
INTRODUCTION
It appears increasingly clear that the stable acquisition of a cancer phenotype involves metabolic remodeling. This echoes the pioneering studies from Otto Warburg and can be achieved through redirecting glucose and non glucose-dependent pathways toward anabolic generation of macromolecules, a crucial requirement for cancer cells[1, 2]. As a proof of this, multilayered modulation of metabolic enzymes by known oncogenes and tumor suppressors has been recently unveiled, with more detailed data available regarding the c-MYC-mediated modulation of glycolysis and glutamine metabolism in cancer cells [3]. Resistance to therapy is an inherent part of the pro-tumorigenic program and, almost invariably, an adverse prognostic factor for solid and non-solid tumors. Emergence within the tumor mass, of distinct chemoresistant cell populations has been recognized as an important mechanism for chemoresistance, hence tumor relapse. We and others have characterized chemoresistant cell subpopulations from breast and mesothelioma cell lines and shown that those cells are endowed with Epithelial-To-Mesenchymal (EMT) features, exhibit a precursor-like phenotype and possess high levels of Aldehyde Dehydrogenase (ALDH) activity [4, 5]. ALDH belongs to a class of detoxifying enzymes whose expression is linked to cancer chemoresistance [6, 7] and, by virtue of those high levels of ALDH activity, chemoresistant cell subpopulations can be tracked by FACS (ALDHbright cells). We and others have shown that breast, ALDHbright-enriched cancer cell subpopulations are resistant in vitro to campthotecin, cisplatin, etoposide, topotecan [5] and docetaxel (in vivo) [8]. Tanei et al have reported that ALDH1+ cells are increased in a group of 78 breast cancer patients after neoadjuvant chemotherapy [9] and such phenomenon has been shown to occur in early passage colon cancer xenograft tumors as well [10]. Consequently, ALDH expression can be an important prognostic factor [6, 11]. Little is known regarding the metabolic features of the ALDHbright chemoresistant cell subpopulations. Here we explore which are the metabolic features of the chemoresistant ALDHbright cells and whether their metabolic characteristics reflect their functional properties. This may add precious knowledge to the mechanisms of tumor relapse and its modulation, to achieve anticancer effects. With regard to this latter point, metformin, an oral anti-diabetic biguanide, has been shown to target chemoresistant putative cancer stem cells from a variety of solid tumors, including lung, prostate, ovary cancer and glioma [12-14]. We and others have shown that metformin interferes with tumor engraftment and synergizes with chemotherapy in mouse xenografts, with both effects that suggest the targeting of chemoresistant, tumor initiating cell populations within the tumor mass. Additionally, we have shown a metabolic anticancer effect of metformin on unfractionated breast cancer cells lines which is partially dependent on DICER-mediated microRNA modulation [15]. However, to our knowledge, no studies have detailed the effect of metformin on purified chemoresistant cells in terms of metabolic modulation and microRNA modulation, in particular whether the metabolic effect of metformin are similar or different from those we described on unfractionated cell populations. Thus, here we studied the metabolic features of ALDHbright cells isolated from three histologically different breast cancer cell lines. We first show that ALDHbright cells are metabolically different from ALDHlow cells. Subsequently, we describe how metformin treatment affects ALDHbright cell metabolism by reducing the differences between the chemoresistant ALDHbright and the chemosensitive ALDHlow cells through targeting pyruvate metabolism, glycolysis, glutathione metabolism, pentose phosphate pathway, HIF-1α and the insulin signalling pathways. Additionally, we show that metformin treatment largely modulated the microRNA expression profile of ALDHbright cells and did so by broadly modulating microRNAs predicted to impinge on cell metabolism and to target the mentioned pathways. Finally, we show that metformin modulated master cancer metabolic modulators such as c-MYC and IRS2 in the ALDHbright cells and this correlated with modulation of the microRNA-33a which is known to target both the abovementioned factors. We believe this study adds on what is known on the anticancer metabolic actions of metformin and further provides a rationale for its potential use combined with chemotherapy to prevent tumor relapse, through targeting of chemoresistant, residual cell subpopulations.
RESULTS
FACS-based isolation of breast cancer ALDHbright and ALDHlow cells (Fig.1).
We determined by FACS the percentage of cells endowed with high ALDH activity (ALDHbright cells) from MCF-7, BT-474 and SUM-159 cell lines. ALDH enzymatic activity was detected by following the accumulation of a fluorescent ALDH substrate, with the mean fluorescence of cells pretreated with a known inhibitor of the ALDH activity (DEAB) set as background (Fig 1A-B). We found that all the breast cancer cell lines in our collection contained readily detectable ALDHbright cells, although with some variability among each cell line. Given the involvement of the ALDHbright cells in mediating cancer chemoresistance, we treated MCF-7, BT-474 and SUM159 cells with cisplatin and doxorubicin, two commonly used chemotherapy agents for breast cancer and we evaluated the percentage of ALDHbright cells among the surviving cell populations. We found that, upon chronic treatment with cisplatin (40µM, 72hrs) or doxorubicin (0.1µM, 72hrs) the percentage of breast cancer ALDHbright cells increased in BT-474 and SUM-159 cells and remained unchanged or only slightly decreased in the MCF-7 cells (Fig. 1A-B and Suppl. Fig.1). Interestingly, a barely detectable number of ALDHbright cells was found in the dysplastic, non-transformed breast cells MCF10A (1.4 ± 0.4 %)(Fig.1B and Suppl. Fig.1). This is in agreement with previous work [5]. However, the MCF-10A ALDHbright cells became undetectable upon cisplatin and doxorubicin treatment (Fig.1B and Suppl. Fig.1), in agreement with the chemosensitivity of the cell line[16]. Clonogenic assays with FACS-purified ALDHbright and ALDHlow cells confirmed that the ALDHbright cells in the transformed breast cancer cell lines represented the main chemoresistant cell subpopulation as compared to the ALDHlow cells (the latter representing most of the cells in unsorted cell lines) (Fig. 1C-D). The availability of the described experimental system prompted us to characterize the metabolic profile of the purified ALDHbright chemoresistant cell subpopulations.
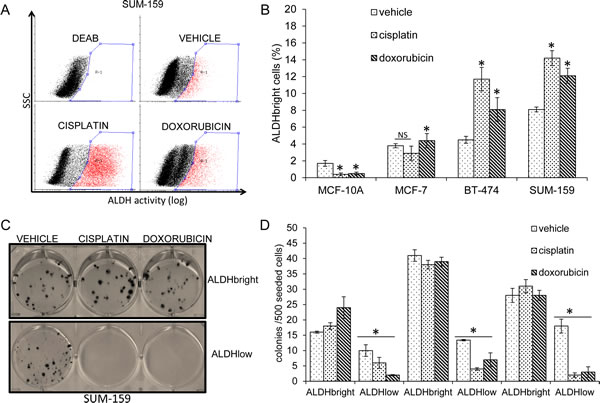
Fig.1: Breast cancer ALDHbright cells are chemoresistant. A. Representative FACS dot plots of SUM-159 cells assayed for Aldhehyde Dehydrogenase (ALDH) activity. Cells enriched for ALDH activity (ALDHbright) were gated (red-r1). To set the background, the percentage of ALDHbright cells was determined in the presence of DEAB, an inhibitor of the ALDH enzyme (upper left plot). B. Histogram showing the percentage of ALDHbright cells in the indicated cell lines treated with vehicle, cisplatin or doxorubicin for 72hrs. Mean ± SE values of two independent experiments were reported. NS: not significant. *: p<0.05 (when compared to vehicle-treated samples). Additional representative dot plots relative to the graph in 1B are available in Suppl. Fig. 1. C. Clonogenic assay. Representative micrographs of the colonies formed by SUM-159 cells pulsed with vehicle, cisplatin and doxorubicin for 16hrs and seeded at clonal density. Colonies stained with crystal violet 9 days later. D. Histogram showing the absolute number of colonies formed by the indicated cell lines treated as in 1C. Please note that the untransformed MCF-10A were not assayed because non clonogenic. Mean ± SE values of two independent experiments were reported. *: p<0.05 (as compared to ALDHbright cells).
Table 1: Metabolites contributing to the difference between ALDHbright and ALDHlow, determined by the analysis of O-PLS-DA loadings. |
||
Metabolite |
ALDHbright vs. ALDHlow* |
Related KEGG Pathway Maps** |
3-Hydroxy-butyrate |
High (p) |
Synthesis and degradation of ketone bodies |
Alanine |
High (p) |
Alanine, aspartate and glutamate metabolism |
Lactate |
High (p) |
Glycolysis/Gluconogenesis |
Pyruvate metabolism |
||
Acetate |
Low (p) |
Glycolysis/Gluconogenesis |
Pyruvate metabolism |
||
Succinate |
Low (c) |
Citrate cycle (TCA cycle) |
Oxidative phosphorylation |
||
Alanine, aspartate and glutamate metabolism |
||
Tyrosine metabolism |
||
Phenylalanine metabolism |
||
Carbon metabolism |
||
Glucose |
Low (c) |
Glycolysis/Gluconogenesis |
Pentose phosphate pathway |
||
Amino sugar and nucleotide sugar metabolism |
||
HIF-1 signaling pathway |
||
Insulin signaling pathway |
||
Formate |
High (p) |
Pyruvate metabolism |
Carbon metabolism |
||
*“High” indicates that the metabolite was at a higher concentration in medium samples from ALDHbright cells; “Low” indicates that the metabolite was at a lower concentration in medium samples from ALDHbright cells; “c” and “p” for each metabolite indicate consumption or production, respectively.
** The metabolites are mapped to their respective biochemical pathways as delineated in the Kyoto Encyclopedia of Genes and Genomes (Release 69.0, January 1, 2014; KEGG, http://www.genome.jp/kegg).
The metabolic profile of ALDHbright cells is different from that of ALDHlow cells (Fig. 2).
The conditioned media from FACS sorted ALDHbright and ALDHlow MCF-7, BT-474 and SUM-159 cells, were analysed using 1H-NMR spectroscopy. NMR profiles identified at the beginning of observation were compared to those representing the end of the experiment (after 24 hours). In this way the resulting positive net balances indicated release or production of metabolites, whereas negative net balances indicated uptake or consumption of metabolites. We analyzed the NMR data by using Principal Component Analysis (PCA) carried out on untreated samples. This unsupervised method allows orthogonal decomposition of variance associated with the analyzed metabolites. Six components (PC 1-6) were sufficient to explain 65% of total variability of the system. In Fig. 2A, the PC1 (the most important metabolic component explaining 21% of total variance of the samples) and PC2 (the second most important metabolic component explaining 13% of total variance of the samples) discriminated between the conditioned media of the BT-474, MCF-7 and SUM-159-PT cell lines. Being the three breast cancer cell lines histologically and biologically very different, this was partially expected. To a minor extent, the PC1/PC2 plane was also capable of discriminating the subpopulation state (ALDHbright or ALDHlow). To detail more the previous observations we carried out an ANOVA test on the component scores relative to the BT-474, MCF-7 and SUM-159-PT samples by establishing, as sources of variation, the cell subpopulation (ALDHbright and ALDHlow cells) and the cell line (MCF-7, BT-474 and SUM-159-PT) (Suppl. Table 1).This analysis showed that the cell line differences acted as a major factor affecting the distribution of the samples (F statistics = 98.42 and 432.77 for PC1 and PC2, respectively). However, the difference among the cell subpopulations (ALDHbright vs ALDHlow) common to all cell lines was detectable by the PC1 and PC4 components (F statistics = 35.54 and 28.08 for PC1 and PC4, respectively). The ALDHbright vs ALDHlow difference, cell line dependent, was detectable by the PC1 and PC3 components (F statistics = 20.76 and 18.39 for PC1 and PC3, respectively). The detectable impact of the ALDHbright vs ALDHlow condition on the whole metabolic profile of the samples analyzed prompted us to exclude the major effect of the cell line and to focus on the differences between ALDHbright and ALDHlow cell samples by using the orthogonal projections to latent structures-discriminant analysis (OPLS-DA) (Fig. 2B). The OPLS-DA model generated one Latent Variable (LV) that explained 55% of the X-variance and 100% of Y-variance with a cumulative predicted fraction of the joint X and Y variation of 85%. A clear separation between the ALDHlow and ALDHbright populations was found in the first component (LV1) independently from cell type (while the metabolic difference between the cell lines were observed on LV0) (Fig. 2B). From the analysis of the O-PLS-DA loadings we observed that 3-hydroxy-butyrate, alanine, lactate, acetate, succinate, glucose and formate were the main discriminatory metabolites responsible for the difference between ALDHbright and ALDHlow cell subpopulations (Table 1).
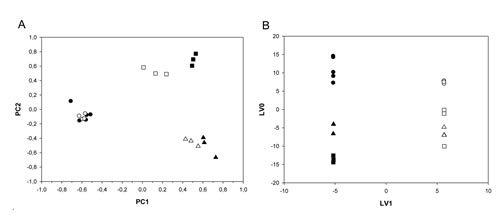
Figure 2: A-B ALDHbright and ALDHlow cells are metabolically distinguishable cell subpopulations. Overview of the PCA (A) and OPLS-DA (B) models built on the NMR datasets of medium samples collected from ALDHlow (open) and ALDHbright (filled) populations of MCF-7 (triangle), BT-474 (square) and SUM-159 (circle) cell lines. A. The obtained PCA model of ALDH subpopulations was dominated by the effect of cell line differences acting as major order parameter: PC1 and PC2 components are mainly responsible for among cell lines differences (see also Table 1). B. OPLS-DA analysis of the same samples as from A shows a clear separation between the ALDHlow and ALDHbright populations, independently from cell type, was found in the first component.
Metformin-induced changes in the metabolic phenotype of ALDHbright and ALDHlow cells (Fig. 3).
Next, we tested the effect of metformin on the metabolic processes of the ALDHbright and ALDHlow cell subpopulations. More specifically, we detailed the 1H-NMR profile of the conditioned media from the ALDHbright and ALDHlow subpopulations of BT-474, MCF-7 and SUM-159 cells treated with vehicle (PBS1X) or metformin at a non cytotoxic concentration/length of treatment combination (0.5mM-24hrs) (Suppl. Fig. 2). This was done to avoid interference of apoptotic processes with the 1H-NMR analysis.
PCA analysis of the vehicle- and metformin- treated ALDHbright and ALDHlow cells from all three breast cancer cell lines was very complex and revealed, again, a predominant effect of the differences among the parental cell lines (thus masking the effect of metformin treatment)(Suppl. Table 2). Therefore, we analyzed the effect of metformin treatment on the ALDHbright and ALDHlow cell subpopulations for each cell line independently. The PCA results for each dataset are depicted in Figure 3A-C, where the score plots for the first two model components are shown. A clear separation among control and metformin-treated samples for each subpopulation was obtained. This revealed that the PC1 (for the BT-474 and SUM-159 cells), and the PC2 (for the MCF-7) were mainly responsible for the differences between vehicle- and metformin-treated ALDHbright and ALDHlow cell subpopulations. The quantitative PCA approach revealed that, when considering the same metabolic process for both cell subpopulations, the effects of metformin were more evident in the ALDHbright as compared to the ALDHlow cells. The maximum difference between the metformin- and vehicle-treated groups (thus, the maximum drug effect) was observed for the BT-474 and SUM-159 ALDHbright cells on the PC1 and for the MCF-7 ALDHbright cells on the PC2 (Suppl. Table 3). Notably, we found that the treatment with metformin reduced the difference between ALDHbright and ALDHlow cells. More specifically, we found that, for each cell line, the Euclidean distance (arrows) between the centroids (i.e, the barycenter of the group of samples)(stars) (Fig. 3A-C) of the treated ALDHlow and ALDHbright samples was smaller than the distance between the treated ALDHlow and the untreated ALDHbright samples, suggesting that Metformin changed the metabolism of ALDHbright towards a phenotype more similar to that of metformin-treated ALDHlow cells This was very evident for the SUM-159 cell line.

Figure 3A-C: PCA analysis of vehicle- and metformin-treated ALDHbright and ALDHlow cells. PCA models built on the NMR datasets of medium samples collected from ALDHlow (red) and ALDHbright (black) subpopulations of vehicle- (open) and metformin-treated (filled) MCF-7 (triangle), BT-474 (square) and SUM-159(circle) cells). Please note that filled samples (metformin-treated cells) occupy a very similar location in the component space. In each line, the Euclidean distance (arrows) between the centroids (stars) of the two treated subpopulations is smaller than the distance between the treated ALDHlow and untreated ALDHbright cells, suggesting that the metabolism of ALDHbright and ALDHlow cells became more similar upon metformin treatment.
Metabolomic signatures of Metformin treated ALDHbright cells (Fig. 4).
Next, we detailed the metabolic profiles relative to the vehicle- and metformin- treated ALDHbright cells (Fig. 4A-E). For each cell line, the PCA produced solutions with two significant components, explaining about 52%, 65% and 47% of the total variability of the system for the MCF-7, BT-474, and SUM-159 cells, respectively (data not shown). This highlighted significant differences between the two groups on the PC1. Additionally, despite some differences between the cell lines, we observed similarities in the correlation patterns of metabolite loadings (Fig. 4A-E). For each cell line, the PC1 included the following variables with the highest correlation levels: glutamine and glucose consumption and pyroglutamate production with positive loadings as well as lactate production with negative loadings. Therefore, the PC1 analysis indicated that metformin treatment induced higher consumption of glutamine and glucose as well as higher production of lactate (positive correlation with PC1) compared to untreated cells (Fig. 4C-E).
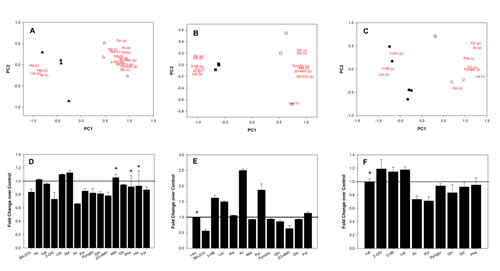
Figure 4: A-F: Metabolomic analysis of metformin treated ALDHbright cells. Upper panels. Overview of the PCA model built on the NMR dataset of media samples of control (open) and metformin-treated (filled) ALDHbright cells from MCF-7 (triangle), BT-474 (square) and SUM-159 (circle) cells. The score and loading plots of the first two components (PC1 versus PC2) are shown superimposed. The score plot shows the differentiation between untreated and metformin-treated samples, while the loading plot highlights which metabolites are responsible in separating control and metformin-treated samples. Lower panels. Histograms indicate the relative levels of the metabolites considered in the score/loading plots for MCF-7 (right), BT-474 (middle) and SUM-159 (left). Loading values are represented using the abbreviation of metabolites: leucine, Leu; valine, Val; 3-Methyl-2-oxovalerate, 3M-2OV; 3-hydroxy-butyrate, 3-HB; lactate, Lac; alanine, Ala; acetate, Ac; methionine, Met; glutamine, Gln; glutamate, glu; pyruvate, Pyr; pyroglutamate, Pyroglu; 2-Oxo-4-methylvalerate, 2O-4MV; glucose, Glc; histidine, His, phenylalanine, Phe ;formate, For.
The negative correlation between glutamine and glucose consumption and lactate production in the loading plots of the ALDHbright cells from all the analysed cell lines suggested higher fluxes through glycolysis or glutaminolysis in the treated cells compared to untreated controls. Previous studies using NMR analysis with [1,2-13C]-glucose revealed that, in metformin-treated cells, lactate is more produced from glutaminolysis rather than from glycolysis therefore suggesting that the net effect of metformin consists of a reduction of the glycolytic flux. A lower production of pyroglutamate upon metformin treatment was also observed (negative correlation with PC1). A lower excretion of pyroglutamate suggested a reduced level of intracellular glutathione. In facts, pyroglutamate, also known as 5-oxoproline, is converted to glutamate by 5-Oxoprolinase. As glutamate is required in the first step of GSH synthesis, the lower production of pyroglutamate observed in treated cells suggest a minor level of intracellular glutathione. Moreover, in MCF-7 and BT-474 cell lines, we observed that glucose and glutamine consumption correlated also with the production of alanine (opposite loadings) suggesting a higher activation of alanine aminotransferases in the metformin-treated cells. However, the fact that acetate was a strong negative loader on PC1 for BT-474, demonstrated that the alanine aminotransferase pathway was also used to provide precursors needed for fatty acid synthesis. Nevertheless, the higher excretion of acetate into media of the metformin-treated cells reflected the smaller availability of acetyl-CoA units for fatty acid synthesis. For the MCF-7 and BT-474 cells, the negative correlation of 3-methyl-2-oxovalerate and 2-oxo-4-methylvalerate loadings within the metformin-treated cells as opposed to the control-treated cells indicated a higher metabolic flux through the branched-chain amino acid aminotransferase pathway. This strongly suggests the use of branched amino acids for energy production instead of its use for macromolecule biosynthesis in the metformin-treated cells. Analysis of the identified metabolites with the KEEG pathways indicated that, in all three ALDHbright cell subpopulations treated with metformin, we observed perturbations of the glycolysis, pyruvate metabolism, glutathione metabolism, purine and pyrimidine metabolism, alanine, aspartate, glutamate, arginine and proline metabolism , pentose phosphate pathway, amino sugar and nucleotide sugar metabolism, HIF-1α and the insulin signalling pathways in all cell lines (Table 2).
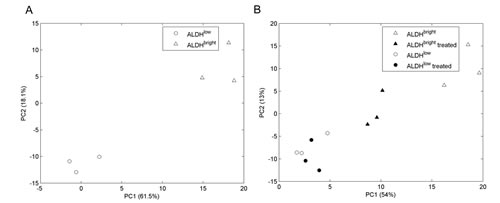
Figure 5: A. ALDHbright and ALDHlow cells differ in their microRNA expression profile. PCA of the microRNA expression profile of the untreated ALDHbright and ALDHlow cell subpopulations derived from the SUM-159 cells (497 microRNAs expressed/analyzed). B. Metformin modulates microRNAs in the ALDHbright and ALDHlow cells. PCA of 125 miRNAs modulated by metformin in ALDHbright and ALDHlow SUM-159 cells. Principal Component Analysis. Percentage of the explained variance is indicated for the first two components.
Table 2. Metabolic pathways perturbed by Metformin in all the analyzed breast cancer cell lines. |
||
Metabolite |
Metformin vs. control* |
Related KEGG Pathway Maps** |
Lactate |
High (p) |
Glycolysis/Gluconogenesis |
Pyruvate metabolism |
||
Pyroglutamate |
Low (p) |
Glutathione metabolism |
Glutamine |
Low (c) |
Purine metabolism |
Pyrimidine metabolism |
||
Alanine, aspartate and glutamate metabolism |
||
Arginine and Proline metabolism |
||
Glucose |
Low (c) |
Glycolysis/Gluconogenesis |
Pentose phosphate pathway |
||
Amino sugar and nucleotide sugar metabolism |
||
HIF-1 signaling pathway |
||
Insulin signaling pathway |
||
*“High” indicates that the metabolite was at a higher concentration in medium samples from metformin-treated cells; “Low” indicates that the metabolite was at a lower concentration in medium samples from metformin-treated cells; “c” and “p” for each metabolite indicate consumption or production, respectively.
** The metabolites are mapped to their respective biochemical pathways as delineated in the Kyoto Encyclopedia of Genes and Genomes (Release 69.0, January 1, 2014; KEGG, http://www.genome.jp/kegg).
Metformin-mediated modulation of microRNAs sustains its metabolic effects on the ALDHbright cells (Fig. 5 and 6). We previously demonstrated that metformin exerts anticancer metabolic effect on unsorted breast cancer cell populations by broadly modulating the metabolic pathways, at least partially in a DICER-dependent microRNA modulation[15]. We verified whether a similar mechanism would take place in the sorted ALDHbright cells and whether this may explain the metabolic changes observed in the treated cells. Therefore, we analysed the microRNA expression profile of vehicle- and metformin- treated, FACS sorted ALDHbright cells (Fig. 5). This first revealed that, at steady state, the microRNA expression profile of ALDHlow and ALDHbright was very different, mirroring the observed difference in their metabolic profile. In facts, the unsupervised PCA analysis of 497 expressed microRNAs in the ALDHlow and ALDHbright cell subpopulations revealed a clear separation of the two cell subpopulations on both the PC1 and the PC2 (61.5% and 18.1% of the total variance, respectively)(Fig.5A). Next, we evaluated the microRNA expression profile of vehicle- and metformin- treated ALDHbright and ALDHlow cells derived from the SUM-159 cells. This provided us with two observations: first, metformin treatment induced changes in microRNA expression levels in both ALDH bright and ALDHlow cells (Fig.5B), with a slightly more evident effect on the ALDHbright cells; second, metformin treatment reduced the distance in the microRNA expression profile between ALDHbright and ALDHlow cells (Fig. 5B). This was reminiscent of the metabolic effects of the drug on the ALDHbright and ALDHlow cells, which were consistent with the acquisition of a more similar metabolic profile between the two cell subpopulations. Altogether, these observations suggest that metformin treatment of breast cancer ALDHbright cells partially reverted a chemoresistant and clonogenic phenotype to a chemosensitive, more differentiated one. In order to support this observation, we detailed the microRNA modulation in the metformin treated-ALDHbright cells (Fig.6A) Interestingly, a large fraction (89/125) of the microRNAs significantly modulated in the metformin-treated ALDHbright cells (Suppl. Table 4) was predicted to target metabolic pathways (Table 3) and caused a strong separation of the samples on the PC1 (Fig. 6A) We found that the pathways found deregulated by metformin in the 1H-NMR studies were represented within those predicted by the microRNA analysis, thus establishing a correlation between the effect of metformin on the microRNAs and its metabolic activity(Table 3).
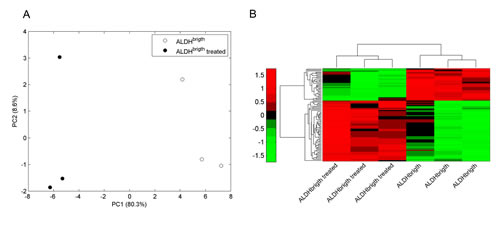
Figure 6: Metformin modulates microRNAs associated with metabolic functions in the ALDHbright cells. A. PCA of 89 miRNAs modulated by metformin and involved in metabolic pathways in SUM-159 cells. Principal Component Analysis. Percentages of the explained variance is indicated for the first two components. B. Heat Map of 89 miRNAs modulated by metformin and involved in metabolic pathways. Unsupervised Hierarchical Clustering. Red indicates higher expression and green lower expression of the indicated miRNAs for each sample. The list of the 89 microRNAs used for the heat map can be found in Suppl. Table 4.
Table 3. Main metabolic functions predicted to be modulated by the metformin-regulated microRNAs.
insulin path |
miR-335-5p,miR-124-3p,miR-98,miR-16-5p,miR-155-5p,miR-26b-5p,miR-1,miR-7-5p,miR-21-5p,let-7b-5p,miR-192-5p,miR-128,miR-148b-3p,miR-375,miR-30a-5p,miR-130b-3p,miR-125a-5p,miR-103a-3p,miR-93-5p,miR-10a-5p,miR-34a-5p,miR-24-3p,miR-19b-3p,miR-193b-3p,miR-122-5p,miR-186-5p,let-7,miR-126-3p,miR-181a-5p,miR-29c-3p,miR-101-3p,miR-141-3p,miR-145-5p,miR-214-3p,miR-590-3p,miR-33a-5p,miR-769-5p,miR-200a-3p,miR-32-5p,miR-132-3p,miR-27a-3p,miR-149-3p,miR-217,miR-200c-3p,mir-199a,miR-615-3p,mir-199a*,miR-22-3p,miR-374b-5p,miR-429,miR-9-5p,miR-140-3p,miR-18a-5p,miR-200b-3p,miR-421,miR-96-5p' |
alanine aspartame |
'miR-335-5p,miR-155-5p,miR-16-5p,miR-26b-5p,miR-34a-5p,miR-192-5p,miR-128,miR-24-3p,miR-27a-3p,miR-21-5p,miR-1,miR-124-3p,miR-590-3p,miR-101-3p,miR-96-5p,miR-186-5p,miR-30b-5p,miR-7-5p,miR-183-5p,miR-98,miR-32-5p' |
amino sugar |
'miR-26b-5p,miR-124-3p,miR-155-5p,miR-30a-5p,miR-375,miR-30b-5p,miR-1,miR-34a-5p,miR-335-5p,miR-106b-5p,mir-30,miR-98,miR-16-5p,miR-32-5p,miR-9-5p,let-7b-5p,mir-199a,miR-21-5p,miR-101-3p,miR-193b-3p,miR-128,miR-590-3p,miR-24-3p,miR-192-5p,miR-148b-3p,miR-122-5p' |
aminoacyl trna biosint |
'miR-16-5p,miR-155-5p,miR-26b-5p,let-7b-5p,miR-30a-5p,miR-93-5p,miR-101-3p,miR-124-3p,miR-19b-3p,miR-192-5p,miR-1,miR-21-5p,miR-98,miR-130b-3p' |
cysteine metab |
'miR-26b-5p,miR-375,miR-193b-3p,miR-155-5p,miR-335-5p,miR-29b-3p,let-7b-5p,miR-29c-3p,miR-369-5p,miR-34a-5p,miR-29a-3p,miR-16-5p,miR-33a-5p,miR-191-5p,mir-199a,mir-30,miR-186-5p,miR-548b-3p,miR-30a-5p,miR-152,let-7d-5p,miR-106b-5p,mir-148,miR-1,miR-148b-3p,miR-7-5p,miR-192-5p,miR-124-3p' |
D-glutamine and glutamate |
'miR-335-5p,miR-7-5p' |
glycolisis |
'miR-335-5p,miR-124-3p,miR-155-5p,miR-30a-5p,mir-17-92,miR-375,miR-34a-5p,miR-1,miR-7-5p,miR-16-5p,miR-26b-5p,mir-199a,miR-122-5p,mir-30,miR-148b-3p,let-7b-5p,miR-98,miR-24-3p,miR-22-3p,miR-132-3p,miR-145-5p,miR-133b,miR-192-5p,miR-181a-5p,miR-33a-5p,miR-133a,miR-27a-3p,miR-128,miR-9-5p' |
pentose phospate |
'miR-124-3p,miR-335-5p,miR-1,miR-26b-5p,miR-148b-3p,miR-375,miR-34a-5p,edited-hsa-mir-376a-5p,mir-1b,let-7b-5p,miR-192-5p,mir-30,miR-142-3p,miR-30a-5p,miR-373-3p' |
pyruvate |
'miR-335-5p,miR-34a-5p,miR-155-5p,miR-26b-5p,let-7b-5p,miR-16-5p,miR-30a-5p,miR-124-3p,miR-24-3p,miR-7-5p,miR-1,miR-33a-5p,miR-133b,miR-27a-3p,miR-375,miR-133a,miR-21-5p,miR-22-3p,miR-122-5p,miR-23b-3p,miR-193b-3p,miR-148b-3p,miR-98,miR-192-5p,miR-181a-5p' |
'purine' |
'miR-124-3p,miR-335-5p,let-7b-5p,miR-26b-5p,miR-98,miR-155-5p,miR-30a-5p,miR-1,miR-16-5p,miR-193b-3p,miR-7-5p,miR-34a-5p,miR-192-5p,mir-30,miR-24-3p,miR-96-5p,miR-128,miR-375,miR-19b-3p,miR-21-5p,miR-103a-3p,miR-196a-5p,miR-148b-3p,let-7d-5p,miR-142-3p,miR-93-5p,miR-133b,miR-186-5p,edited-hsa-mir-376a-5p,miR-130b-3p,miR-133a,miR-9-5p,miR-421,miR-122-5p,mir-132/mir-212,miR-340-5p,miR-18a-5p' |
'pyrimidine' |
'miR-26b-5p,let-7b-5p,miR-34a-5p,miR-124-3p,miR-155-5p,miR-193b-3p,miR-192-5p,miR-1,miR-7-5p,miR-335-5p,miR-30a-5p,miR-16-5p,miR-24-3p,miR-98,mir-30,miR-186-5p,miR-148b-3p,miR-375,miR-21-5p,miR-196a-5p,miR-101-3p,miR-128,miR-103a-3p,miR-142-3p,miR-122-5p,miR-421,miR-9-5p,miR-10a-5p,mir-199a*,miR-96-5p,miR-590-3p,let-7d-5p,mir-132/mir-212,miR-374b-5p' |
Sinthesis of ketone Bodies |
'miR-21-5p,miR-192-5p,miR-18a-5p,miR-1,miR-335-5p,miR-23b-3p,miR-155-5p,miR-19b-3p,miR-26b-5p,miR-186-5p,miR-375,miR-124-3p,miR-96-5p' |
'valine' |
'miR-124-3p,miR-26b-5p,miR-192-5p,miR-1,miR-155-5p,miR-34a-5p,miR-335-5p,miR-16-5p,miR-32-5p,miR-193b-3p,miR-21-5p,miR-9-5p,miR-19b-3p,miR-128,let-7b-5p,miR-186-5p,miR-96-5p,miR-7-5p,miR-18a-5p,miR-27a-3p,miR-23b-3p' |
Metformin targets c-MYC and IRS-2 via mir-33a modulation (Fig. 7). We and others have recently demonstrated that the anticancer metabolic activity of metformin toward unsorted breast cancer cells is partially due to a mir-33a-dependent modulation of c-MYC levels [15]. In facts, C-MYC is a central modulator of cancer cell metabolism, alone or in cooperation with HIF-1α [17, 18]. To assess whether a similar mechanism would operate in the ALDHbright cancer cells as well, we evaluated the levels of microRNA 33a and its target c-MYC in ALDHbright cells, treated with vehicle or metformin, respectively. By performing quantitative PCR, we observed a strong modulation of mir-33a levels which anti-correlated with those of c-MYC. Additionally, we found that the levels of IRS-2, a central modulator of insulin signalling in normal and cancer cells and a target of microRNA33a as well [19] were modulated by metformin (Fig. 7). A working model that generally resumes the collected observations is shown in Fig. 8 (Fig. 8).
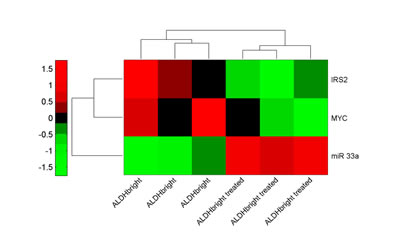
Figure 7: Metformin inversely modulates mir-33a and its targets. The heat map shows opposite regulation of the microRNA-33a and its gene targets (c-MYC, IRS-2)(Normalized intensity values) in metformin treated ALDHbright cells. Triplicate experiments.
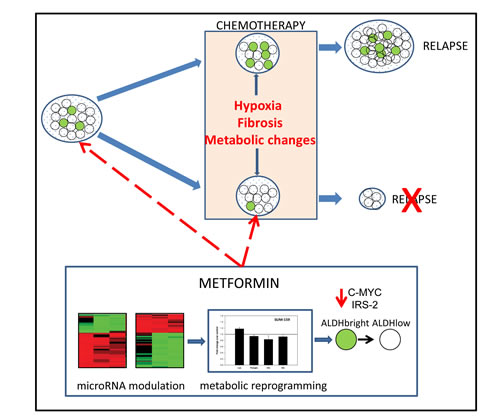
Figure 8: Representative working model. Metformin modulates the microRNA profile of ALDHbright cells thereby impacting on their metabolic properties. This makes the ALDHbright cells more similar to the ALDHlow cells and affect chemoresistance of the tumor, potentially reducing its relapse. Metformin proposed action may take place either before or in conjunction with chemotherapy. Dashed arrows indicate speculated actions of the drug.
DISCUSSION
We have recently shown that a complex interplay of microRNA modulation and pathway signalling underlies the anticancer effects of metformin, an anti-diabetic agent which epidemiological evidence suggest to play an important role in cancer prevention [15, 20, 21]. With the aim of detailing our previous studies, in this work we have studied the metabolic changes taking place in breast ALDHbright cells, the main cell subpopulation responsible for the chemoresistance of breast tumors. This because understanding the metabolism of chemoresistant cell subpopulations may aid the identification of the basic mechanism sustaining their chemoresistance, a major factor shaping the prognosis of breast cancer patients. Our experimental systems included three breast cancer cell lines, differing for histological origin and growth rate. Indeed, our metabolomics analysis identified the cell line specificity as a major determinant of the differences between the samples, thus confirming the efficiency of the approach. Despite this, the OPLS-DA analysis allowed us to focus on the differences between vehicle- and metformin- treated ALDHbright cells. First, we found that the metabolic profile of the purified ALDHbright cell subpopulations was significantly different from that of the ALDHlowlow cells. When focusing on the metabolic changes induced by metformin treatment and common to the ALDHbright cells from all the cell lines examined (MCF-7, BT-474, SUM-159-ALDHbright cells), an increase in glucose and glutamine uptake and lactate production as well as the decrease in pyroglutamate production were observed upon metformin treatment. Interestingly, a reduced excretion of pyroglutamate from the metformin-treated cells indicated reduced levels of intracellular glutathione. This is in line with recent findings showing that metformin affected the glutathione (GSH) homeostasis of breast cancer cells, with the reduced glutathione biosynthesis correlating with a blockage of de novo purine/pyrimidine synthesis[22]. Our observations suggest that negative modulation of the glutathione homeostasis in the ALDHbright cell subpopulations may underlie the anticancer action of metformin. In facts, higher levels of intracellular glutathione have been described in chemo- and radio-resistant “cancer stem cell –like (CSCs)” cell subpopulations derived from MMTV-WNT1 breast tumors[23]. Additionally, this correlated with a broad modulation of the mRNA levels of the genes involved in the glutathione metabolism. We speculate that metformin treatment may reduce the glutathione pool in the breast ALDHbright cell subpopulations thereby abating their resistance to therapy. Interestingly, recent findings suggest that blockage of c-MYC downstream effectors affected glycolysis and glutathione biosynthesis in MYC-driven mouse lymphoma models[24]. In line with this, we have found that metformin downregulated the c-MYC levels in ALDHbright cells, possibly by upregulating the mir-33a levels. C-MYC activation in ALDHbright cells may strongly cooperate with the activation of HIF-1 metabolic effectors, such as several glycolytic enzymes [3, 25, 26]. The latter phenomenon was observed by our 1H-NMR analysis. Additionally, IRS-2 is downregulated by metformin and, like for c-MYC, this inversely correlated with the levels of mir-33a in the metformin treated cells. IRS-2 is an important modulator of aerobic glycolysis in mouse mammary cancer cells [27]. Notably, modulation of c-MYC and IRS-2 mRNA levels was already shown by us in unfractionated, metformin treated breast cells lines [15]. What we believe is remarkable here is that the treatment with metformin drastically reduced the metabolic difference between ALDHbright and ALDHlow cells. A very similar effect was observed on the microRNA expression profile of metformin treated ALDHbright and ALDHlow cells. This is very interesting since the ALDHlow cells represent the chemosensitive fraction of solid tumors and it indicates the potential of metformin for metabolic reprogramming of chemoresistant cell subpopulations. This represents a further mechanism of cancer interference by this promising compound (Fig. 8).
METHODS
Reagents.
Metformin (1,1-Dimethylbiguanide-hydrochloride) Cisplatin and and Doxorubicin-hydrochloride were dissolved according to the manufacturer’s instructions. (Sigma, St Louis, MO, USA)
Cell culture conditions.
MCF-7 and BT-474 breast cancer cell lines were grown in DMEM/F12 supplemented with 10% non-heat inactivated FBS (Invitrogen-GIBCO, Carlsbad, CA, USA); the MCF10A and SUM159 cells were grown in DMEM/F-12 supplemented with 5% FBS, Insulin 5µgr/ml (SIGMA) and Hydrocortisone 0.5 µgr/mL (SIGMA).
ALDH activity assay.
ALDH activity was detected by FACS CALIBUR instrument (BD Biosciences, Franklin Lakes, New Jersey, USA). ALDEFLUOR kit (Stem Cell Technologies, Vancouver, Canada) was used according to the manufacturer’s instructions. ALDHbright cells were defined as the cells that displayed greater fluorescence compared with a control staining reaction containing the ALDH inhibitor, DEAB (diethylaminobenzaldehyde). The analysis was performed by using FlowingSoftware 2.0 (Cell Imaging Core, University of Turku, Finland).
Cell Sorting.
ALDEFLUOR kit was used according to the manufacturer’s instructions to detect the ALDH activity. Cells were filtered through a 40µM mesh to obtain a single cell suspension . Cell sorting was performed with a MoFLO cell sorter (DAKOCYTOMATION ,Fort Collins, Colorado, USA)
Cell Viability.
The SYTOX® Orange Dead Cell Stain( Invitrogen-Molecular Probes, Carlsbad, CA, USA) was used to assess cell viability as per manufacturer’s instructions.
Clonogenic assays.
The breast cancer cell lines were grown to 70% confluence and pulse treated with the indicated drugs. 16hrs later, cells were detached(Accutase-GIBCO) and seeded at 500-1500 cells/well into 6-well dishes (Corning-Costar, Tewksbury, MA, USA) in drug-free media. Fresh medium (25%) was added every three days. Colonies were stained with crystal violet (SIGMA) and colonies (>50 cells) counted after 7- 14 days
NMR sample preparation
Extracellular metabolites were separated by ultrafiltration (AMICON) before methanol/chloroform/water extraction (2/2/1.8) to extract non polar metabolites[28, 29].
1H-NMR Spectroscopy.
For 1H-NMR analysis, polar extracts were dissolved in 600μl of 1 mM TSP [(trimethylsilyl)-propionic-2,2,3,3-d4 acid] and 10 mM NaN3 solution in D2O 0.1 M phosphate buffer (pH=7.4), while non-polar extracts were dissolved in 600μl of CDCl3 containing 0.03% TMS (tetramethylsilane) / CD3OD solution (2:1, v/v).
All 2D 1H J-resolved (JRES) NMR spectra were acquired on a 500 MHz DRX Bruker Avance spectrometer (Bruker, Germany) using a double spin echo sequence with 8 transients per increment for a total of 32 increments. These were collected into 16k data points using spectral widths of 6 kHz in F2 and 40 Hz in F1. There was a 3.0 s relaxation delay. The water resonance was suppressed using presaturation. Each FID was Fourier transformed after a multiplication with sine-bell window functions in both dimensions. JRES spectra were tilted by 45°, symmetrised about F1, referenced to TSP at δH = 0.0 ppm and the proton-decoupled skyline projections (p-JRES) exported using Bruker’s XWIN-NMR software. Metabolites were identified using an in-house NMR database and literature data and confirmed by 2D homo- and heteronuclear NMR spectroscopy. 1H-NMR spectra pre-processing. All p-JRES exported were aligned and then reduced into spectral bins with widths ranging from 0.01 to 0.02 ppm by using the ACD intelligent bucketing method (ACD/Labs, Canada) that sets the bucket divisions at local minima (within the spectra) to ensure that each resonance is in the same bin throughout all spectra. After this procedure, the 1H spectra were divided into n bins. The area within each spectral bin was integrated to yield a n-component vector, where each component value was represented by the integral value corresponding to the specific spectral bin. To compare the spectra, the integrals derived from the bucketing procedure were normalized to the total integral region, following exclusion of bins corresponding to solvent (residual water/HDO, δ4.76-4.82 ppm; CDCl3, δ 7.45-7.50 ppm; CD3OD,δ 3.33-3.37 ppm), TSP, TMS (δ -0.5-0.5 ppm) and metformin peaks (δ3.03-3.06 ppm). Data from extracellular media were expressed in terms of net balances at 0 and 24hrs. This to establish consumption (c) or production (p) of the selected metabolites in time.
Univariate statistical data analysis.
Analysis of variance (ANOVA) is a technique for analysing experimental data in which one or more (or dependent) variables are measured under various conditions, identified by one or more classification variables. The response is separated into variation attribuTable to differences between the classification variables and variation attribuTable to random errors. An analysis of variance constructs tests to determine the significance of the classification effects. A typical goal is to compare means of the response variables for various combinations of the classification variables (interaction effects).
In our study, we considered metabolite consumption and production as a dependent variable while type of subpopulation, type of cell line and treatment effect as the classification variables. The resulting data was used as input for univariate and multivariate analysis Principal Component Analysis (PCA)[30] and Orthogonal Projections to Latent Structures-Discriminant Analysis (OPLS-DA)[31]. PCA and OPLS-DA were conducted using SIMCA-P+ version 12 (Umetrics, Umea, Sweden).
Principal Component Analysis.
Principal Component Analysis (PCA) is a projection method used for exploiting the information embedded in multidimensional data sets. The data is reduced to a few latent variables (or principal components) collecting the information implicit in the original variables correlation structure. The presence of correlations between the original variables allows for the reduction of dimensionality of the data set in the new space without noticeable loss of information. The extracted components (PCs) are each orthogonal and ordered in terms of percentage of explained variation, with the first components collecting the ‘signal’ (correlated) portion of information, while minor components can be considered as ‘noise’ components. Because the principal components are, by construction, orthogonal to each other, a clear-cut separation of the different and independent features characterizing the data set is made possible. Each statistical unit is assigned a score relative to each extracted component, while the correlation coefficient between each original variable and extracted components (loading) gives a meaning to the PCs. The output from the PCA analysis consists of score plots, which provide an indication of the differences between the classes in terms of metabolic similarity, and loading plots. These loading plots give an indication of which metabolite net balances are important with respect to the classification obtained in the score plots.
Orthogonal projections to latent structures discriminant analysis (OPLS-DA)
Orthogonal projections to latent structures discriminant analysis (OPLS-DA) is a supervised pattern recognition technique widely used in the field of metabolomics to interpret large multivariate data sets describing differences between the groups under study in a straightforward and accurate way.
OPLS-DA separates the systematic variation in the matrix X (spectroscopic data) into two parts, one linearly related (variation of interest) to the matrix Y (the classification variables) and one orthogonally related (so called orthogonal variation or structured noise) to the matrix Y. This partitioning of the X-data improved the interpretation of the model.
In our study, we considered consumption and production of metabolites as X matrix and type of subpopulation as Y matrix. The influence of the original variables on the obtained model was determined using loading values.
RNA processing and hybridization
Total RNA from the FACS sorted ALDHbright and ALDHlow cells (98 ± 0.4% and 97 ± 1.4% purity, respectively, as assessed by re-FACS) was extracted by TRI Reagent lysis reagent (Life Technologies-AMBION Carlsbad, CA, USA) according to the manufacturer’s instructions. The microRNA Complete Labeling and Hyb Kit (AGILENT Santa Clara, CA, USA) was used to generate fluorescent miRNA, according to manufacturer’s instructions. Scanning and image analysis were performed using the Agilent DNA Microarray Scanner (P/N G2565BA). Feature Extraction Software (V-10.5) was used for data extraction from raw microarray image files using the miRNA_105_Dec-09FE protocol.
Analysis of microRNA expression.
Pearson’s correlation coefficient was calculated in order to assess quality of replicates. ANOVA test α=5%) was performed to assess statistical significance of the observed differences in microRNA profiling (Fig.S2).
Preprocessing.
The signal of 851 human miRNAs was processed by MATLAB (The MathWorks Inc.) in in house-built routines. All background-subtracted intensity values lower than 1 were considered below detection and thresholded to 1. The arrays were quantile-normalized forcing each slide to assume the same mean distribution and log2-trasformed.
Feature selection. Clusters in data were identified by unsupervised Hierarchical Clustering and Principal Component Analysis. A permutation t-test and bootstrap test were used to select deregulated miRNAs after treatment. A false discovery rate (FDR) procedure was also applied for multiple comparisons. All tests were two-tailed and considered significant if both p-value and q-value (FDR) were less than 0.05.
miRNA-pathway assignment prediction.
A list of predicted miRNAs in pathway experimentally identified from metabolomics was extracted using miRWalk (http://www.umm.uni-heidelberg.de/apps/zmf/mirwalk/)[32]. Predicted miRNAs with significant modulation of the signal after treatment in the array experiment were considered for further investigations.
cDNA synthesis and gene expression.
The first-strand cDNA was synthesized according to manufacturer’s instructions (M-MLV Reverse Transcriptase, Life Techonlogies). Gene expression was measured by real-time PCR using the FAST SYBRGreen master mix (Applied Biosystems, Foster City, CA, USA) on a StepOne instrument (Applied Biosytems).
Sequences of the Q-PCR primers are: IRS-2 Fw: ACGCCAGCATTGACTTCTTGT, Rv :GCCAGACAGATCTTCACTCTTTCA; ACTIN Fw: GGCATGGGTCAGAAGGATT, Rv: CACACGCAGCTCATTGTAGAAG; C-MYC Fw: CTCCTGGCAAAAGGTCAGAG, Rv: TCGGTTGTTGCTGATCTGTC.ACTIN was used as endogenous control
Statistical Analysis.
Generally, Student’s t-test was used to assess significance of the data generated except where otherwise specified.
ACKNOWLEDGEMENTS
We acknowledge the support of Ministero Salute Progetto Tevere to GB and PM, FIRB to GB and PM, AIRC/ROC to GB.
REFERENCES
1. Dang CV, Lewis BC, Dolde C, Dang G and Shim H. Oncogenes in tumor metabolism, tumorigenesis, and apoptosis. Journal of bioenergetics and biomembranes. 1997; 29(4):345-354.
2. Hirsch HA, Iliopoulos D and Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proceedings of the National Academy of Sciences of the United States of America. 2012.
3. Dang CV, Le A and Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(21):6479-6483.
4. Canino C, Mori F, Cambria A, Diamantini A, Germoni S, Alessandrini G, Borsellino G, Galati R, Battistini L, Blandino R, Facciolo F, Citro G, Strano S, Muti P, Blandino G and Cioce M. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene. 2012; 31(26):3148-3163.
5. Cioce M, Gherardi S, Viglietto G, Strano S, Blandino G, Muti P and Ciliberto G. Mammosphere-forming cells from breast cancer cell lines as a tool for the identification of CSC-like- and early progenitor-targeting drugs. Cell cycle. 2010; 9(14):2878-2887.
6. Marcato P, Dean CA, Giacomantonio CA and Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell cycle. 2011; 10(9):1378-1384.
7. Abdullah LN and Chow EK. Mechanisms of chemoresistance in cancer stem cells. Clinical and translational medicine. 2013; 2(1):3.
8. Ginestier C, Liu S, Diebel ME, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan JL, Dontu G and Wicha MS. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. The Journal of clinical investigation. 2010; 120(2):485-497.
9. Tanei T, Morimoto K, Shimazu K, Kim SJ, Tanji Y, Taguchi T, Tamaki Y and Noguchi S. Association of breast cancer stem cells identified by aldehyde dehydrogenase 1 expression with resistance to sequential Paclitaxel and epirubicin-based chemotherapy for breast cancers. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(12):4234-4241.
10. Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J and Gurney AL. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PloS one. 2008; 3(6):e2428.
11. Kaur H, Mao S, Li Q, Sameni M, Krawetz SA, Sloane BF and Mattingly RR. RNA-Seq of Human Breast Ductal Carcinoma In Situ Models Reveals Aldehyde Dehydrogenase Isoform 5A1 as a Novel Potential Target. PloS one. 2012; 7(12):e50249.
12. Iliopoulos D, Hirsch HA and Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer research. 2011; 71(9):3196-3201.
13. Sato A, Sunayama J, Okada M, Watanabe E, Seino S, Shibuya K, Suzuki K, Narita Y, Shibui S, Kayama T and Kitanaka C. Glioma-initiating cell elimination by metformin activation of FOXO3 via AMPK. Stem cells translational medicine. 2012; 1(11):811-824.
14. Shank JJ, Yang K, Ghannam J, Cabrera L, Johnston CJ, Reynolds RK and Buckanovich RJ. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecologic oncology. 2012; 127(2):390-397.
15. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S, Manetti C, Citro G, Muti P and Strano S. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nature communications. 2012; 3:865.
16. Lim S, Becker A, Zimmer A, Lu J, Buettner R and Kirfel J. SNAI1-mediated epithelial-mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PloS one. 2013; 8(6):e66558.
17. Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer research. 2010; 70(3):859-862.
18. Gordan JD, Thompson CB and Simon MC. HIF and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer cell. 2007; 12(2):108-113.
19. Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suarez Y, Lai EC, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(22):9232-9237.
20. Barba M and Muti P. Is it time to test metformin in breast cancer prevention trials? A reply to the authors. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2009; 18(9):2565; author reply 2565.
21. Muti P, Berrino F, Krogh V, Villarini A, Barba M, Strano S and Blandino G. Metformin, diet and breast cancer: an avenue for chemoprevention. Cell cycle. 2009; 8(16):2661.
22. Corominas-Faja B, Quirantes-Pine R, Oliveras-Ferraros C, Vazquez-Martin A, Cufi S, Martin-Castillo B, Micol V, Joven J, Segura-Carretero A and Menendez JA. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging. 2012; 4(7):480-498.
23. Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, Gurney A, Lewicki J and Clarke MF. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem cells. 2008; 26(2):364-371.
24. Doherty JR, Yang C, Scott KE, Cameron MD, Fallahi M, Li W, Hall MA, Amelio AL, Mishra JK, Li F, Tortosa M, Genau HM, Rounbehler RJ, Lu Y, Dang CV, Kumar KG, et al. Blocking Lactate Export by Inhibiting the Myc Target MCT1 Disables Glycolysis and Glutathione Synthesis. Cancer research. 2013.
25. Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, Rodriguez-Enriquez S and Moreno-Sanchez R. HIF-1alpha modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini reviews in medicinal chemistry. 2009; 9(9):1084-1101.
26. Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Foundation symposium proceedings. 2007; (4):35-53.
27. Pankratz SL, Tan EY, Fine Y, Mercurio AM and Shaw LM. Insulin receptor substrate-2 regulates aerobic glycolysis in mouse mammary tumor cells via glucose transporter 1. The Journal of biological chemistry. 2009; 284(4):2031-2037.
28. Tiziani S, Emwas AH, Lodi A, Ludwig C, Bunce CM, Viant MR and Gunther UL. Optimized metabolite extraction from blood serum for 1H nuclear magnetic resonance spectroscopy. Analytical biochemistry. 2008; 377(1):16-23.
29. Bligh EG and Dyer WJ. A rapid method of total lipid extraction and purification. Canadian journal of biochemistry and physiology. 1959; 37(8):911-917.
30. Lindon JC and Nicholson JK. Spectroscopic and statistical techniques for information recovery in metabonomics and metabolomics. Annual review of analytical chemistry. 2008; 1:45-69.
31. Wiklund S, Johansson E, Sjostrom L, Mellerowicz EJ, Edlund U, Shockcor JP, Gottfries J, Moritz T and Trygg J. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Analytical chemistry. 2008; 80(1):115-122.
32. Dweep H, Sticht C, Pandey P and Gretz N. miRWalk--database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. Journal of biomedical informatics. 2011; 44(5):839-847.