ significantly from the MIR34A unmethylated cases with respect to age, sex, clinical stage, LDH, performance score or IPI (Table 1), which was also the case for MIR34B/C methylated vs. unmethylated cases (data not shown).
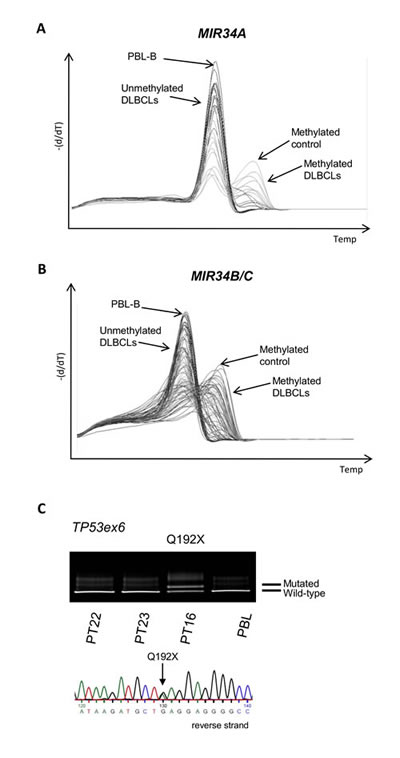
Figure 3: MIR34A, MIR34B/C and TP53 deregulation in primary DLBCLs. (A) MIR34A methylation in primary DLBCL by MS-MCA. (B) MIR34B/C methylation in primary DLBCL (MS-MCA). (C) Representative image of DGGE gel and Sanger sequence of TP53 mutation in primary DLBCL.
TP53 and MIR34A mutations, and the MDM2 SNP309 GG genotype in DLBCL
DGGE-based screening of exons 5-9 of the TP53 gene revealed mutations in 24 out of 150 (16%) of the primary DLBCL cases (Figure 3C). These mutations comprised both missense and non-sense mutations (Supplemental Table 2), and were distributed throughout the investigated exons. One case carried two mutations suggesting they may be biallelic. No mutations or SNPs were detected in the MIR34A expressed sequence, although 2 SNPs have previously been reported in the region (www.genome.ucsc.edu). Since studies in CLL suggest that the MDM2 SNP309 GG genotype associate with low miR34A expression and poor prognosis, we investigated the role of this SNP in a subset (120) of these primary DLBCLs, but observed no prognostic impact (see Supplemental Material).
A subset of de novo DLBCL carries concomitant methylation of MIR34A and TP53 mutation
By combining the above data we identified a subset of nine primary DLBCL cases that carried combined TP53 mutation and MIR34A promoter methylation at diagnosis. The TP53 mutations were both loss of function mutations (4) and missense mutations in the DNA binding domain (5) (Supplemental Table 2). A total of 9 of the 24 (38%) TP53 mutant cases also had MIR34A methylation and 9 of the 42 (21%) MIR34A methylated cases also had TP53 mutation (P=0.19), indicating that these alterations do occur independently of each other. These data support our findings from the cell lines, that a subset of DLBCL has combined loss of p53 and miR34A function. We observed that the MIR34A methylated and TP53 mutated patients were associated with aggressive disease with Ann Arbor stage III-IV (P=0.030), elevated LDH level (P=0.015), and IPI group III-IV (P=0.023). In the cohorte of patients treated with Rituximab the MIR34A methylated and TP53 mutated cases were also significantly associated with Ann Arbor stage III-IV (P=0.025), elevated LDH level (P=0.002), and IPI group III-IV (P=0.027) (Table 1).
Table 1a: Clinical characteristics of all DLBCL patients at time of diagnosis (n=150, 62 Rituximab and 88 non-Rituximab treated DLBCL patients) |
|||||
|
n |
TP53wt and MIR34A unmeth |
TP53mut or MIR34A meth |
TP53mut and MIR34A meth |
P-value |
Mean Age (yrs) |
150 |
59 |
63 |
65 |
0.270 |
Sex |
147 |
|
|
|
0.495 |
F |
|
39 |
23 |
4 |
|
M |
|
55 |
21 |
5 |
|
Stage |
145 |
0.030 |
|||
I+II |
46 |
24 |
1 |
||
III+IV |
48 |
18 |
8 |
||
Performance score |
144 |
|
|
|
0.129 |
0+1 |
|
79 |
35 |
5 |
|
2+3 |
|
14 |
6 |
4 |
|
S-LDH |
139 |
0.015 |
|||
Normal |
46 |
26 |
1 |
||
Elevated |
45 |
14 |
7 |
||
IPI |
144 |
|
|
|
0.023 |
L+LI |
|
65 |
30 |
2 |
|
H+HI |
|
25 |
10 |
6 |
|
Table 1b: Clinical characteristics of Rituximab treated DLBCL patients at time of diagnosis (n=62) |
|||||
|
n |
TP53wt and MIR34A unmeth |
TP53mut or MIR34A meth |
TP53mut and MIR34A meth |
P-value |
Mean Age (yrs) |
62 |
62 |
63 |
60 |
0.850 |
Sex |
62 |
|
|
|
0.208 |
F |
|
18 |
6 |
3 |
|
M |
|
23 |
9 |
3 |
|
Stage |
62 |
0.025 |
|||
I+II |
18 |
8 |
0 |
||
III+IV |
23 |
7 |
6 |
||
Performance score |
62 |
|
|
|
0.226 |
0+1 |
|
38 |
14 |
4 |
|
2+3 |
|
3 |
1 |
2 |
|
S-LDH |
62 |
0.002 |
|||
Normal |
26 |
11 |
0 |
||
Elevated |
15 |
4 |
6 |
||
IPI |
62 |
|
|
|
0.027 |
L+LI |
|
30 |
10 |
1 |
|
H+HI |
|
11 |
5 |
5 |
|
Overall Survival
We initially analyzed the prognostic impact on overall survival of each individual molecular alteration. The Kaplan-Meier estimate of overall survival for cases with methylation of MIR34B/C did not differ from cases with unmethylated MIR34B/C (P=0.803). Cases with TP53 mutations had significantly poorer survival (P=0.004), whereas cases with MIR34A methylation showed a tendency towards poorer survival (P=0.077). However, the 9 cases with disruption of both TP53 and MIR34A showed an exceedingly poor survival of only median 9.4 months (P<0.0001) (Figure 4A). Interestingly, 6 of the 9 “double-hit” cases were treated with Rituximab in addition to combination chemotherapy, which did not seem to improve survival (Figure 4B).
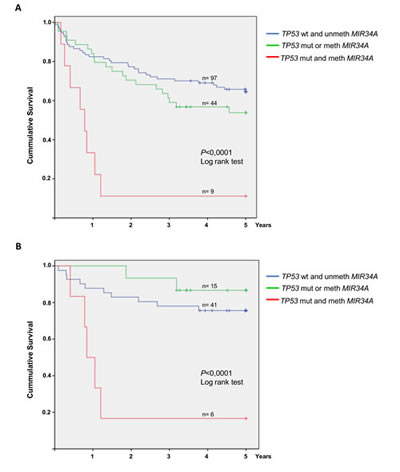
Figure 4: Overall survival of DLBCL patients with and without MIR34A methylation and TP53 mutation. Patients are divided into 3 groups: Patients with no alterations of TP53 or MIR34A, patients with either MIR34A methylation or TP53 mutation only, and patients with concomitant MIR34A methylation and TP53 mutation. (A) 5 year overall survival for the three groups in the entire cohorte (n=150). The group with concomitant MIR34A methylation and TP53 mutation (n=9) shows significantly poor overall survival (P<0.0001). (B) Among the Rituximab treated patients (n=62) concomitant MIR34A methylation and TP53 mutation (n=6) leads to significantly inferior survival as well (P<0.0001).
To compare the relative impact on survival of either TP53 mutation or MIR34A methylation alone to that of concomitant TP53 mutation and MIR34A methylation, we performed a multivariate Cox regression analyses. Baseline risk factors included in the model were: age, LDH, Ann Arbor stage, performance status and IPI. The model revealed that TP53 mutation or MIR34A methylation alone are not independent factors for survival. However, combined MIR34A methylation and TP53 mutation is an independent factor for survival both in the entire cohorte (P = 0.0002) and in the Rituximab treated patients (P=0.021). Furthermore, elevated LDH, age, performance status and lack of Rituximab are independent prognostic factors in the entire cohorte, while only LDH and age retain negative prognostic impact in Rituximab treated patients (Table 2).
Table 2 |
|||||
Multivariate analysis |
Hazard ratio |
95% Hazard Ratio Confidence Limits Lower Upper |
P |
||
Rituximab treated patients (n=62) |
|
|
|
|
|
MIR34A unmeth and TP53wt |
1.000 |
|
|
|
|
MIR34A meth or TP53mut |
0.550 |
0.120 |
2.530 |
0.4433 |
|
MIR34A meth and TP53mut |
4.364 |
1.245 |
15.302 |
0.0213 |
|
LDH (normal/elevated) |
3.699 |
1.081 |
12.663 |
0.0372 |
|
Age |
1.044 |
1.008 |
1.081 |
0.0156 |
|
All patients (n=150) |
|
|
|
|
|
MIR34A unmeth and TP53wt |
1.000 |
|
|
|
|
MIR34A meth or TP53mut |
1.449 |
0.800 |
2.800 |
0.2035 |
|
MIR34A meth and TP53mut |
6.518 |
2.461 |
17.264 |
0.0002 |
|
LDH (normal/elevated) |
3.094 |
1.621 |
5.903 |
0.0006 |
|
Performance status (0-1/2-4) |
2.135 |
1.151 |
3.963 |
0.0162 |
|
Age |
1.044 |
1.023 |
1.064 |
<.0001 |
|
Immunotherapy (No/yes) |
2.449 |
1.300 |
4.611 |
0.0055 |
|
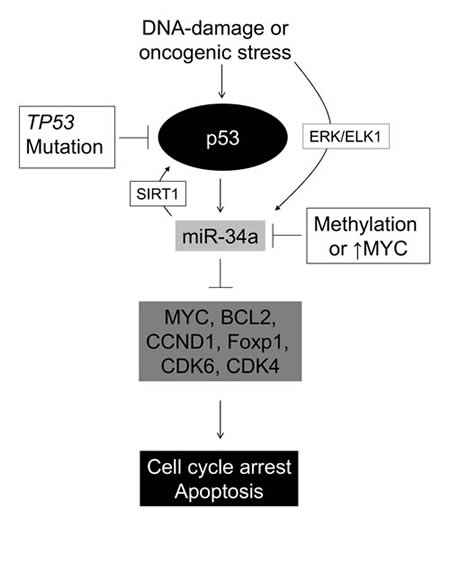
Figure 5: A schematic model of the p53-miR34 axis. Oncogenic stress or DNA damage may activate p53 leading to cell cycle arrest, senescense and/or apoptosis. This is mediated via its downstream effectors miR34A -B and -C, which in turn downregulate known DLBCL proto-oncogenes. Our data indicate that DLBCL cells with concomitant TP53 mutation and MIR34A methylation have a growth advantage compared to “single-hit” tumors, suggesting that in DLBCL miR-34A may also be upregulated independently of p53 by oncogenic stress, probably via the ERK/ELK pathway as suggested by Christoffersen et al [17].
DISCUSSION
Within the last decade, miRs have been recognized as important players in tumorigenesis and many miRs may serve as markers of disease. In DLBCL, miRs may be used in disease classification and as predictors of outcome [282013;30], and some miRs may be directly involved in DLBCL pathogenesis [31, 32].
Disruption of the individual miRs in the miR34 family has been demonstrated in a variety of cancers, and has got attention due to the direct involvement in the p53 and MYC pathways. Overexpression of miR34 in p53 deficient cells can reinstate p53 functions such as senescence, apoptosis and cell cycle arrest [16, 33]. The miR34 family inhibits cell cycle progression at the G1/S transition checkpoint by targeting the mRNAs of several proto-oncogenes that act in the p16 and ARF/p53 pathways including N- and C-MYC, CDK6, CDK4, and CCND1 [162013;19, 34]. Furthermore, miR34A may create a positive feedback loop on p53 by downregulating the deacetylase SIRT1, leading to p53 acetylation and activation [35], and miR34A has also been shown to target pro-apoptotic BCL-2 for downregulation. The miRs 34B/C have been shown to repress MYC in response to DNA damage [34, 36], and in Burkitts’ lymphoma miR34B/C downregulation was suggested to be a more common mechanism for MYC overexpression than IGH/MYC translocation [34]. In addition, overexpression of miR34A was shown to disrupt normal B-lymphocyte development by targeting FOXP1 [37], which acts as an oncogene in DLBCL when miR34A is downregulated [21]. Importantly, miR34 mimics have been shown to abrogate growth and induce apoptosis in DLBCL in vivo [38].
In the present study we initially analyzed the role of the individual members of the miR34 family in normal PBL-B and reactive lymph nodes. Interestingly, miR34A-5p seems to play by far the most dominant role in normal reactive lymph nodes in vivo. In line with other tumor suppressors it is expressed at low levels in normal PBL-B, but is upregulated significantly in activated B-cells and reactive lymph nodes. The high expression of miR34A-5p in reactive cells may be an important limitation to measuring expression of this miR in primary DLBCLs, which are typically infiltrated by reactive B-cells. In normal PBL-B the MIR34B/C promoter is enriched for the bivalent silencing mark H3K27me3/H3K4me3, and miR34B and miR34C are not expressed. No significant induction of¨miR34A-3p, miR34B or miR34C was observed during B-cell activation or in reactive lymph nodes. Taken together, these results indicate that among the miR34s, miR34A-5p plays a dominant role in regulating normal B-cell function.
Several in vitro studies show that deregulation of miR34s is important in lymphomagenesis [15, 34]. In line with what has been shown for other tumor suppressors, we observe that MIR34B/C undergo an epigenetic switch from polycomb mediated gene silencing to permanent gene silencing by DNA methylation during lymphomagenesis [392013;41]. In addition, concurrent DNA methylation of MIR34B/C is present in all but two of the MIR34A methylated DLBCLs. This suggests that under certain circumstances e. g. oncogenic stress, these molecules may potentially compensate for each other, and that a cellular growth advantage is only acquired when all three molecules are efficiently downregulated.
Surprisingly, our DLBCL cell line data shows that methylation of approximately half of the investigated alleles at the reported MIR34A TSS [33], is associated with miR34A-5p downregulation. By contrast, cell lines with a completely unmethylated MIR34A promoter show a high miR34A-5p expression. A previous study showed that many cancer cell lines carry both methylated and unmethylated MIR34A alleles, and that the expression is significantly reduced in cells with both methylated and unmethylated alleles, as compared to cells with completely unmethylated alleles [42]. As opposed to the previous study, we analyzed the miR34A-5p expression in the DLBCL cell lines by real time quantitative PCR, and could show that cells with both methylated and unmethylated MIR34A alleles have extremely low miR34A-5p expression (corresponding to ~2 copies of miR34A-5p per cell). Thus, our data suggest that mechanisms different than promoter methylation may keep the unmethylated alleles silenced , which is a subject for further investigation.
Two previous studies focus on the role of MIR34A methylation in lymphomagenesis. Analyses of a small sample (32 miscellaneous types of NHL, no DLBCLs analyzed) showed promoter hypermethylation primarily in NK/T-cell NHL [43], and a second study showed that miR34A is downregulated by promoter hypermethylation (4 of 7 analyzed DLBCLs) and/or MYC overexpression during the transformation of gastric MALT type lymphoma to aggressive gastric DLBCL [21]. However, in both studies only a limited amount of cases are investigated and none of them address the prognostic impact of MIR34A methylation. For the MDM2 SNP309 our observations are in line with previous studies [44, 45], which also show that the outcome in DLBCL is independent of the GG-phenotype.
The prognostic role of TP53 mutations in DLBCL is still debated [3, 44, 462013;48] however a recent, large study shows that TP53 disruption is still a negative prognostic factor for survival after the implementation of Rituximab[48]. In the present, relatively small study (n=62), none of the Rituximab treated patients with TP53 mutation only had died (Supplemental Figure 2), while all but one of those with concommitant disruption of TP53 and MIR34A died within the first 13 months from diagnosis. The only patient with “double hit” lymphoma that is alive after more than 5 years, is a 38 years old woman, which on revision had a primary mediastinal large B-cell lymphoma, (PMBL). This was the only patient in the “double-hit” group to achieve an etoposide containing regimen (R-CHOEP). Interestingly, PMBL was recently shown to have particularly good outcome when treated with the same drugs (dose-adjusted EPOCH-R) [49].
These data are supported by our multivariate analysis which show that neither methylation of MIR34A nor TP53 mutation alone influence survival , while the MIR34A/TP53 “doublehit” is an independent factor for survival. This is quite surprising given that studies in CLL have indicated that miR34A acts in concert with p53 and MDM2, and may serve as a surrogate marker for disruption of these molecules. Furthermore, it has been suggested that the apoptosis inducing function of miR34A is p53 dependent [50]. However, studies of the role of miR34A in cellular senescence have shown that miR34A may be activated independently of p53 to inhibit MYC in an alternative pathway that involves the ETS transcription factor ELK1 [17] (Figure 5). This is in support of the current observations, which show that patients with “double-hit” DLBCL have a particularly poor outcome.
Treatment of tumors with loss of p53 is challenging since conventional chemotherapy targets both proliferating normal cells and cancer cells. One promising new treatment strategy is cyclotherapy, which allows targeting p53 deficient cancer cells while shielding normal cells with intact p53 from cytotoxicity [512013;53]. Alternative treatment options include miR34 mimics, which retain apoptotic pathways in p53 deficient cancer cells. It has been demonstrated that the restoration of miR34A expression can inhibit the growth of TP53-mutant gastric carcinoma cells [54], and that miR34A mimics inhibit the growth of DLBCL and pulmonary carcinoma in in vivo mouse models [38, 55]. Interestingly, a recent study showed that miR34A may activate p21 downstream of p53 by downregulation of HDAC1 [56]. However, an important limitation to in vivo pharmacological targeting of miRs is systemic delivery, although novel advances such as lipophilic nanoparticles are promising. [57]. An additional concern is that the function of individual miRs may be dependent on the particular cellular context. Anti-miR34s have been shown to improve myocardial function [58], thus overexpression may lead to unforeseen side effects. However, for miR34A these obstacles may be overcome: Studies of many different cancer cell types including the present study suggest that miR34s for the larger part are downregulated by promoter hypermethylation, and we show that miR34A-5p can be upregulated by a demethylating agent in DLBCL cells with a methylated MIR34A promoter in cells with and without TP53 mutations.
Thus we believe, we have identified a novel rare, aggressive “double-hit” DLBCL that may be targeted by demethylating therapy prior to or in combination with conventional immunochemotherapy.
MATERIALS AND METHODS
Specimens
Pre-treatment DLBCL biopsies were obtained from 150 patients, and reactive lymph nodes from 6 individuals with inflammatory disease and CD19+ peripheral blood B-lymphocytes (PBL-B), and CD3+T-cells from 6 healthy donors were used as controls. All patients were treated with anthracycline containing regimens (CHOP or CHOP-like). Sixty-two of the patients also received Rituximab (R). The clinical data were obtained from the patient files and from the Danish lymphoma registry, LYFO. Approval of this study was obtained from the ethical committee.
In vitro activation of normal B-cells
PBL-B purified from healthy donors by RoboSep human CD19 positive selection kit (Stemcell) were plated 5 x 105 cells/ml and cultured in 20% HIFCS RPMI1640 with 100 units/ml penicillin, 100µg/ml streptomycin, 50 ug/ml lipopolysaccarides from Escherichia coli O55:B5 (LPS E.coli, Sigma Aldrich), and 0,6 ng/mL IL-1α (Genescript) to stimulate IgM and IgG3. The activated B cells were harvested on day 4.
Cell lines and treatment
DLBCL cell lines: DB1, Toledo, Farage, DOHH2, HT, and U2932 were cultured as above. HT and U2932 cells were seeded 2x105 cells/ml 24 hrs prior to treatment with 0.1µM and 0.5 µM of the DNA methyltransfrease inhibitor 5-Aza-2-deoxycytidine (5-Aza-CdR;Sigma-Aldrich). The medium was changed and the drug removed after 24 hrs treatment, and cells were collected 1 day and 5 days later. Untreated cells and cells treated with cytosine arabinoside (AraC; Sigma-Aldrich) 20nM or 100nM were grown and harvested under similar conditions as control.
MIR34A and MIR34B/C chromatin immunoprecipitation (ChIP)
PBL-B were crosslinked, nuclear extracts sonicated, and mixed with 10 µg of antibodies H3 (Abcam ab791), H3K27me3 (Millipore 17-622) and 4 µg of antibody H3K4me3 (Active Motif 39159). Parallel preimmune control precipitation was performed by normal IgG (Cell Signaling 2729). DNA precipitated in the ChIP experiments was amplified by RT-qPCR at the promotors, transcription start sites (TSS) and genomic regions for MIR34A and MIR34B/C using the primers in Supplemental Table 1 a/b.
Detection of TP53 mutations
The coding sequences and splice sites of exons 5-9 of the TP53 gene were scanned for mutations by PCR and denaturing gradient gel electrophoresis (DGGE) as desribed [59]. All mutations were confirmed in a second round of PCR from the original sample.
Promoter hypermethylation of MIR34A and MIR34B/C
One µg of each DNA sample was bisulfite converted using the EZ DNA Methylation kit (Zymo Research). The methylation status of the MIR34A and the MIR34B/C promoters was examined using methylation-specific melting curve analysis (MS-MCA) [60]. The DNA sequences analyzed for promoter hypermethylation locate to the promoter CpG islands at the TSSs as identified by others [33, 61]. For both segments, amplification was carried out using the LightCycler 480 instrument (Roche diagnostics) as described [62]. For primers see Supplemental Table 1c. The melting peaks were calculated using the LightCycler 480 Software Release 1.5.0SP3. SssI treated DNA (Millipore) was bisulfite converted and used as positive control. Bisulfite converted DNA from PBL-B was used as normal control.
Bisulfite sequencing
To analyze the methylation status of individual MIR34A alleles PCR products were cloned into the pCR2.1 vector using the TOPO-TA cloning kit (Invitrogen). Colonies were screened for the respective inserts, amplified using TempliPhi (Amersham Biosciences) and 10 individual colonies were sequenced. For primers see Supplemental Table 1d.
MiR34A, miR34B, and miR34C expression
Fifty ng of RNA was reverse transcribed (RT) using miRCURY LNA™ Universal RT (Exiqon). RT-qPCR for miR34A-5p, miR34A-3p, miR34B-5p, miR34B-3p, miR34C-5p and miR34C-3p was performed using miRCURY LNA™ Universal RT primer sets (nomenclature according to miRbase18), and U44 RNA was used for normalization (prod. No. 204486, 204318, 204005, 204424, 204373, 204407, 203902, respectively). The RT-qPCRs were performed on the LightCycler 480 instrument (Roche diagnostics) using the conditions recommended by Exiqon. Absolute quantification was performed using a miR-34A-5p mimic in final amounts of 1x108 copies to 10 copies per RT reaction.
Statistics
Differences in clinical characteristics of patients with or without methylation of MIR34A, MIR34B/C and mutation of TP53 were assessed using the Pearson chi-square or Fisher’s exact tests. Overall survival was estimated using the Kaplan-Meier method and log-rank test. For assessment of independent predictors of survival a multivariate Cox regression hazard model with backward stepwise (likelihood ratio) entry was applied. Effects not meeting a p-value < 0.05 were removed from the model. Statistical analyses were performed in SPSS 19.0 for Windows (SPSS Inc.) and SAS version 9.20 (Cary, NC). Any differences were considered to be statistically significant when the p-value was <0.05.
ACKNOWLEDGEMENTS
This study was supported by The Novo Nordisk Foundation, Rigshospitalets Research Foundation, The Danish Cancer Society, and University of Copenhagen.
Authorship and Disclosures:
Conceived and designed the experiments: FA CH KG. Performed the experiments: FA CH GK MTP HMN AP UR. Analyzed the data: FA CH GK MTP AP ER PB KG. Contributed reagents/materials/analysis tools: MBM ER PB KG. Wrote the paper: FA CH PB KG. All authors have read and approved the final manuscript. The authors have nothing to disclose.
References
1. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Lu L, et al.: Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000; 403: 503–511.
2. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, Müller-Hermelink HK, Campo E, Braziel RM, Jaffe ES, Pan Z, Farinha P, Smith LM, Falini B, Banham AH, Rosenwald A, et al.: Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004; 103: 275–282.
3. Grønbæ K, Brown PDN, Møller MB, Nedergaard T, Ralfkiaer E, Møller P, Zeuthen J, Guldberg P: Concurrent disruption of p16 INK4a and the ARF-p53 pathway predicts poor prognosis in aggressive non-Hodgkin ’ s lymphoma. Leukemia 2000; 14: 1727–1735.
4. Lenz G, Wright GW, Emre NCT, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, Powell J, Rosenwald A, Ott G, Muller-Hermelink HK, Gascoyne RD, Connors JM, et al.: Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A 2008; 105: 13520–13525.
5. Tagawa H, Suguro M, Tsuzuki S, Matsuo K, Karnan S, Ohshima K, Okamoto M, Morishima Y, Nakamura S, Seto M: Comparison of genome profiles for identification of distinct subgroups of diffuse large B-cell lymphoma. Blood 2005; 106: 1770–1777.
6. Jardin F, Jais JP, Molina TJ, Parmentier F, Picquenot JM, Ruminy P, Tilly H, Bastard C, Salles GA, Feugier P, Thieblemont C, Gisselbrecht C, de Reynies A, Coiffier B, Haioun C, Leroy K: Diffuse large B-cell lymphomas with CDKN2A deletion have a distinct gene expression signature and a poor prognosis under R-CHOP treatment: a GELA study. Blood 2010; 116: 1092–1104.
7. Johnson NA, Savage KJ, Ludkovski O, Ben-Neriah S, Woods R, Steidl C, Dyer MJS, Siebert R, Kuruvilla J, Klasa R, Connors JM, Gascoyne RD, Horsman DE: Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood 2009; 114: 2273–2279.
8. Aukema SM, Siebert R, Schuuring E, van Imhoff GW, Kluin-Nelemans HC, Boerma EJ, Kluin PM: Double-hit B-cell lymphomas. Blood 2011; 117: 2319–2331.
9. Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, Coetzee GA, Jones PA: Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006; 9: 435–443.
10. Baer C, Claus R, Plass C: Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res 2013; 73: 473–477.
11. Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, Ozturk A, Hicks GG, Hannon GJ, He L: miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol 2011; 13: 1353–1360.
12. Hermeking H: The miR-34 family in cancer and apoptosis. Cell Death Differ 2010; 17: 193–9.
13. Schirmer U, Doberstein K, Rupp A, Bretz NP, Kiefel H, Breunig C, Fiegl H, Müller-holzner E, Zeillinger R, Schuster E, Zeimet AG, Sültmann H: Role of miR-34a as a suppressor of L1CAM in endometrial carcinoma. Oncotarget 2014; 5, 462–472.
14. Siemens H, Jackstadt R, Kaller M, Hermeking H: Repression of c-Kit by p53 is mediated by miR-34 and is associated with reduced chemoresistance , migration and stemness. Oncotarget 2013; 4: 1399–1415.
15. Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE, Valeri N, Calore F, Sampath D, Fanini F, Vannini I, Musuraca G, et al.: Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA 2011; 305: 59–67.
16. He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ: A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130–1134.
17. Christoffersen NR, Shalgi R, Frankel LB, Leucci E, Lees M, Klausen M, Pilpel Y, Nielsen FC, Oren M, Lund AH: p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ 2010; 17: 236–245.
18. Wei JS, Song YK, Durinck S, Chen QR, Cheuk a TC, Tsang P, Zhang Q, Thiele CJ, Slack A, Shohet J, Khan J: The MYCN oncogene is a direct target of miR-34a. Oncogene 2008; 27: 5204–5213.
19. Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X: Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett 2008; 582: 1564–1568.
20. Asslaber D, Piñón JD, Seyfried I, Desch P, Stöcher M, Tinhofer I, Egle A, Merkel O, Greil R: microRNA-34a expression correlates with MDM2 SNP309 polymorphism and treatment-free survival in chronic lymphocytic leukemia. Blood 2010; 115: 4191–4197.
21. Craig VJ, Cogliatti SB, Imig J, Renner C, Neuenschwander S, Rehrauer H, Schlapbach R, Dirnhofer S, Tzankov A, Müller A: Myc-mediated repression of microRNA-34a promotes high-grade transformation of B-cell lymphoma by dysregulation of FoxP1. Blood 2011; 117: 6227–6236.
22. Dufour A, Palermo G, Zellmeier E, Mellert G, Duchateau-Nguyen G, Schneider S, Benthaus T, Kakadia PM, Spiekermann K, Hiddemann W, Braess J, Truong S, Patten N, Wu L, Lohmann S, Dornan D, et al. Inactivation of TP53 correlates with disease progression and low miR-34a expression in previously treated chronic lymphocytic leukemia patients. Blood 2013; 121: 3650–3657.
23. Wong KY, Huang X, Chim CS: DNA methylation of microRNA genes in multiple myeloma. Carcinogenesis 2012; 33: 1629–1638.
24. Wong KY, Yim RLH, So CC, Jin DY, Liang R, Chim CS: Epigenetic inactivation of the MIR34B/C in multiple myeloma. Blood 2011; 118: 5901–5904.
25. Zhu Y, Monni O, Franssila K, Elonen E, Vilpo J, Joensuu H, Knuutila S: Deletions at 11q23 in different lymphoma subtypes. Haematologica 2000; 85: 908–912.
26. Tagawa H, Tsuzuki S, Suzuki R, Karnan S, Ota A, Kameoka Y, Suguro M, Matsuo K, Yamaguchi M, Okamoto M, Morishima Y, Nakamura S, Seto M: Genome-wide array-based comparative genomic hybridization of diffuse large B-cell lymphoma: comparison between CD5-positive and CD5-negative cases. Cancer Res 2004; 64: 5948–5955.
27. Amini RM, Berglund M, Rosenquist R, Von Heideman A, Lagercrantz S, Thunberg U, Bergh J, Sundström C, Glimelius B, Enblad G: A novel B-cell line (U-2932) established from a patient with diffuse large B-cell lymphoma following Hodgkin lymphoma. Leuk Lymphoma 2002; 43: 2179–2189.
28. Lawrie CH, Soneji S, Marafioti T, Cooper CDO, Palazzo S, Paterson JC, Cattan H, Enver T, Mager R, Boultwood J, Wainscoat JS, Hatton CSR: MicroRNA expression distinguishes between germinal center B cell-like and activated B cell-like subtypes of diffuse large B cell lymphoma. Int J Cancer 2007; 121: 1156–1161.
29. Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, Banham AH, Pezzella F, Boultwood J, Wainscoat JS, Hatton CSR, Harris AL: Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Br J Haematol 2008; 141: 672–675.
30. Lawrie CH, Chi J, Taylor S, Tramonti D, Ballabio E, Palazzo S, Saunders NJ, Pezzella F, Boultwood J, Wainscoat JS, Hatton CSR: Expression of microRNAs in diffuse large B cell lymphoma is associated with immunophenotype, survival and transformation from follicular lymphoma. J Cell Mol Med 2009; 13: 1248–1260.
31. Pedersen IM, Otero D, Kao E, Miletic AV, Hother C, Ralfkiaer E, Rickert RC, Grønbæk K, David M: Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med 2009; 1: 288–95.
32. Costinean S, Sandhu SK, Pedersen IM, Tili E, Trotta R, Perrotti D, Ciarlariello D, Neviani P, Harb J, Kauffman LR, Shidham A, Croce CM: Src homology 2 domain-containing inositol-5-phosphatase and CCAAT enhancer-binding protein beta are targeted by miR-155 in B cells of Emicro-MiR-155 transgenic mice. Blood 2009; 114: 1374–1382.
33. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT: Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26: 745–752.
34. Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C, van Rijk A, Nyagol J, Byakika B, Lazzi S, Tosi P, van Krieken H, Leoncini L: MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol 2008; 216: 440–450.
35. Yamakuchi M, Ferlito M, Lowenstein CJ: miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 2008; 105: 13421–13426.
36. Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, Elia A, Kress TR, Dickens M, Clemens MJ, Heery DM, Gaestel M, Eilers M, Willis AE, Bushell M: p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci U S A 2010; 107: 5375–5380.
37. Rao DS, O’Connell RM, Chaudhuri AA, Garcia-Flores Y, Geiger TL, Baltimore D: MicroRNA-34a perturbs B lymphocyte development by repressing the forkhead box transcription factor Foxp1. Immunity 2010; 33: 48–59.
38. Craig VJ, Tzankov a, Flori M, Schmid C a, Bader a G, Müller a: Systemic microRNA-34a delivery induces apoptosis and abrogates growth of diffuse large B-cell lymphoma in vivo. Leukemia 2012; 26: 2421–2424.
39. Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A: Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A 2008; 105: 12979–12984.
40. Mills AA: Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer 2010; 10: 669–682.
41. Asmar F, Punj V, Christensen J, Pedersen MT, Pedersen A, Nielsen AB, Hother C, Ralfkiaer U, Brown P, Ralfkiaer E, Helin K, Grønbæk K: Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 2013; 98: 1912–1920.
42. Lodygin D, Tarasov V, Epanchintsev A, Berking C, Knyazeva T, Körner H, Knyazev P, Diebold J, Hermeking H: Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle 2008; 7: 2591–2600.
43. Chim CS, Wong KY, Qi Y, Loong F, Lam WL, Wong LG, Jin DY, Costello JF, Liang R: Epigenetic inactivation of the miR-34a in hematological malignancies. Carcinogenesis 2010; 31: 745–750.
44. Zainuddin N, Berglund M, Wanders A, Ren Z-P, Amini R-M, Lindell M, Kanduri M, Roos G, Rosenquist R, Enblad G: TP53 mutations predict for poor survival in de novo diffuse large B-cell lymphoma of germinal center subtype. Leuk Res 2009; 33: 60–66.
45.Bittenbring J, Parisot F, Wabo A, Mueller M, Kerschenmeyer L, Kreuz M, Truemper L, Landt O, Menzel A, Pfreundschuh M, Roemer K: MDM2 gene SNP309 T/G and p53 gene SNP72 G/C do not influence diffuse large B-cell non-Hodgkin lymphoma onset or survival in central European Caucasians. BMC Cancer 2008; 8: 116.
46. Ichikawa A, Kinoshita T, Watanabe T, Kato H, Nagai H, Tsushita K, Saito H, Hotta T: Mutations of the p53 gene as a prognostic factor in aggressive B-cell lymphoma. N Engl J Med 1997; 337: 529–534.
47. Young KH, Leroy K, Møller MB, Colleoni GWB, Sánchez-Beato M, Kerbauy FR, Haioun C, Eickhoff JC, Young AH, Gaulard P, Piris MA, Oberley TD, Rehrauer WM, Kahl BS, Malter JS, Campo E, et al.: Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood 2008; 112: 3088–3098.
48. Xu-Monette ZY, Wu L, Visco C, Tai YC, Tzankov A, Liu W, Montes-Moreno S, Dybkaer K, Chiu A, Orazi A, Zu Y, Bhagat G, Richards KL, Hsi ED, Zhao XF, Choi WWL, et al.: Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012; 120: 3986–3996.
49. Dunleavy K, Pittaluga S, Maeda LS, Advani R, Chen CC, Hessler J, Steinberg SM, Grant C, Wright G, Varma G, Staudt LM, Jaffe ES, Wilson WH: Dose-adjusted EPOCH-rituximab therapy in primary mediastinal B-cell lymphoma. N Engl J Med 2013; 368: 1408–1416.
50. Merkel O, Asslaber D, Piñón JD, Egle A, Greil R: Interdependent regulation of p53 and miR-34a in chronic lymphocytic leukemia. Cell Cycle 2010; 9: 2764–2768.
51. Van Leeuwen IMM: Cyclotherapy: opening a therapeutic window in cancer treatment. Oncotarget 2012; 3: 596–600.
52. Blagosklonny M V: Sequential activation and inactivation of G2 checkpoints for selective killing of p53-deficient cells by microtubule-active drugs. Oncogene 2002; 21: 6249–6254.
53. Rao B, van Leeuwen IMM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S: Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1: 639–650.
54. Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D, Xu L: Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer 2008; 8: 266.
55. Wiggins JF, Ruffino L, Kelnar K, Omotola M, Patrawala L, Brown D, Bader AG: Development of a lung cancer therapeutic based on the tumor suppressor microRNA-34. Cancer Res 2010; 70: 5923–5930.
56. Zhao J, Lammers P, Torrance CJ, Bader AG: TP53-independent function of miR-34a via HDAC1 and p21(CIP1/WAF1.). Mol Ther 2013; 21: 1678–86.
57. Bader AG: miR-34 – a microRNA replacement therapy is headed to the clinic. Front Genet 2012.
58. Bernardo BC, Gao X-M, Winbanks CE, Boey EJH, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du X-J, Lin RCY, McMullen JR: Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci 2012; 109: 17615–17620.
59. Guldberg P, Nedergaard T, Nielsen HJ, Olsen AC, Ahrenkiel V, Zeuthen J: Single-step DGGE-based mutation scanning of the p53 gene: application to genetic diagnosis of colorectal cancer. Hum Mutat 1997; 9: 348–355.
60. Worm J, Aggerholm A, Guldberg P: In-tube DNA methylation profiling by fluorescence melting curve analysis. Clin Chem 2001; 47: 1183–1189.
61. Toyota M, Suzuki H, Sasaki Y, Maruyama R, Imai K, Shinomura Y, Tokino T: Epigenetic silencing of microRNA-34b/c and B-cell translocation gene 4 is associated with CpG island methylation in colorectal cancer. Cancer Res 2008; 68: 4123–4132.
62. Guldberg P, Worm J, Grønbaek K: Profiling DNA methylation by melting analysis. Methods 2002; 27: 121–127.