
INTRODUCTION
During the last years, intensive basic research has led to an increasing number of treatment options for cancer patients. This in turn has created an urgent need for biomarkers to predict response to targeted therapy and to monitor emergent drug resistance. Therapy decision for many tumour types is based on biomarkers that are evaluated at the primary tumour by the classic biopsy approach; however, classic biopsy is not allowing monitoring of primary tumours evolution during time, while repeated sampling of metastatic sites is not always possible for practical reasons. “Liquid biopsy” based on minimally-invasive blood-based tests has the potential to be a strong complement to the classic biopsy approach; Circulating Tumour Cells (CTCs) and circulating tumour DNA (ctDNA) are the major players in liquid biopsy analysis, since they can both provide an alternative approach that enables a sensitive, dynamic and specific serial tumour sampling during the course of treatment, and an early marker of response to systemic therapy [1–3]. An important requisite for liquid biopsy technologies is that CTCs or ctDNA are present in blood, and that they can be successfully isolated and analyzed. One major question for liquid biopsy technologies is whether CTCs and ctDNA composition is representative of the patient’s tumour and whether they provide comparable or complementary information.
Nowadays, the detection and molecular characterization of CTCs is one of the most active and hot areas of translational cancer research [3, 4]. Therefore there is a considerable interest in CTCs research and CTCs assays development since these cells are well-defined targets for understanding tumour cell dissemination [5, 6]. The clinical importance of CTCs enumeration has been shown and is FDA-cleared for metastatic breast, colorectal and prostate cancer already a decade ago [7]. Especially in breast cancer CTCs detection and enumeration is associated with poor outcome and a number of clinical trials are now further evaluating its clinical importance [8]. Many groups so far, even by using completely different experimental approaches have clearly shown that understanding CTCs biology could be a key issue in favor of cancer patients [9].
On the other hand, the verified presence of extractable amounts of ctDNA circulating in serum and plasma of cancer patients, suggests that ctDNA carrying tumour specific alterations has a very strong potential as a liquid biopsy biomarker as well [10, 11]. The analysis of ctDNA in plasma is an extremely promising tool to identify mutations relevant to certain targeted therapies and monitor tumour evolution during disease progression [10, 11]. Moreover, ctDNA is a relatively stable biological material, much easier to isolate and analyze than CTCs. The recent identification of mutations, translocations, or copy number variations previously found in resected tumours by the classic biopsy approach in ctDNA represents a quite simple liquid biopsy approach. However even ctDNA analysis seems simpler to perform than CTCs, we must have in mind that it requires very sensitive techniques since ctDNA is just a very small percentage of cell free DNA (cfDNA) that is circulating in plasma [12, 13]. In a very limited number of breast cancer patients, it was recently shown that ctDNA provided the earliest measure of treatment response in metastatic breast cancer [14].
DNA methylation plays a fundamental role in the development and progression of many types of cancer, mainly through the inactivation of certain tumour-suppressor genes [15]. Moreover, DNA methylation is considered to be an early event in the process of cancer development and progression since tumour suppressor genes are frequently inactivated at very early stages. Detection of tumour-specific DNA methylation alterations in ctDNA could provide important information for the clinical assessment of breast cancer patients [16]. Our group has shown for the first time that tumour suppressor and metastasis suppressor genes are epigenetically silenced in CTCs isolated from peripheral blood of breast cancer patients [17]. Moreover we have recently shown that SOX17 promoter is highly methylated in primary breast tumours, in CTCs isolated both from patients with early and metastatic breast cancer, and in corresponding ctDNA samples [18].
The primary goal of our present study was to investigate to what extent DNA isolated from CTCs and plasma is representing the primary tumour and whether is carries similar information in terms of DNA methylation biomarkers. To address this question we performed a direct comparison study of the methylation status of SRY (sex-determining region Y)-box 17 (SOX17), Cystatin M (CST6) and Breast cancer metastasis suppressor 1 (BRMS1) in primary tumours, EpCAM-positive pooled CTC-fractions and ctDNA in well characterized matched clinical samples of breast cancer patients.
RESULTS
An outline of our study is shown in Figure 1.
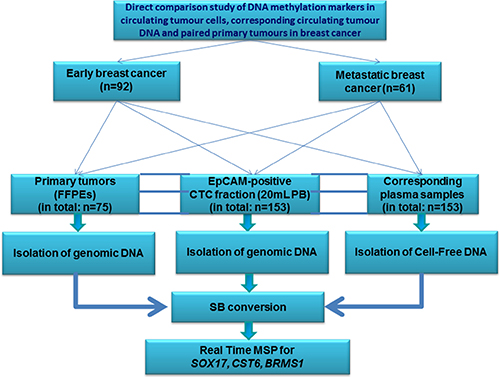
Figure 1: Workflow of the study.
Analytical sensitivity and specificity of real time MSP assays
Evaluation of the analytical sensitivity and specificity of the real time MSP assays used for the evaluation of SOX17, CST6 and BRMS1 methylation was crucial, since ctDNA is only a fraction of cfDNA circulating in plasma, while CTCs are highly heterogeneous and only a subgroup of cells is expected to carry DNA methylated sequences. The analytical sensitivity of the developed real-time MSP assays for SOX17, CST6 and BRMS1 was evaluated by using the above described synthetic control mixtures. According to our results, the developed real-time MSP assays for SOX17, CST6 and BRMS1 could specifically and reliably detect methylated sequences when present at 0.1%. The analytical specificity of the developed real-time MSP assays for SOX17, CST6 and BRMS1, was validated by initially testing all primers in silico and then in PCR, using SB-modified human placental gDNA (0% methylated) and unconverted DNA; no amplification of the SOX17, CST6 and BRMS1 promoter was observed.
SOX17, CST6 and BRMS1 promoter methylation in primary breast tumours (FFPEs)
FFPE samples were available for 75 out of these 153 patients (Table 1). SOX17 was detected in 26/42 (61.9%) patients diagnosed with early breast cancer and in 21/33 (63.6%) patients that developed metastasis. CST6 promoter methylation was observed in 11/31 (35.5%) patients with early breast cancer and 16/32 (50.0%) patients with metastasis. BRMS1 promoter methylation was detected in 19/42 (45.2%) patients with early breast cancer and 5/32 (15.6%) metastatic patients. There was no correlation between the methylation status of these genes and tumour size, number of lymph nodes, tumour grade, tumour stage, PR and ER receptors, HER2 status and age (data not shown). The diagnostic specificity of real-time MSP assays for each gene was further evaluated in non-cancerous breast tissues (mammoplasties) and fibroadenomas used as controls. SOX17 promoter was methylated in 3/29(10.3%) non-cancerous breast tissues, and in 2/9 (22.2%) breast fibroadenomas. CST6 promoter was methylated in 3/29 (10.3%) non-cancerous breast tissues (mammoplasties) and in 1/10 (10.0%) breast fibroadenomas. BRMS1 promoter methylation was detected neither in 29 non-cancerous breast tissues nor in the 10 fibroadenoma samples tested.
Table 1: SOX17, CST6 and BRMS1 gene promoter methylation in the EpCAM positive CTC-fraction, paired ctDNA and corresponding FFPE samples
Samples |
SOX17 |
CST6 |
BRMS1 |
|---|---|---|---|
Early breast cancer group (n = 92) |
|||
EpCAM-positive CTC-fraction |
19/92 (20.7%) |
24/92 (26.1%) |
20/92 (21.7%) |
paired ctDNA |
24/92 (26.1%) |
33/92 (35.9%) |
22/92 (23.9%) |
FFPEs* |
26/42 (61.9%) |
11/31 (35.5%) |
19/42 (45.2%) |
Metastatic breast cancer group (n = 61) |
|||
EpCAM-positive CTC-fraction |
26/61 (42.6%) |
21/61 (34.4%) |
31/61 (50.8%) |
paired ctDNA |
22/61 (36.1%) |
26/61 (42.6%) |
4/61 (6.6%) |
FFPEs* |
21/33 (63.6%) |
16/32 (50.0%) |
5/32 (15.6%) |
Control group |
|||
EpCAM-positive CTC-fraction (healthy individuals) |
1/23 (4.3%) |
1/23 (4.3%) |
2/23 (8.7%) |
Plasma, cfDNA |
1/49 (2.0%) |
2/49 (4.1%) |
2/49 (4.1%) |
Fibroadenomas (FFPEs) |
2/9 (22.2%) |
1/10 (10.0%) |
0/10 (0%) |
Non-cancerous breast tissues (FFPEs - mammoplasties) |
3/29(10.3%) |
3/29 (10.3%) |
0/29 (0%) |
*: FFPEs were not available for all clinical samples studied at the peripheral blood level (CTCs and ctDNA).
SOX17, CST6 and BRMS1 promoter methylation in DNA isolated from the EpCAM-positive CTC-fraction
We evaluated SOX17, CST6, and BRMS1 promoter methylation in the EpCAM-positive CTC-fraction of 153 DNA samples isolated from 92 patients with early and 61 patients with metastatic breast cancer (Table 1). SOX17 promoter was methylated in 19/92 (20.7%) patients with early and in 26/61 (42.6%) patients with metastatic breast cancer. CST6 promoter was methylated in 24/92 (26.1%) patients with early, and in 21/61 (34.4%) patients with metastatic breast cancer. BRMS1 promoter was methylated in 20/92 (21.7%) patients with early, and in 31/61 (50.8%) patients with metastatic breast cancer. According to our findings there was no correlation between the promoter methylation status of these genes in the EpCAM-positive CTC-fraction and tumour size, number of lymph nodes, tumour grade, tumour stage, the presence of progesterone (PR) and estrogen receptors (ER), HER2 status, and age (data not shown). The diagnostic specificity of real-time MSP assays for each gene was further evaluated by analyzing the EpCAM-positive CTC-fraction of 23 healthy individuals used as control group revealing that promoter methylation of SOX17 was observed in 1/23 (4.3%), CST6 in 1/23 (4.3%) and BRMS1 in 2/23 (8.7%) healthy individuals.
SOX17, CST6 and BRMS1 promoter methylation in ctDNA isolated from matched plasma samples
We further evaluated the methylation status of SOX17, CST6, and BRMS1 promoter methylation in ctDNA isolated from matched plasma samples from the same 153 patients as above and 49 healthy individuals (Table 1). Promoter methylation of SOX17 was observed in 24/92 (26.1%) patients with early breast cancer, in 22/61 (36.1%) patients with metastatic disease and in 1/49 (2.0%) healthy individuals. Promoter methylation of CST6 was observed in 33/92 (35.9%) patients with early, 26/61 (42.6%) patients with metastatic breast cancer and 2/49 (4.1%) healthy individuals. Promoter methylation of BRMS1 was observed in 22/92 (23.9%) patients with early and 4/61 (6.6%) patients with metastatic breast cancer and 2/49 (4.1%) healthy individuals. Finally, there was no correlation between the methylation status of these genes in ctDNA and tumour size, number of lymph nodes, tumour grade, tumour stage, the presence of PR and ER receptors, HER2 status, and age (data not shown).
Direct comparison between SOX17, CST6, and BRMS1 methylation in the EpCAM-positive CTC-fraction, ctDNA and corresponding primary tumours
We further directly compared the methylation status of SOX17, CST6, and BRMS1 promoter in the EpCAM-positive CTC-fraction, corresponding ctDNA and paired FFPEs and investigated whether there is a direct association.
Association between EpCAM-positive CTC-fraction and corresponding ctDNA
We found a concordance between the EpCAM-positive CTC-fraction and ctDNA in both early and metastatic breast cancer only for SOX17 promoter methylation but not for CST6 and BRMS1 (Table 2). There was a clear association for SOX17 promoter methylation both for patients with early (P = 0.001, 71/92, 77.2% cases) and metastatic breast cancer (P = 0.046, 39/61, 63.9% cases).
Table 2: Comparison between SOX17, CST6 and BRMS1 gene promoter methylation in the EpCAM-positive CTC-fraction and corresponding paired ctDNA samples in early (n = 92) and metastatic breast cancer patients (n = 61)
Early breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
EpCAM-positive CTC-fraction |
ctNA |
TOTAL |
p value |
κ |
% concordance |
|
U |
M |
||||||
SOX17 |
U |
60 |
13 |
73 |
0.001 |
0.3653 |
71/92 (77.2%) |
M |
8 |
11 |
19 |
||||
TOTAL |
68 |
24 |
92 |
||||
CST6 |
U |
44 |
24 |
68 |
nsa |
0.0197 |
53/92 (57.6%) |
M |
15 |
9 |
24 |
||||
TOTAL |
59 |
33 |
92 |
||||
BRMS1 |
U |
56 |
16 |
72 |
nsa |
0.0751 |
62/92 (67.4%) |
M |
14 |
6 |
20 |
||||
TOTAL |
70 |
22 |
92 |
||||
Metastatic breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
EpCAM positive CTC-fraction |
ctNA |
TOTAL |
p value |
κ |
%concordance |
|
U |
M |
||||||
SOX17 |
U |
26 |
9 |
35 |
0.046 |
0.2478 |
39/61 (62.9%) |
M |
13 |
13 |
26 |
||||
TOTAL |
39 |
22 |
61 |
||||
CST6 |
U |
25 |
15 |
40 |
nsa |
0.1408 |
36/61 (59.0%) |
M |
10 |
11 |
21 |
||||
TOTAL |
35 |
26 |
61 |
||||
BRMS1 |
U |
28 |
2 |
30 |
nsa |
/ |
30/61 (49.2%) |
M |
29 |
2 |
31 |
||||
TOTAL |
57 |
4 |
61 |
||||
a: non significant.
Association between the EpCAM-positive CTC-fraction and corresponding FFPEs
In early breast cancer we found no association between the EpCAM-positive CTC-fraction and 42 available paired primary tumours in respect to the promoter methylation status of any of the three genes studied (Table 3); concordance for SOX17 promoter methylation was observed only in 15/42 (35.7%) cases, while the concordances were very low for CST6, and BRMS1. In the metastatic setting, there was a concordance for 21/33 (63.6%) of cases for SOX17 promoter methylation, while there was no concordance for CST6 and BRMS1.
Table 3: Comparison between SOX17, CST6 and BRMS1 gene promoter methylation in the EpCAM-positive CTC-fraction and corresponding paired FFPE samples, and in ctDNA and corresponding FFPEs in early and metastatic breast cancer patients
EpCAM-positive CTC-fraction versus corresponding paired FFPE samples: Early breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
EpCAM positive CTC-fraction |
FFPEs |
TOTAL |
p value |
κ |
% concordance |
|
U |
M |
||||||
SOX17 |
U |
12 |
23 |
35 |
nsa |
/ |
15/42 (35.7%) |
M |
4 |
3 |
7 |
||||
total |
16 |
26 |
42 |
||||
CST6 |
U |
17 |
8 |
25 |
nsa |
0.1367 |
20/31 (64.5%) |
M |
3 |
3 |
6 |
||||
total |
20 |
11 |
31 |
||||
BRMS1 |
U |
19 |
17 |
36 |
nsa |
/ |
21/42 (50%) |
M |
4 |
2 |
6 |
||||
total |
23 |
19 |
42 |
||||
EpCAM-positive CTC-fraction versus corresponding paired FFPE samples: Metastatic breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
EpCAM- positive CTC-fraction |
FFPEs |
TOTAL |
p value |
κ |
% concordance |
|
U |
M |
||||||
SOX17 |
U |
8 |
8 |
16 |
nsa |
0.2667 |
21/33 (63.6%) |
M |
4 |
13 |
17 |
||||
total |
12 |
21 |
33 |
||||
CST6 |
U |
10 |
11 |
21 |
nsa |
/ |
15/32 (46.9%) |
M |
6 |
5 |
11 |
||||
total |
16 |
16 |
32 |
||||
BRMS1 |
U |
17 |
2 |
19 |
nsa |
0.1390 |
20/32 (62.5%) |
M |
10 |
3 |
13 |
||||
total |
27 |
5 |
32 |
||||
ctDNA versus corresponding paired FFPE samples: Early breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
ctDNA |
FFPEs |
TOTAL |
p value |
κ |
% concordance |
|
U |
M |
||||||
SOX17 |
U |
11 |
18 |
29 |
nsa |
19/42 (45.2%) |
|
M |
5 |
8 |
13 |
||||
total |
16 |
26 |
42 |
||||
CST6 |
U |
15 |
4 |
19 |
nsa |
w |
22/31 (71.0%) |
M |
5 |
7 |
12 |
||||
total |
20 |
11 |
31 |
||||
BRMS1 |
U |
20 |
16 |
36 |
nsa |
0.0292 |
23/42 (54.8%) |
M |
3 |
3 |
6 |
||||
total |
23 |
19 |
42 |
||||
ctDNA versus corresponding paired FFPE samples: Metastatic breast cancer group |
|||||||
|---|---|---|---|---|---|---|---|
Gene promoter methylation |
ctDNA |
FFPEs |
TOTAL |
p value |
κ |
% concordance |
|
U |
M |
||||||
SOX17 |
U |
8 |
12 |
20 |
nsa |
0.0833 |
17/33 (51.5%) |
M |
4 |
9 |
13 |
||||
total |
12 |
21 |
33 |
||||
CST6 |
U |
11 |
7 |
18 |
nsa |
0.250 |
20/32 (62.5%) |
M |
5 |
9 |
14 |
||||
total |
16 |
16 |
32 |
||||
BRMS1 |
U |
26 |
4 |
30 |
nsa |
0.2157 |
27/32 (84.4%) |
M |
1 |
1 |
2 |
||||
total |
27 |
5 |
32 |
||||
a: non significant.
Association between ctDNA and corresponding FFPEs
In early breast cancer, we found no association between ctDNA and 42 available paired primary tumours in respect to the promoter methylation status of these three genes (Table 3); concordance for SOX17 promoter methylation was observed in 19/42 (45.2%) cases while the concordances for CST6, and BRMS1 were very low. In the metastatic setting, for SOX17 there was a concordance for 17/33 (51.5%) cases and there was no concordance for CST6, and BRMS1.
Association between CK-19 mRNA expression and SOX17, CST6 and BRMS1 promoter methylation in the EpCAM-positive CTC-fraction
We further examined CK-19 mRNA expression in all EpCAM-positive CTC-fractions, since we have extensively used this epithelial marker to verify the presence of CTCs (19–21). Using the same procedure, none of 60 healthy individual samples tested (0%) was found positive for CK-19 expression. We further studied the association between CK-19 mRNA expression and SOX17, CST6 and BRMS1 promoter methylation in the EpCAM- positive CTC-fraction. Results are shown in heat maps for each individual patient in all cases (Figure 2).
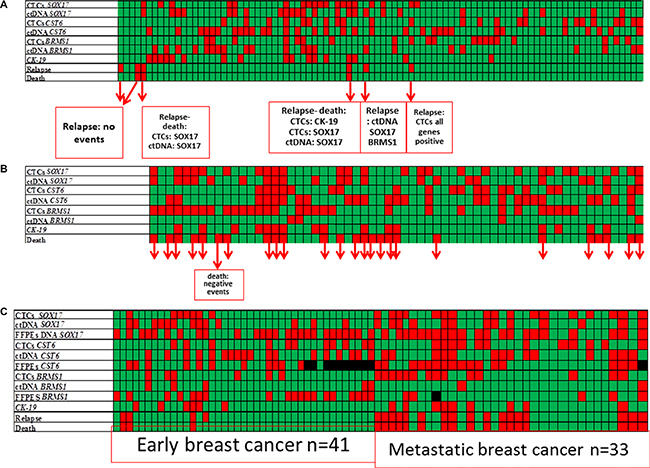
Figure 2: Heat map of SOX17, CST6 and BRMS1 promoter methylation and CK-19 expression in the EpCAM-positive CTC-fraction, and ctDNA in matched samples of patients with: (A) operable breast cancer (n = 92), (B) verified metastasis (n = 61), and (C) all samples including available FFPEs (n = 75). Color code: green: non-methylated/no-relapse/alive, red: methylated/relapse/dead.
Early breast cancer
In early breast cancer, SOX17 methylation status in the EpCAM-positive CTC-fraction was found to be correlated with CK-19 mRNA expression (P = 0.006). Results are shown as a heat maps for each individual patient in Figure 2A.
More specifically, 17/92 (18.5%) samples were positive for CK-19 mRNA while 19/92 (20.6%) samples were positive for SOX17 methylation; 8/92 (8.7%) samples were positive for both SOX17 promoter methylation and CK-19 mRNA and 9/92 (9.8%) samples were positive for CK-19, but were not carrying SOX17 promoter methylation. It is highly remarkable that SOX17 promoter methylation was also identified in the EpCAM-positive CTC-fraction of 11 patients who were negative for CK-19 mRNA expression. However, in the same group CST6 and BRMS1 methylation status was not correlated with CK-19 mRNA expression. More specifically, only 4/92 (4.3%) samples were positive for both CST6 promoter methylation and CK-19 mRNA expression, 13/92 (14.1%) samples were positive for CK-19, but not carrying CST6 promoter methylation, while CST6 promoter methylation was identified in the EpCAM-positive CTC-fraction of 20 patients who were negative for CK-19 mRNA. In the same group only 2/92 (2.1%) samples were positive for both BRMS1 promoter methylation and CK-19 mRNA expression, 16/92 (17.4%) samples were positive for CK-19 but not carrying BRMS1 promoter methylation, while BRMS1 promoter methylation was identified in 17 patients who were negative for CK-19 mRNA expression.
Metastatic disease
In this group 6/61 (26.2%) samples were found positive for CK-19 expression in the EpCAM-positive CTC-fraction. Results are shown as a heat maps for each individual patient in Figure 2B. SOX17, CST6 and BRMS1 promoter methylation were not individually correlated with CK-19 expression. However, in the EpCAM-positive CTC-fraction of the 45 patients who were negative for CK-19 mRNA expression, SOX17 promoter methylation was identified in 18/45 (40.0%), CST6 promoter methylation in 14/45 (31.1%) and BRMS1 promoter methylation in 20/45 (44.4%).
Association between CK-19 mRNA expression and SOX17, CST6 and BRMS1 promoter methylation in corresponding ctDNA
We further studied the association between CK-19 mRNA expression and SOX17, CST6 and BRMS1 promoter methylation in corresponding ctDNA. Results are shown in heat maps for each individual patient in all cases (Figure 2).
Early breast cancer
In this group, there was a statistically significant association between DNA methylation of one or more of these genes in ctDNA and CK-19 expression in the corresponding EpCAM-positive CTC-fraction (P = 0.014). Results are shown as a heat maps for each individual patient in Figure 2A. In 58/92 (63.0%) of patients at least one of the genes studied was found methylated in ctDNA. There were 32/92 (34.8%) cases where all markers were negative, 15/92 (16.3%) cases positive for both DNA methylation in ctDNA and CK-19 mRNA expression, while there were only 2/92 (2.2%) cases positive for CK-19 and negative for SOX17, CST6 and BRMS1 promoter methylation. It is highly remarkable that DNA methylation was identified in ctDNA of 43 patients who were negative for CK-19 mRNA expression.
Metastatic disease
Results are shown as a heat maps for each individual patient in Figure 2B. In this group, SOX17 promoter methylation in ctDNA was highly correlated with CK-19 expression (P = 0.040). CST6 and BRMS1 promoter methylation in ctDNA were not individually correlated with CK-19 expression. However samples that were found to have at least two gene promoters methylated in ctDNA were also CK-19 mRNA positive (P = 0.021).
Association of SOX17, CST6, and BRMS1 promoter methylation in CTC, ctDNA and corresponding primary tumours with clinical outcome
We further evaluated whether there is any association between SOX17, CST6, and BRMS1 promoter methylation in CTC, ctDNA and corresponding primary tumours and the clinical outcome of patients. Results are shown as a heat maps for each individual patient in Figure 2.
Early breast cancer group
After a median follow-up period of 40 months (range 6–121), 6/92 (6.5%) patients with early breast cancer relapsed and 3/92 (3.3%) of them died as a consequence of disease progression. In 4/6 patients that relapsed SOX17 gene promoter was found highly methylated in the primary tumour, in the corresponding CTC-fraction and in ctDNA (Figure 2A). It is worth mentioning that in one patient that relapsed very early, her blood specimen was found methylated in the EpCAM-positive CTC-fraction, for all genes tested, while it was CK-19 negative. The direct comparison study of SOX17, CST6 and BRMS1 promoter methylation in corresponding FFPEs (n =41) is shown in Figure 2C. Kaplan–Meier estimates of the cumulative Disease Free Interval (DFI) were significantly different in favor of patients with non-methylated BRMS1 promoter (Figure 3A; Ρ = 0.042), as we have previously reported for a different cohort of breast cancer patients (27). Concerning CTCs, patients with non-methylated SOX17 promoter in the EpCAM-positive CTC-fraction had a significantly better median overall survival (OS) compared to patients with methylated SOX17 promoter (Figure 3B; P = 0.044).
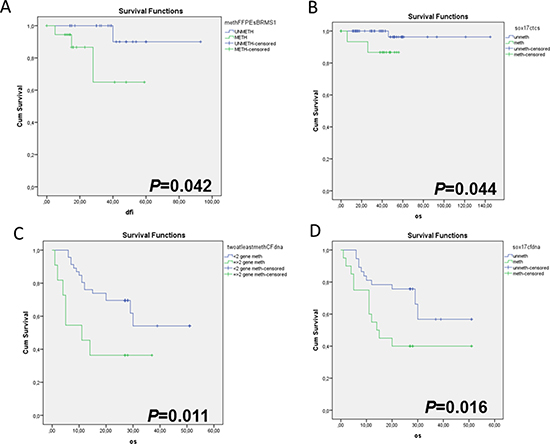
Figure 3: Kaplan-Meier estimates of (A) Disease-free interval (DFI) in months for early breast cancer patients in respect to BRMS1 promoter methylation status in FFPEs (P = 0.042), (B) Overall survival (OS) in months for early breast cancer patients with clinically confirmed metastasis, in respect to SOX17 promoter methylation status in CTCs (P = 0.044), (C) Overall survival (OS) in months for breast cancer patients with clinically confirmed metastasis, in respect to the methylation of at least two genes in ctDNA (P = 0.011), (D) Overall survival (OS) in months for breast cancer patients with clinically confirmed metastasis, in respect to SOX17 promoter methylation status in ctDNA (P = 0.016).
Metastatic breast cancer group
During the follow up, 25/61 (40.9%) patients with metastatic disease died as a consequence of disease progression. The direct comparison study of SOX17, CST6 and BRMS1 promoter methylation in corresponding FFPEs (n = 33) is shown in Figure 2C. In this group, the incidence of deaths was higher when at least two gene promoters were methylated in ctDNA in respect to patients where one or none gene promoter was methylated (Figure 3C; P = 0.011). It is interesting to note that the Kaplan–Meier estimates of the OS were significantly different in favor of patients with non-methylated SOX17 promoter (Figure 3D; P = 0.016).
DISCUSSION
In the present study we performed for the first time a direct comparison study of the DNA methylation status of SOX17, CST6 and BRMS1 in primary tumours, corresponding plasma ctDNA and in matched cell pools of EpCAM-positive CTCs, in order to investigate whether the tumour’s “liquid phase” reflects the status of the primary tumour. We also wanted to evaluate to what extend these markers yield complementary or additional information about prognosis and disease progression. By using specimens derived from the same patients and identical blood draws, and by applying identical and analytically validated methodologies for the MSP assays used, we minimized most pre-analytical parameters that could affect this comparison. The procedure that we have followed for CTCs enrichment and analysis in the present study has been validated in previous studies and has been compared with commercially available molecular assays [20–23].
DNA methylation of these three gene promoters has been extensively studied so far mainly in primary tissues of various types of cancer. We and others have already shown that detection of CST6 promoter methylation in primary tissues provides important prognostic information in patients with operable breast cancer [24, 25]. Furthermore, we reported for the first time that CST6 promoter is methylated in ctDNA and is promising as a highly specific tumour biomarker for early breast cancer patients since it was not detected in plasma of healthy individuals [26].
We have recently shown that BRMS1 promoter methylation in the primary tumour was associated with poor DFS while our study on BRMS1 protein expression by immunofluorescence revealed CTCs heterogeneity even in the same patient [22]. BRMS1 is a predominantly nuclear protein that differentially regulates expression of multiple genes, leading to suppression of metastasis without blocking orthotopic tumour growth [27, 28]. BRMS1 mRNA expression has been shown to be markedly reduced in melanoma, breast cancer, ovarian cancer and non-small cell lung cancer cell lines, while stable overexpression of BRMS1 in these cell lines significantly inhibited their metastatic potential [29–31].
SOX17 plays a critical role in the regulation of development and stem/precursor cell function, at least partly through repression of the canonical Wnt/β-catenin signaling pathway [32, 33]. We have shown already in a limited number of patients that SOX17 promoter is highly methylated in CTCs isolated from patients with breast cancer [17], and in corresponding ctDNA samples [17, 18].
According to our findings, there was no concordance between the EpCAM-positive CTC-fraction and paired primary tumours in respect to the promoter methylation status of any of the genes studied, neither in early, nor in the metastatic setting. A lack of concordance was also found for the same DNA methylation markers between ctDNA and matched primary tumours. A possible explanation for this finding could be based on tumour heterogeneity and rapid evolution through time, indicating that the liquid biopsy is reflecting the actual phase of tumour evolution. It is now clear that CTCs are not only rare but heterogeneous at the same time, even within the same patient [34, 35]. This could partially explain the differences seen in our study.
There are very few studies so far comparing DNA methylation markers in primary tumours, CTCs and paired ctDNA samples [17, 18, 22]. In all these previous studies we have already shown that DNA methylation markers can be detected in EpCAM-positive CTC-fractions but not in the primary tumour, and that EpCAM-positive CTC-fractions and ctDNA do not give identical but highly correlated information. This is the first study where so many samples are analyzed in the primary tumour setting in EpCAM-positive CTCs and in corresponding plasma using the same blood draw, identical methodologies and three genes. A possible explanation for the discrepancies detected between primary tumours, CTCs and ctDNA could be that in the primary tumours the amount of methylated cancer cells could be a small subgroup between millions non-methylated cancer cells. Because of the heterogeneity of the tumour tissue, the tumour tissue biopsy section may contain cells that are not representative for the entire cancer volume. As a result, the methylation information of the whole tissue is more difficult to interpret because we need samples from different sites of the cancer tissue in order to characterize the whole volume. Methylated DNA sequences detected in ctDNA can be released from necrotic and apoptotic cells, while methylated DNA sequences detected in the EpCAM positive CTC-fractions are indicative of an active-metastasis condition.
Our present data indicate a direct association between SOX17 promoter methylation in CTCs and ctDNA in patients with breast cancer. We report for the first time that in the EpCAM-positive CTC-fraction, SOX17 promoter methylation was associated with CK-19 mRNA expression both in patients with early and metastatic breast cancer. In addition, our findings revealed that methylation of SOX17 promoter in ctDNA is associated with unfavorable prognosis in patients with metastatic breast cancer and that in patients with early breast cancer, methylation of SOX17 promoter in the EpCAM-positive CTC-fraction is associated with decreased DFI. We did not find any association between the methylation status of CST6 and BRMS1 in CTCs, ctDNA and corresponding primary tumours. This could be attributed to tumour evolution over time and to the Epithelial Mesenchymal Transition (EMT) phase of CTCs [36].
In the EpCAM-positive CTC-fraction, we found that all these three genes were methylated at significantly higher percentages in the metastatic setting than in early stages of the disease. This could be explained by the higher tumour load in the metastatic setting. It is also worth mentioning that in plasma the number of samples found positive for SOX17 and CST6 promoter methylation were higher than in the EpCAM-positive CTC-fraction while non-specific methylation was detected at even lower percentages in all cases. This could possibly indicate a release of methylated DNA from apoptotic cells as well. Non-specific methylation events were detected at a very low percentage in all our control samples.
It is also interesting to note that in most early breast cancer patients that relapsed early, SOX17 gene promoter was found highly methylated in the primary tumour, in the EpCAM-positive CTC-fraction and in ctDNA. It is also worth mentioning that the blood specimen of one early breast cancer patient that relapsed very early was found methylated in the EpCAM-positive CTC-fraction, for all genes tested, while it was CK-19 negative. We plan to verify this finding in the future by analysing a larger number of patients. It is also remarkable to note that samples found positive for SOX17, CST6 and BRMS1 promoter methylation in the EpCAM-positive CTC-fraction were negative for CK-19 mRNA expression. If CTCs positivity would be based only on CK-19 expression these samples would have been characterized as CTC-negative. This indicates the importance of multi-parametric testing in CTC analysis.
In the metastatic setting, much higher detection rates for CK-19 are usually expected, as we have previously shown using the same RT-qPCR assay [19]. However, the lower percentages found for CK-19 in the present study can be explained, because only samples for which we had available primary tumours and corresponding plasma for ctDNA isolation were selected for analysis.
Molecular characterization of single CTCs combined with massive parallel sequencing (MPS) technologies performed both in single CTC and paired plasma could elucidate further the association between CTCs and ctDNA [37, 38]. Very recent studies have clearly shown that the isolation, ex vivo culture, and characterization of CTCs is a very promising approach to monitor continuously and in a non-invasive way the changing patterns of drug susceptibility in individual patients as their tumours acquire new mutations [39, 40]. However, the processing of blood specimens for CTC analysis is complicated and time consuming. By comparison, ctDNA analysis has recently gained a lot of attendance since it has the potential to be convenient and relatively simple to process in a short time period [41, 42]. Recently the feasibility and potential utility of plasma ctDNA as an alternative to metastatic biopsies for mutational analysis in breast cancer using NGS has been shown [43]. However in this study, discordances between primary tumours and plasma were also reported. Recent results demonstrate also that exome sequencing on cfDNA is a very powerful tool for disease monitoring of metastatic cancers [44].
Concerning the comparison between CTCs and ctDNA, the most important parameter is correlation of the biomarker under evaluation with the clinical outcome. As an example, in a very recent study, Madic et al. used the high prevalence of TP53 mutations in triple negative breast cancer (TNBC) to compare ctDNA and CTCs detection rates and evaluate their prognostic value in metastatic TNBC patients [45]. They used different markers and different methodologies to compare ctDNA and CTCs detection rates, the presence of TP53 for ctDNA as verified by NGS, and the CellSearchTM FDA cleared system for the enumeration of CTC. They report an absence of prognostic impact of baseline ctDNA level suggesting that mechanisms of ctDNA release in metastatic TNBC may involve, beyond tumour burden, biological features that do not dramatically affect patient outcome [45].
CTCs molecular analysis when compared to ctDNA and exosomes as a liquid biopsy approach has the clear advantage that its prognostic significance has already been shown through numerous studies [5–9], and that it addresses viable cells that are not simply carriers of individual tumour biomarkers. Moreover, CTCs represent live tumour cell entities that can give information on emerging tumour sub-clones with altered mutational and drug sensitivity profiles and can be targetable by specific drugs. From this point of view, CTCs molecular characterization can offer an essential component of personalized cancer treatment [4].
In conclusion our findings indicate a direct association only between SOX17 promoter methylation in CTCs and ctDNA both in patients with early breast cancer and in patients with metastatic disease but not for the other genes studied. We could not identify a direct association between the methylation status of these genes in primary tumours CTCs and ctDNA, possibly because tumours are evolving and the liquid biopsy is reflecting the actual state of tumour evolution. Combination of DNA methylation analysis of tumour suppressor and metastasis suppressor genes in EpCAM-positive CTCs and matched ctDNA provides significant prognostic value. We plan to extend this direct comparison study and verify these findings in larger cohorts and for a wide range of gene promoters, using Massive Parallel Sequencing technologies. We will evaluate the clinical significance of these findings in an independent study, in respect to the clinical characteristics of the patients and additional markers already studied by our group on CTCs (methylation, mutations, miRNAs and gene expression) in a large number of patients with a known clinical outcome and a longer follow up.
MATERIALS AND METHODS
Patients
We directly compared SOX17, CST6 and BRMS1 promoter methylation in ctDNA and in the EpCAM-positive CTC-fraction of 153 breast cancer patients; 92 with early and 61 with metastatic breast cancer. Peripheral blood was isolated from all these patients for CTCs and ctDNA analysis. In early breast cancer cases peripheral blood was collected at least two weeks after surgery and before the initiation of adjuvant chemotherapy, while in the metastasis group samples were collected before the initiation of first line treatment. For 75 of these patients the corresponding FFPEs samples from the primary tumours were available. In all cases the primary tumor of patients that relapsed after several months or years had been analysed. More specifically we analysed: a) EpCAM-positive CTC-fraction samples isolated from peripheral blood of all patients (n = 153); for all these samples, information on the expression of CK-19 in the EpCAM-positive CTC-fraction was also available, b) ctDNA samples isolated from corresponding plasma of the same patients during the same venepuncture (n = 153) and c) 75 corresponding primary breast cancer formalin-fixed paraffin-embedded tissues (FFPE) (42 and 33 from patients with early and metastatic breast cancer, respectively). As a control population, peripheral blood was collected from 60 healthy individuals, while 29 non-cancerous breast tissues (mammoplasties) and 10 breast fibroadenomas were used as non-cancerous breast tissue controls. All study participants signed an informed consent form to participate in the study, which was approved by the ethics and scientific committees of our institutions.
DNA isolation from FFPEs
Tissue sections of 10 μm containing 80% tumour cells were used for DNA extraction. DNA 100% methylated (DNA methylation standard, Zymo Research) was used as a positive control in MSP for the detection of SOX17, CST6 and BRMS1 promoter methylation. Genomic DNA (gDNA) from FFPEs was isolated using the QIAamp DNA FFPE Tissue Kit (Qiagen, Germany). DNA concentration was determined in the Nanodrop ND-1000 spectrophotometer (Nanodrop Technologies, USA).
Isolation of the EpCAM-positive CTC-fraction
The EpCAM-positive CTC-fraction was isolated from 20 mL peripheral blood as previously described [17, 18, 46]. All DNA isolation and handling steps took place in a dedicated area and in a laminal flow hood. gDNA was extracted from the EpCAM-positive CTC-fraction as previously described [17, 18]. Total RNA isolation from the EpCAM-positive CTC-fraction was performed using Trizol (Invitrogen, USA) as previously described [46]. All RNA preparation and handling steps took place in a dedicated area in a laminar flow hood, under RNase-free conditions. mRNA was isolated from the total RNA by use of the Dynabeads mRNA purification Kit (Invitrogen, USA) according to the manufacturer’s instructions. cDNA synthesis was performed using the High-Capacity RNA-to-cDNA kit (Applied Biosystems, USA) and was used for CK-19 expression studies in CTCs as previously described [46].
Isolation of ctDNA from plasma
Peripheral blood in EDTA was collected and processed immediately for plasma isolation. All samples were centrifuged at 1600 g (10 min), and plasma was carefully transferred into 2 mL tubes and stored at −20°C until ctDNA isolation. The High Pure Viral nucleic acid kit (Roche Diagnostics) was used to extract ctDNA from plasma (200 μL) as previously described [17, 18].
Sodium bisulfite conversion
Preparation of sodium bisulfite (SB) conversion reaction was performed in a laminar flow hood, in a dedicated separate room. For all clinical samples, before proceeding to SB conversion and MSP reaction steps, the integrity of isolated gDNA was assessed by amplifying exon 20 of the BRCA1 gene as previously described [47]. gDNA extracted from both isolated EpCAM-positive CTC-fractions and plasma and gDNA from FFPEs was SB-modified using the EZ DNA methylation–Gold kit (Zymo Research, USA) as previously described [17, 18]. In each set of SB reactions, deionized water and DNA 100% methylated (DNA methylation standard, Zymo Research) were included as negative and positive control, respectively.
Real time methylation specific PCR (MSP)
For high throughput analysis of our samples we designed highly specific and sensitive real Time MSP assays for each gene of interest. All experiments were performed in the LightCycler 2.0 (IVD instrument, Roche, Germany). All oligonucleotides were de-novo in-silico designed for each gene by using the PrimerPremier 5 software (Premier Biosoft International, USA), and synthesized by IDT (Intergraded DNA technologies, USA) [26, 48]. For each gene, primer pairs and LNA probes are given in Supplementary Table 1. Each reaction was performed in a total volume of 10 μL, and 1 μL of SB-converted DNA was added to a 9 μL reaction mixture [17, 18, 22].
Quality control of real time MSP
To verify that we could specifically detect only the targeted methylated sequences, in each MSP reaction we used the following controls: a) gDNA not submitted to SB-conversion (unconverted DNA) and placental DNA submitted to SB-conversion (placental converted DNA, 0% methylated), were included as negative controls and b) SB-converted DNA from the DNA methylation standard (100%) were included in every run as positive control. To exclude false negative results, all SB-converted samples found negative by real-time MSP, were checked for DNA quality by using a specifically designed primer set that equally amplifies in the same genomic region both methylated and non-methylated SB-converted target sequences.
To evaluate the analytical sensitivity of the developed real-time MSPs we prepared synthetic control mixtures by serial dilutions of SB-converted DNA control samples (0% and 100% methylated). More specifically the SB-converted DNA methylation standard (100%) was spiked in SB-converted placental DNA (0% methylated) in a concentration range of 0%, 1%, 25%, and 50%. For the evaluation of analytical sensitivity, 1 μL of these synthetic samples were used in the Real- Time MSP reactions.
Statistical analysis
We used the χ2 test of independence for data analysis and for the evaluation of the significance of differences between groups. We used the Fisher exact test and Cohen’s kappa coefficient, for the evaluation of agreement between methylation of each gene in respect to its presence in CTCs and ctDNA and in respect to CK-19 gene expression [49]. Correlations between methylation status and clinico-pathological features of the patients were assessed by using the Chi-square test. Disease free interval (DFI), Progression Free Survival (PFS) and overall survival (OS) curves were calculated by using the Kaplan Meier method and comparisons were performed using the long rank test. P values < 0.05 were considered statistically significant. Statistical analysis was performed by using the SPSS Statistics 22.0 Windows program (SPSS Inc., Chicago, IL).
ACKNOWLEDGMENTS AND FUNDING
This work has received funding from the European Union’s Seventh Framework Programme (FP7/2007-2013) for the Innovative Medicine Initiative under grant agreement no (115749). It was also partly funded by “Onco-Seed diagnostics” grant, under the Sinergasia 2009 program that was co-funded by the European Regional Development Fund and National Resources (General Secretariat of Research and Technology in Greece), Project code: Onco-Seed diagnostics. We would like to thank Dr. G. Kalergi (Lab of Tumour Biology, University of Crete) for kindly donating the MCF-7 cell line. MC, AS, performed the experiments, NM, SK and VG provided the patients’ samples and all the clinical data needed, ES conceived and supervised the whole study and wrote the manuscript.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Alix-Panabières C, Pantel K. Circulating tumor cells: liquid biopsy of cancer. Clin Chem. 2013; 59:110–18. https://doi.org/10.1373/clinchem.2012.194258.
2. Lianidou ES, Mavroudis D, Georgoulias V. Clinical challenges in the molecular characterization of circulating tumour cells in breast cancer. Br J Cancer. 2013; 108:2426–32. https://doi.org/10.1038/bjc.2013.265.
3. Schlange T, Pantel K. Potential of circulating tumor cells as blood-based biomarkers in cancer liquid biopsy. Pharmacogenomics. 2016; 17:183–86. https://doi.org/10.2217/pgs.15.163.
4. Lianidou ES. Molecular characterization of circulating tumor cells: holy Grail for personalized cancer treatment? Clin Chem. 2014; 60:1249–51. https://doi.org/10.1373/clinchem.2014.230144.
5. Lianidou ES, Strati A, Markou A. Circulating tumor cells as promising novel biomarkers in solid cancers. Crit Rev Clin Lab Sci. 2014; 51:160–71. https://doi.org/10.3109/10408363.2014.896316.
6. Lianidou ES, Mavroudis D, Pantel K. Advances in circulating tumor cells (ACTC): from basic research to clinical practice. Breast Cancer Res. 2013; 15:319. https://doi.org/10.1186/bcr3582.
7. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004; 351:781–91. https://doi.org/10.1056/NEJMoa040766.
8. Bidard FC, Fehm T, Ignatiadis M, Smerage JB, Alix-Panabières C, Janni W, Messina C, Paoletti C, Müller V, Hayes DF, Piccart M, Pierga JY. Clinical application of circulating tumor cells in breast cancer: overview of the current interventional trials. Cancer Metastasis Rev. 2013; 32:179–88. https://doi.org/10.1007/s10555-012-9398-0.
9. Zhang L, Riethdorf S, Wu G, Wang T, Yang K, Peng G, Liu J, Pantel K. Meta-analysis of the prognostic value of circulating tumor cells in breast cancer. Clin Cancer Res. 2012; 18:5701–10. https://doi.org/10.1158/1078-0432.CCR-12-1587.
10. Marzese DM, Hirose H, Hoon DS. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Expert Rev Mol Diagn. 2013; 13:827–44. https://doi.org/10.1586/14737159.2013.845088.
11. Diaz LA Jr, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014; 32:579–86. https://doi.org/10.1200/JCO.2012.45.2011.
12. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012; 4:136ra68. https://doi.org/10.1126/scitranslmed.3003726.
13. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108–12. https://doi.org/10.1038/nature12065.
14. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–209. https://doi.org/10.1056/NEJMoa1213261.
15. Esteller M. Epigenetics in cancer. N Engl J Med. 2008; 358:1148–59. https://doi.org/10.1056/NEJMra072067.
16. Fackler MJ, Lopez Bujanda Z, Umbricht C, Teo WW, Cho S, Zhang Z, Visvanathan K, Jeter S, Argani P, Wang C, Lyman JP, de Brot M, Ingle JN, et al. Novel methylated biomarkers and a robust assay to detect circulating tumor DNA in metastatic breast cancer. Cancer Res. 2014; 74:2160–70. https://doi.org/10.1158/0008-5472.CAN-13-3392.
17. Chimonidou M, Strati A, Tzitzira A, Sotiropoulou G, Malamos N, Georgoulias V, Lianidou ES. DNA methylation of tumor suppressor and metastasis suppressor genes in circulating tumor cells. Clin Chem. 2011; 57:1169–77. https://doi.org/10.1373/clinchem.2011.165902.
18. Chimonidou M, Strati A, Malamos N, Georgoulias V, Lianidou ES. SOX17 promoter methylation in circulating tumor cells and matched cell-free DNA isolated from plasma of patients with breast cancer. Clin Chem. 2013; 59:270–79. https://doi.org/10.1373/clinchem.2012.191551.
19. Stathopoulou A, Vlachonikolis I, Mavroudis D, Perraki M, Kouroussis C, Apostolaki S, Malamos N, Kakolyris S, Kotsakis A, Xenidis N, Reppa D, Georgoulias V. Molecular detection of cytokeratin-19-positive cells in the peripheral blood of patients with operable breast cancer: evaluation of their prognostic significance. J Clin Oncol. 2002; 20:3404–12. https://doi.org/10.1200/JCO.2002.08.135.
20. Stathopoulou A, Ntoulia M, Perraki M, Apostolaki S, Mavroudis D, Malamos N, Georgoulias V, Lianidou ES. A highly specific real-time RT-PCR method for the quantitative determination of CK-19 mRNA positive cells in peripheral blood of patients with operable breast cancer. Int J Cancer. 2006; 119:1654–59. https://doi.org/10.1002/ijc.22017.
21. Xenidis N, Ignatiadis M, Apostolaki S, Perraki M, Kalbakis K, Agelaki S, Stathopoulos EN, Chlouverakis G, Lianidou E, Kakolyris S, Georgoulias V, Mavroudis D. Cytokeratin-19 mRNA-positive circulating tumor cells after adjuvant chemotherapy in patients with early breast cancer. J Clin Oncol. 2009; 27:2177–84. https://doi.org/10.1200/JCO.2008.18.0497.
22. Chimonidou M, Kallergi G, Georgoulias V, Welch DR, Lianidou ES. Breast cancer metastasis suppressor-1 promoter methylation in primary breast tumors and corresponding circulating tumor cells. Mol Cancer Res. 2013; 11:1248–57. https://doi.org/10.1158/1541-7786.MCR-13-0096.
23. Strati A, Kasimir-Bauer S, Markou A, Parisi C, Lianidou ES. Comparison of three molecular assays for the detection and molecular characterization of circulating tumor cells in breast cancer. Breast Cancer Res. 2013; 15:R20. https://doi.org/10.1186/bcr3395.
24. Kioulafa M, Balkouranidou I, Sotiropoulou G, Kaklamanis L, Mavroudis D, Georgoulias V, Lianidou ES. Methylation of cystatin M promoter is associated with unfavorable prognosis in operable breast cancer. Int J Cancer. 2009; 125:2887–92. https://doi.org/10.1002/ijc.24686.
25. Barekati Z, Radpour R, Lu Q, Bitzer J, Zheng H, Toniolo P, Lenner P, Zhong XY. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer. 2012; 12:244. https://doi.org/10.1186/1471-2407-12-244.
26. Chimonidou M, Tzitzira A, Strati A, Sotiropoulou G, Sfikas C, Malamos N, Georgoulias V, Lianidou E. CST6 promoter methylation in circulating cell-free DNA of breast cancer patients. Clin Biochem. 2013; 46:235–40. https://doi.org/10.1016/j.clinbiochem.2012.09.015.
27. Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000; 60:2764–69.
28. Phadke PA, Vaidya KS, Nash KT, Hurst DR, Welch DR. BRMS1 suppresses breast cancer experimental metastasis to multiple organs by inhibiting several steps of the metastatic process. Am J Pathol. 2008; 172:809–17. https://doi.org/10.2353/ajpath.2008.070772.
29. Shevde LA, Samant RS, Goldberg SF, Sikaneta T, Alessandrini A, Donahue HJ, Mauger DT, Welch DR. Suppression of human melanoma metastasis by the metastasis suppressor gene, BRMS1. Exp Cell Res. 2002; 273:229–39. https://doi.org/10.1006/excr.2001.5452.
30. Yang J, Zhang B, Lin Y, Yang Y, Liu X, Lu F. Breast cancer metastasis suppressor 1 inhibits SDF-1alpha-induced migration of non-small cell lung cancer by decreasing CXCR4 expression. Cancer Lett. 2008; 269:46–56. https://doi.org/10.1016/j.canlet.2008.04.016.
31. Zhang S, Lin QD, Di W. Suppression of human ovarian carcinoma metastasis by the metastasis-suppressor gene, BRMS1. Int J Gynecol Cancer. 2006; 16:522–31. https://doi.org/10.1111/j.1525-1438.2006.00547.x.
32. Zhang W, Glöckner SC, Guo M, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, Ahuja N, Baylin SB. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res. 2008; 68:2764–72. https://doi.org/10.1158/0008-5472.CAN-07-6349.
33. Du YC, Oshima H, Oguma K, Kitamura T, Itadani H, Fujimura T, Piao YS, Yoshimoto T, Minamoto T, Kotani H, Taketo MM, Oshima M. Induction and down-regulation of Sox17 and its possible roles during the course of gastrointestinal tumorigenesis. Gastroenterology. 2009; 137:1346–57. https://doi.org/10.1053/j.gastro.2009.06.041.
34. Powell AA, Talasaz AH, Zhang H, Coram MA, Reddy A, Deng G, Telli ML, Advani RH, Carlson RW, Mollick JA, Sheth S, Kurian AW, Ford JM, et al. Single cell profiling of circulating tumor cells: transcriptional heterogeneity and diversity from breast cancer cell lines. PLoS One. 2012; 7:e33788. https://doi.org/10.1371/journal.pone.0033788.
35. Polzer B, Klein CA. Metastasis awakening: the challenges of targeting minimal residual cancer. Nat Med. 2013; 19:274–75. https://doi.org/10.1038/nm.3121.
36. Bednarz-Knoll N, Alix-Panabières C, Pantel K. Plasticity of disseminating cancer cells in patients with epithelial malignancies. Cancer Metastasis Rev. 2012; 31:673–87. https://doi.org/10.1007/s10555-012-9370-z.
37. Swennenhuis JF, Reumers J, Thys K, Aerssens J, Terstappen LW. Efficiency of whole genome amplification of single circulating tumor cells enriched by CellSearch and sorted by FACS. Genome Med. 2013; 5:106. https://doi.org/10.1186/gm510.
38. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Höfler G, Eisner F, Sill H, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013; 73:2965–75. https://doi.org/10.1158/0008-5472.CAN-12-4140.
39. Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, Wittner BS, Stojanov P, Brachtel E, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014; 345:216–20. https://doi.org/10.1126/science.1253533.
40. Speicher MR, Pantel K. Tumor signatures in the blood. Nat Biotechnol. 2014; 32:441–43. https://doi.org/10.1038/nbt.2897.
41. Luke JJ, Oxnard GR, Paweletz CP, Camidge DR, Heymach JV, Solit DB, Johnson BE, and Cell Free DNA Working Group. Realizing the potential of plasma genotyping in an age of genotype-directed therapies. J Natl Cancer Inst. 2014; 106:106. https://doi.org/10.1093/jnci/dju214.
42. Madhavan D, Wallwiener M, Bents K, Zucknick M, Nees J, Schott S, Cuk K, Riethdorf S, Trumpp A, Pantel K, Sohn C, Schneeweiss A, Surowy H, Burwinkel B. Plasma DNA integrity as a biomarker for primary and metastatic breast cancer and potential marker for early diagnosis. Breast Cancer Res Treat. 2014; 146:163–74. https://doi.org/10.1007/s10549-014-2946-2.
43. Rothé F, Laes JF, Lambrechts D, Smeets D, Vincent D, Maetens M, Fumagalli D, Michiels S, Drisis S, Moerman C, Detiffe JP, Larsimont D, Awada A, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014; 25:1959–65. https://doi.org/10.1093/annonc/mdu288.
44. Klevebring D, Neiman M, Sundling S, Eriksson L, Darai Ramqvist E, Celebioglu F, Czene K, Hall P, Egevad L, Grönberg H, Lindberg J. Evaluation of exome sequencing to estimate tumor burden in plasma. PLoS One. 2014; 9:e104417. https://doi.org/10.1371/journal.pone.0104417.
45. Madic J, Kiialainen A, Bidard FC, Birzele F, Ramey G, Leroy Q, Rio Frio T, Vaucher I, Raynal V, Bernard V, Lermine A, Clausen I, Giroud N, et al. Circulating tumor DNA and circulating tumor cells in metastatic triple negative breast cancer patients. Int J Cancer. 2015; 136:2158–65. https://doi.org/10.1002/ijc.29265.
46. Strati A, Markou A, Parisi C, Politaki E, Mavroudis D, Georgoulias V, Lianidou E. Gene expression profile of circulating tumor cells in breast cancer by RT-qPCR. BMC Cancer. 2011; 11:422. https://doi.org/10.1186/1471-2407-11-422.
47. Kroupis C, Christopoulos K, Devetzoglou M, Tsiagas I, Lianidou ES. Asymmetric real-time PCR detection of BRCA1 5382insC mutation by melting curve analysis in the LightCycler. Clin Chim Acta. 2008; 390:141–44. https://doi.org/10.1016/j.cca.2007.12.024.
48. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002; 18:1427–31. https://doi.org/10.1093/bioinformatics/18.11.1427.
49. Berry CC. The kappa statistic. JAMA. 1992; 268:2513–14. https://doi.org/10.1001/jama.1992.03490180045013.