
INTRODUCTION
Intracellular Ca2+ ([Ca2+]i) signals play critical roles in the modulation of cellular physiology and pathophysiology. In many non-excitable cells, intracellular Ca2+ signals respond to extracellular agonist stimulation in an oscillatory, rather than sustained, manner [1–3]. It is well accepted that two distinct intracellular Ca2+ pools, the inositol-1, 4, 5-trisphosphate (InsP3)-sensitive and ryanodine-sensitive pools, participate in the genesis of oscillatory Ca2+ signals [4]. In 1986, Putney presented a model for capacitive calcium (Ca2+) entry conveying that depletion of endoplasmic reticulum-stored Ca2+ leads to activation of plasma membrane Ca2+ channels that mediate influx of Ca2+ from the extracellular space into cells [5], in a process called store-operated Ca2+ entry (SOCE). Recent molecular studies have identified that stromal interaction molecule (STIM), the Ca2+ sensor of the intracellular compartments, together with Orai, the subunits of Ca2+ permeable channels on the plasma membrane [6], cooperate in regulating multiple cellular functions as diverse as proliferation, differentiation, migration and gene expression, demonstrating that SOC channels are important targets for normal cell function and a variety of diseases [7]. Unfortunately, there are few selective blockers for SOCE. There is a need to develop new compounds for SOCE as pharmacological tools for investigation and as therapeutic agents for diseases.
The membrane-penetrable compound, 2-aminodiphenyl borinate (2-APB) was initially developed as membrane-permeable InsP3 receptor antagonist since 2-APB produced concentration-dependent inhibition of InsP3-induced Ca2+ release from mouse cerebellar membranes without affecting InsP3 binding [8]. After this initial report, numerous studies have reported that 2-APB antagonizes SOCE in different cell types [9, 10]. Thereafter, Mikoshiba’s group further developed 2-APB analogs and demonstrated that 2-APB analogs exhibit more selective inhibition of SOCE compared to inhibition of InsP3-induced Ca2+ release, in either transfected cell lines or DT40 cells [11, 12]. In the present study, we used patch-clamp recording and Ca2+ imaging to test the effects of a novel 2-APB analog, DPB161 (Figure 1) on SOCE in rat submandibular cells. We found that DPB161 inhibited SOCE in a concentration-dependent manner. Thus, this study provides evidence that a 2-APB analog, DPB161 serves as a SOCE blocker to modulate cell Ca2+ signals in rat submandibular cells.
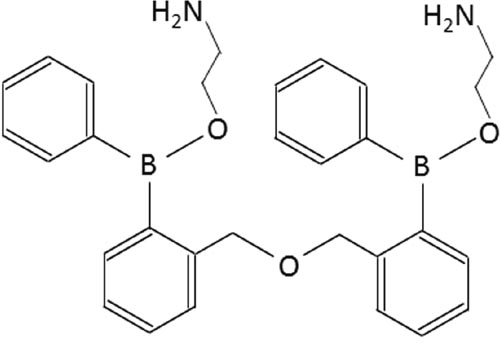
Figure 1: Chemical structure of DPB-161.
RESULTS
Rat submandibular cells model of SOCE
To open SOC channels, intracellular Ca2+ store need to be depleted. Initial experiments were designed to compare two types of cells (mouse pancreatic acinar cells and rat submandibular cells) for agonist-induced emptying of Ca2+ stores. Under free extracellular Ca2+ ([Ca2+]o=0) conditions, continuous bath-application of ACh (20 nM) induced persistent Ca2+ oscillatory responses as measured by the Ca2+-dependent Cl- current using patch-clamp whole-cell recordings in mouse pancreatic acinar cells (Figure 2A). However, in rat submandibular cells under the same experimental conditions, ACh-induced Ca2+ oscillatory responses stopped within minutes, and after washout of ACh with [Ca2+]o=0 solution for minutes, ACh still failed to induce any oscillatory responses (Figure 2B), suggesting an emptied intracellular Ca2+ store that was not refilled with extracellular Ca2+ in a SOCE-dependent manner. These results demonstrate that compared to pancreatic acinar cells, the response of rat submandibular cells to ACh stimulation is more dependent on Ca2+ entering through SOC channels to refill the Ca2+ store. Therefore, we used rat submandibular cells for all further experiments.
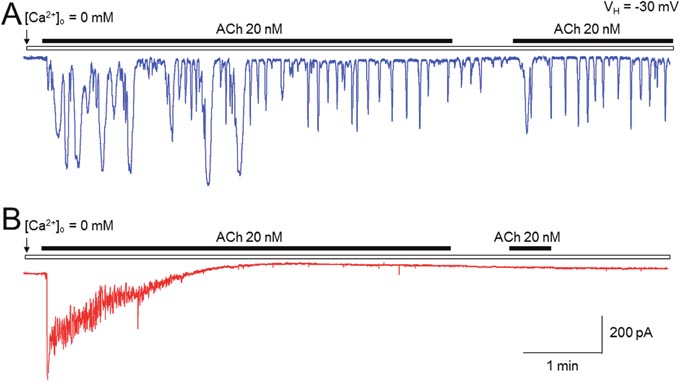
Figure 2: Comparison of ACh-induced Ca2+ oscillations between pancreatic acinar cells and submandibular cells using patch-clamp recordings. Under [Ca2+]o = 0 condition, ACh (20 nM) induced a long-lasting Ca2+ oscillatory response in mouse pancreatic acinar cells (A) but induced a short-lasting Ca2+ oscillatory response in rat submandibular cells (B). Typical traces from 6 cells tested are presented in A (mouse pancreatic acinar cells) and B (rat submandibular cells), respectively.
DPB161 blocks Ca2+ store refilling following ACh-induced depletion of intracellular Ca2+ stores
In these experiments, we characterized the effects of DPB161 on SOCE-dependent intracellular Ca2+ store refilling in rat submandibular cells. Under [Ca2+]o=0 conditions, bath-application of 20 nM ACh for 3-5 min induced an oscillatory response, which likely caused complete release of Ca2+ from the intracellular Ca2+ store since after washout of ACh for one min, reapplication of ACh failed to induce any oscillatory responses. Then, 1 mM Ca2+ was bath-applied to refill the Ca2+ store. Thereafter, we applied ACh again under [Ca2+]o=0 conditions and induced a Ca2+ response, suggesting that under [Ca2+]o=0 condition, the initial 20 nM ACh application emptied the Ca2+ store, and the subsequent bath-perfusion of 1 mM [Ca2+]o successfully refilled Ca2+ store (Figure 3A). When 50 μM DPB161 was included in the bath-perfusion with 1 mM [Ca2+], refilling of the Ca2+ store as prevented. Similar experimental results were obtained from 6 cells tested. These results suggest that DPB161 blocks SOCE (Figure 3B).
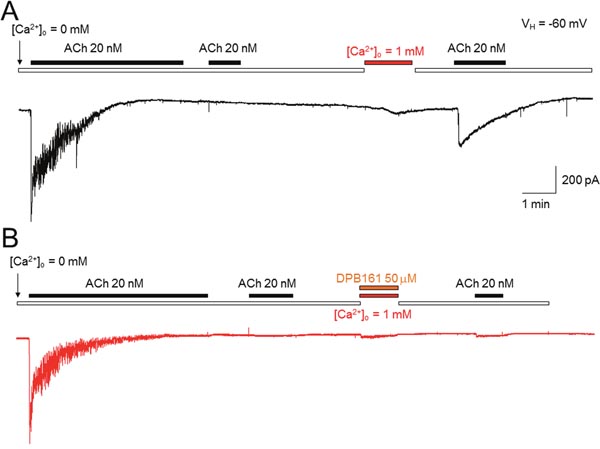
Figure 3: Effects of DPB-161 on [Ca2+]i responses to ACh measured by patch-clamp recordings in rat submandibular cells. (A) Under [Ca2+]o = 0 conditions, bath-application of 20 nM ACh induced Ca2+oscillatory response. Bath-application of 1 mM Ca2+ refills the Ca2+stores through opened SOC channels. (B) Using the same experimental protocol but refilling Ca2+ stores with 1 mM Ca2+ and 50 μM DBP161 prevented the refilling of Ca2+ stores, suggesting that DPB161 blocks SOCE. Typical traces in A and B are presented from 6 submandibular cells tested, respectively.
DPB161 blocks Ca2+ store refilling measured by fura-2 Ca2+ imaging
To further evaluate the role of DPB161 in inhibiting SOCE, we measured intracellular Ca2+ concentrations ([Ca2+]i) using Fura-2 imaging in isolated rat submandibular cells. Application of high concentration (1 μM) of ACh resulted in a spike in [Ca2+]i that declined much faster under [Ca2+]o=0 conditions (Figure 4A) than under 1 mM [Ca2+]o conditions (Figure 4B), suggesting that ACh empties Ca2+ stores and opens SOCE to refill the Ca2+ stores. However, when the bath solution contained DPB161 (50 μM), 1 μM ACh induced [Ca2+]i spike, with the same rise and decline, regardless of whether the bath solutions contained Ca2+ or not (Figure 4C, 4D). Statistical analysis indicated that the ACh-induced current areas (between 4 – 12 min) were 4504.8 ± 409.4 under 1 mM [Ca2+]o condition (n=12, A) and 3542.4 ± 231.4 for the [Ca2+]o=0 conditions (n=13, B). The difference was significant (p<0.05, unpaired t-test). In the presence of 50 μM DPB161, the ACh-induced current areas under 1 mM [Ca2+]o (n=11, C) and [Ca2+]o=0 conditions (n=16, D) were 1575.4 ± 80.2 and 1377.8 ± 190.8 (p>0.05, unpaired t-test). These results support the patch-clamp finding that DPB161 blocks the refilling of the Ca2+ store through SOCE channels.
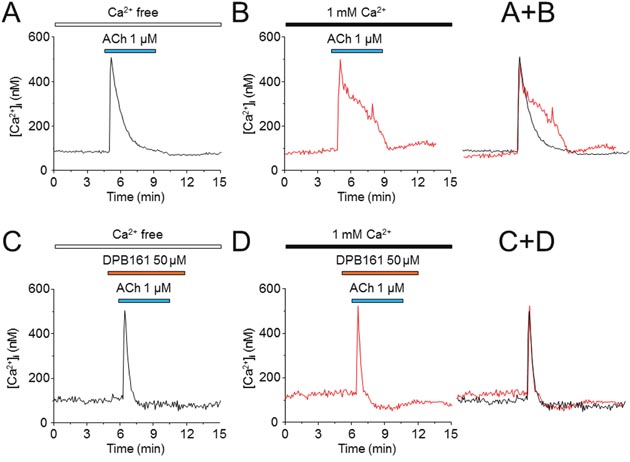
Figure 4: Effects of DPB-161 on [Ca2+]i responses to ACh measured by Ca2+ imaging in rat submandibular cells. (A) In the presence of extracellular Ca2+, the cell was stimulated with 1 μM ACh (4 min). (B) ACh was applied to the cell in a Ca2+-free solution. (C) Before (1 min) and during stimulation with ACh in the Ca2+-free solution, 50 μM DPB-161 was applied. (D) Before (1 min) and during stimulation with ACh in the Ca2+-containing solution, 50 μM DPB-161 was applied. The representative trace of 11-16 experiments is shown in each panel.
DPB161 blocks Ca2+ store refilling in a concentration-dependent manner
To compare the effects of different concentrations of DPB161 on SOCE, we quantitatively measured ACh (1 μM)-induced elevation of [Ca2+]i twice with 5-10 min intervals with or without external Ca2+. As shown in Figure 5A, under [Ca2+]o=0 without external Ca2+ refilling, the second ACh-induced elevation of [Ca2+]i was eliminated, suggesting that the first ACh exposure (1 μM for ~4 min) has already depleted the Ca2+ store. However, with [Ca2+]o perfusion (1 mM for ~ 4 min), the Ca2+ store was able to refill as the second ACh exposure induced [Ca2+]i elevation to a similar extent (Figure 5B). Thereafter, we used this protocol to compare the ratio of the second [Ca2+]i response (S2) and first [Ca2+]i response with different concentrations of DPB161 under conditions of 1 mM [Ca2+]o perfusion. One-way ANOVA showed a statistical difference in S2/S1 ratio after perfusion of DPB161 (F(4)=56.1, p<0.001). Tukey analysis showed little effect of 5 μM DPB161 in prevention of [Ca2+]o entry compared to 0 μM DPB161 (without DPB161, the S2/S1 ratio was normalized as 100%, the 5 μM DPB161 was 87.6±7.1%, p>0.05, n=8, Figure 5C), but 20 μM DPB161 reduced S2/S1 ratio to 64.6±7.0% (p<0.001, n=8), and 50 μM DPB161 reduced S2/S1 ratio to 23.0±4.8% (p<0.001, n=15, Figure 4D). These data demonstrate that DPB161 prevents SOCE in a concentration-dependent manner in rat submandibular cells (Figure 5E).
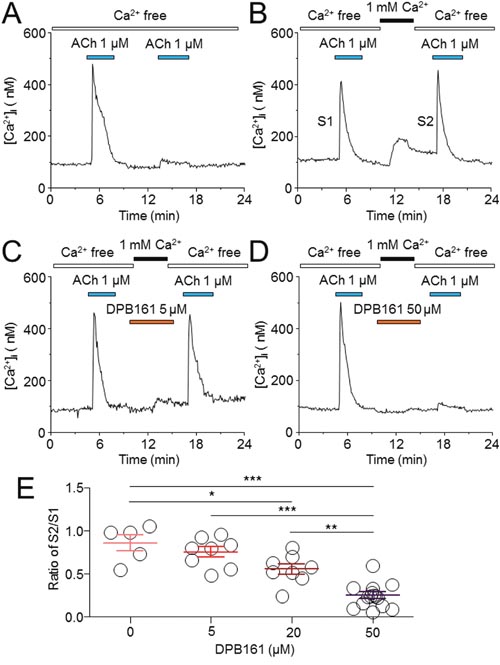
Figure 5: Effects of DPB-161 on Ca2+ store refilling following ACh-induced Ca2+ store depletion in rat submandibular cells. (A) In Ca2+-free solution, the cell was stimulated by 1 μM ACh (4 min). After the first challenge of ACh, a second ACh challenge (4 min) was performed. (B) Between the two ACh challenges, Ca2+-containing solution was applied for 2 min. (C) Before (1 min) and during addition of extracellular Ca2+, 5 μM DPB-161 was applied. (D) Before (1 min) and during addition of extracellular Ca2+, 50 μM DPB-161 was applied. The representative trace of 5-19 experiments is shown in each panel. (E) Concentration-response relationships for DPB-161 inhibition of store-operated Ca2+ entry. Relative sizes of the second ACh responses to the first ones are shown, measured by the area under the curve. Values are means of 5, 8, 8, and 15 cells tested for 0, 5, 20 and 50 μM DPB-161, respectively. Vertical bars indicate SE. Statistical significance was obtained by one-way ANOVA. *: p < 0.05, **: p < 0.01, ***: p < 0.001.
DPB161 blocks SOC channel-dependent [Ca2+]i elevation
Data presented thus far demonstrates that DPB161 blocks ACh-triggered SOCE in rat submandibular cells. Finally, we directly measured SOC channel-mediated [Ca2+]i elevation under conditions of DPB161 treatment. To measure SOC channel-mediated [Ca2+]i elevation, we depleted intracellular Ca2+ store by pre-treating cells with thapsigargin (TG) under [Ca2+]o=0 conditions. Depletion of Ca2+ stores was demonstrated by the inability of ACh application to induce [Ca2+]i elevation (Figure 6A). After depletion of the Ca2+ store, SOC channels were opened, and we were able to record 1 mM external Ca2+-induced [Ca2+]i elevation through SOC channels (Figure 6B). Then, we tried to refill the Ca2+ store with 1 mM [Ca2+]o in the presence of 5 μM (Figure 6C) or 50 μM DPB161 (Figure 6D). At 50 μM, but not 5 μM, DPB161 prevented the SOC channel-mediated [Ca2+]i elevation.
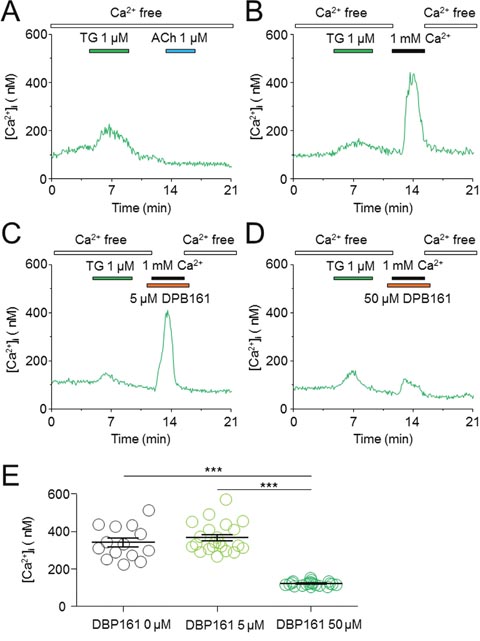
Figure 6: Effects of DPB-161 on the Ca2+ entry following thapsigargin-induced Ca2+ store depletion in rat submandibular gland acinar cells. (A) In Ca2+-free solution, 1 μM thapsigargin (TG) was first applied (4 min), followed by 1 μM ACh. (B) After TG treatment (4 min), the Ca2+-containing solution was applied (2 min). (C) Before (1 min) and during addition of extracellular Ca2+, 5 μM DPB-161 was applied. (D) Before (1 min) and during addition of extracellular Ca2+, 50 μM DPB-161 was applied. (E) Statistical analysis of 14-21 experiments are shown in panels B, C, D.
DISCUSSION
In the present study, we employed both patch-clamp recordings and Fura-2 Ca2+ imaging to examine the effects of a 2-APB analog, DPB161 on SOCE in freshly isolated rat submandibular cells. We demonstrate that DPB161 blocks rat submandibular cell SOCE in a concentration-dependent manner, suggesting that this 2-APB analog can serve as a pharmacological tool to investigate intracellular Ca2+ signaling mechanisms and provide a new therapeutic strategy for treatment of SOCE-associated diseases.
Since previous experiments showed that pharmacological manipulation of SOCE using 2APB analogs in different cell lines [11, 12], in our initial experiments, we sought an appropriate natural cells for studying the effects of DPB161 on SOCE. In accord with our experience of using pancreatic acinar cells [13–17], we first tested the effects of ACh-induced Ca2+ oscillations in mouse pancreatic acinar cells under [Ca2+]o=0 conditions, anticipating that ACh (10-20 nM)-induced Ca2+ oscillations world terminate in the absence of external Ca2+ due to an inability to refill the emptied Ca2+ stores resulting from persistent ACh exposure. However, our data shows that even under [Ca2+]o=0 conditions, ACh-induced Ca2+ oscillations continued for more than 30 min, suggesting that ACh-induced Ca2+ oscillations are not strongly dependent on SOCE to refill Ca2+ stores in pancreatic acinar cells. The precise reason is still unclear, but could be due to the highly expressed Ca2+ pump on the membrane of Ca2+ stores, which may be strong enough to reuptake released Ca2+ back to Ca2+ stores to maintain the persistent Ca2+ oscillations during exposure of ACh. Then, we selected rat submandibular cells based on previous work showing that ACh-induced Ca2+ oscillations in rat submandibular cells are dependent on external Ca2+ entry since ACh only induces a short-lasting Ca2+ oscillations if the external Ca2+ is removed [18, 19]. In the present study, we confirmed this phenomenon and further demonstrate the SOCE dependence of the persistent Ca2+ oscillations in response to ACh stimulation in rat submandibular cells. Compared to mouse pancreatic acinar cells, rat submandibular cells exhibit a much shorter duration of Ca2+ oscillation in response to persistent ACh exposure under [Ca2+]o=0 conditions, suggesting that rat submandibular cells are an excellent cell model to evaluate the pharmacological effects of agents that modulate SOCE.
Under [Ca2+]o=0 conditions, we exposed cells to either ACh or thapsigargin to deplete Ca2+ stores and open SOC channels, and tested the effects of DPB161 on SOCE. Patch-clamp experiments showed that persistent exposure of ACh (20 nM) induces a short-lasting oscillatory response (measured Ca2+-dependent Cl- currents) under [Ca2+]o=0 conditions. This is due to the ACh has released all releasable Ca2+ from Ca2+ stores without extracellular Ca2+ refilling. After emptying Ca2+ stores, exposure to 1 mM external Ca2+ enables the refilling of Ca2+ stores and recovery of the 20 nM ACh-induced oscillatory responses (Figure 3A). We used this experimental protocol to examine the effect of DPB161 on Ca2+ refilling, and found that co-application of 50 μM DPB161 in the presence of 1 mM external Ca2+ prevents the refilling of Ca2+ stores as judged by the lack of a detectable ACh-induced response after attempting to refill Ca2+ stores in the presence of 50 μM DPB161 (Figure 3B). These data suggest that 50 μM DPB161 completely blocks SOCE in rat submandibular cells. Ca2+ imaging experiments comparing [Ca2+]i elevation by ACh in the presence and absence of external Ca2+ (1 mM) indicate that the duration of the [Ca2+]i response is remarkably short in the absence of extracellular Ca2+, suggesting that during ACh depletion of Ca2+ stores, the refilling of Ca2+ stores requires the presence of external Ca2+ (Figure 4A, 4B). Using the same protocol, but comparing 1 mM external Ca2+ plus 50 μM DPB161 perfusion vs. perfusion in the absence of external Ca2+, we find ACh induces similar waveforms of [Ca2+]i elevation when perfused with 50 μM DPB161 plus 1 mM external Ca2+, suggesting that DPB161 blocks SOCE. To qualitatively compare the concentration dependence of DPB161 on SOCE, we applied ACh twice under conditions with and without external Ca2+. Without external Ca2+, the second Ca2+ response is dramatically reduced (Figure 5A), whereas in the presence of external Ca2+, sequential applications of ACh (interval > 10 min) induces [Ca2+]i elevations of similar size (Figure 5B). In the presence of different concentrations of DPB161 during the second ACh application, the concentration-dependent reduction in the ratio of the second to the first peak amplitude of ACh-induced [Ca2+]i elevations under free extracellular Ca2+ conditions demonstrates that DPB161 blocks SOCE in a concentration-dependent manner (Figure 5E). Under [Ca2+]o =0 conditions, thapsigargin depletes Ca2+ stores and opens SOC channels enabling bath perfusion with 1 mM Ca2+ to increase [Ca2+]i through SOC channels. Co-application of DPB161 with 1 mM Ca2+ prevents SOCE-mediated [Ca2+]i elevation. Taken together, these results suggest that the 2APB analog, DPB161 inhibits SOCE in rat submandibular cells.
Previous work demonstrates that the 2APB analogs DPB162 and DPB163 potently block SOCE in cell lines, in which these 2-APB analogs show high affinity for blocking the SOCE [12, 20, 21]. We further demonstrate that another 2APB analog, DPB161 also inhibits SOCE in rat submandibular cells. However, the potency of SOCE inhibition by DPB161 in rat submandibular cells is lower. For example, 5 μM DPB161 fails to block SOCE elicited by ACh- or thapsigargin-induced depletion of Ca2+ stores. This suggests that the structure difference between DPB161 and the other 2APB analogs may partially explain the difference in its potency to block the SOCE. The IC50 values for SOCE inhibition by DPB161 are about 50-fold higher than DPB163 (1.7x10-1 vs. 5.9x10-2 μM) in InsP3 receptor-deficient DT40 cells [11], suggesting a lower affinity of DPB161 compared to DPB163 for inhibition of SOCE, which is consistent with the findings in the present study. However, the affinity of DPB161 for inhibition of the SOCE in native rat submandibular cells is much lower than that in InsP3 receptor-deficient DT40 cells. These lines of evidence suggest a diversity of SOCE in different cell types.
MATERIALS and METHODS
All experiments were performed in accordance with approved guidelines set and all experimental protocols were approved by the First Affiliated Hospital of Zhengzhou University, the Barrow Neurological Institute, and the Shantou University Medical College Ethical Committees.
Preparation of single rat submandibular cells
Acute dissociation of rat submandibular cells was performed as described in our previous publication [19]. In brief, after isoflurane anesthesia, the fragments from adult Wistar rats (both genders) submandibular glands were minced and digested with collagenase (100 units/ml) for 15 min at 37°C. At the end of the collagenase digestion, the cell suspension was gently pipetted to obtain further separation of the cells. The cell suspension was then filtered through a 100 pm platinum mesh, washed twice with the extracellular solution containing 0.2% bovine serum albumin (BSA), and kept in the solution until use. A small amount of the cell suspension was added to a 2 ml-capacity experimental bath on a stage of an inverted microscope (Olympus IX7, Japan). The isolated cells usually adhered to the bottom within 15–20 min and were used for recording within 3 h after preparation. All experiments were performed at room temperature (22 ± 1°C). For the experiments, single cells were usually chosen, but in some cases, small clusters of three to five cells were also used.
Patch-clamp whole-cell recordings
Conventional whole-cell, patch-clamp recording was used to record the Ca2+-activated Cl− currents for monitoring intracellular Ca2+ signal oscillations, as reported previously [13, 17, 19]. The recording pipettes, made from borosilicate glass capillaries (Narishige, Japan), were pulled in two stages by a vertical microelectrode puller (P10, Narishige, Japan) to make patch-clamp electrodes. The tip resistance of the electrodes ranged from 4-6 MΩ when filled with 140 mM KC1-containing internal solution. After reaching a high seal resistance (> 2 GΩ), the whole-cell configuration was established by applying a brief negative suction. Transmembrane currents were recorded with a patch-clamp amplifier (Axopatch 200B; Molecular Devices; Sunnyvale, CA USA) at a holding potential (VH) of −60 mV. No series resistance was compensated in these experiments.
Measurement of intracellular Ca2+ concentration ([Ca2+]i)
Isolated cells were preloaded with a standard external solution containing 2 μM Fura-2/AM (Invitrogen, Oregon, USA) for 40 min at 37°C. Ca2+ images were captured using an inverted fluorescence microscope (Olympus IX70, Japan) and a silicon intensifier target camera (Cool SNAPfx, Roper Scientific), and recorded on a fluorescence-imaging system (LAMBAD 10-2, Sutter Instrument Co, USA). For measurement of [Ca2+]i with Fura-2, the excitation wavelengths were 340 nm and 380 nm, a dichroic mirror of 440 nm and an emission filter of 510 nm were used. [Ca2+]i was calculated from the ratio (R) of the two fluorescence intensities, F340/F380 [22]. The change in [Ca2+]i was calculated by subtracting [Ca2+]i at the peak response level from that at the pre-stimulated level.
Experimental solutions
The standard extracellular (bath) solution contained (in mM) NaCl 130, KCl 14.7, CaC12 1.0, MgC12 1.13, HEPES 10, and glucose 10, titrated with NaOH to pH 7.2. To make a Ca2+-free external solution ([Ca2+]o = 0 mM), CaCl2 was removed from and 1 mM EGTA was routinely added to the standard extracellular solution. The standard intracellular (pipette) solution contained (in mM) KC1 140, MgCl2 1.13, HEPES 10, glucose 10, ATP 1.0, and EGTA 0.25, titrated with KOH to pH 7.2.
Application of drugs
Cells were perfused continuously with a stream of the extracellular solution (~0.5 ml/min). A computer-controlled U-tube system was used for drug application (Huang et al., 2016). The reagents used for the present study were acetylcholine and thapsigargin (Sigma-Aldrich, St. Louis, MO), collagenase (Wako Chemicals, Japan), and Fura-2/AM (Invitrogen, Oregon, USA). The 2-APB analog DPB161 (Figure 1) was a gift from Professor Katsuhiko Mikoshiba.
Data analysis and statistics
For patch-clamp experiments, the Ca2+-activated Cl- current responses were presented as net charge (current area/Cm/min), then the drug-induced changes were normalized to the current response (induced by ACh) before testing drug (e.g., DPB161) exposure (Baseline). If data were obtained from the same recorded cell and the changes of ACh response were compared prior to, during and after testing drug exposure, a paired t-test was used. To analyze multiple effects (e.g., different concentrations), one-way ANOVA with Tukey’s post hoc tests were used.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
Authors thank Professor Stanley Lin for his comments and carefully correction and edition of English.
REFERENCES
1. Thorn P, Lawrie AM, Smith PM, Gallacher DV, Petersen OH. Ca2+ oscillations in pancreatic acinar cells: spatiotemporal relationships and functional implications. Cell Calcium. 1993; 14:746-57.
2. Petersen OH, Petersen CC, Kasai H. Calcium and hormone action. Annu Rev Physiol. 1994; 56:297-319.
3. Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993; 361:315-25.
4. Petersen OH, Tepikin A, Park MK. The endoplasmic reticulum: one continuous or several separate Ca(2+) stores? Trends Neurosci. 2001; 24:271-6.
5. Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986; 7:1-12.
6. Stathopulos PB, Ikura M. Cell Calcium. 2017; 63:3-7.
7. Putney JW, Steinckwich-Besancon N, Numaga-Tomita T, Davis FM, Desai PN, D'Agostin DM, Wu S, Bird GS. The functions of store-operated calcium channels. Biochim Biophys Acta. 1864; 6:900-6.
8. Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1, 4, 5)P3-induced Ca2+ release. J Biochem. 1997; 122:498-505.
9. Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002; 16:1145-50.
10. Iwasaki H, Mori Y, Hara Y, Uchida K, Zhou H, Mikoshiba K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1, 4, 5-trisphosphate receptors. Receptors Channels. 2001; 7:429-39.
11. Zhou H, Iwasaki H, Nakamura T, Nakamura K, Maruyama T, Hamano S, Ozaki S, Mizutani A, Mikoshiba K. 2-Aminoethyl diphenylborinate analogues: selective inhibition for store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007; 352:277-82.
12. Suzuki AZ, Ozaki S, Goto J, Mikoshiba K. Synthesis of bisboron compounds and their strong inhibitory activity on store-operated calcium entry. Bioorg Med Chem Lett. 2010; 20:1395-8.
13. Wu J, Kamimura N, Takeo T, Suga S, Wakui M, Maruyama T, Mikoshiba K. 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca(2+) signals mediated by inositol 1, 4, 5-trisphosphate-sensitive Ca(2+) stores in single pancreatic acinar cells of mouse. Mol Pharmacol. 2000; 58:1368-74.
14. Wu J, Takeo T, Kamimura N, Wada J, Suga S, Hoshina Y, Wakui M. Thimerosal modulates the agonist-specific cytosolic Ca2+ oscillatory patterns in single pancreatic acinar cells of mouse. FEBS Lett. 1996; 390:149-52.
15. Wu J, Takeo T, Suga S, Kanno T, Osanai T, Mikoshiba K, Wakui M. 2-aminoethoxydiphenyl borate inhibits agonist-induced Ca2+ signals by blocking inositol trisphosphate formation in acutely dissociated mouse pancreatic acinar cells. Pflugers Arch. 2004; 448:592-5.
16. Huang ZB, Wang HY, Sun NN, Wang JK, Zhao MQ, Shen JX, Gao M, Hammer RP Jr, Fan XG, Wu J. Congo red modulates ACh-induced Ca(2+) oscillations in single pancreatic acinar cells of mice. Acta Pharmacol Sin. 2014; 35:1514-20.
17. Huang Z, Wang H, Wang J, Zhao M, Sun N, Sun F, Shen J, Zhang H, Xia K, Chen D, Gao M, Hammer RP, Liu Q, et al. Cannabinoid receptor subtype 2 (CB2R) agonist, GW405833 reduces agonist-induced Ca(2+) oscillations in mouse pancreatic acinar cells. Sci Rep. 2016; 6:29757.
18. Zhang W, Fukushi Y, Nishiyama A, Wada J, Kamimura N, Mio Y, Wakui M. Role of extracellular Ca2+ in acetylcholine-induced repetitive Ca2+ release in submandibular gland acinar cells of the rat. J Cell Physiol. 1996; 167:277-84.
19. Takeo T, Suga S, Wu J, Dobashi Y, Kanno T, Wakui M. Kinetics of Ca2+ release evoked by photolysis of caged InsP3 in rat submandibular cells. J Cell Physiol. 1998; 174:387-97.
20. Goto J, Suzuki AZ, Ozaki S, Matsumoto N, Nakamura T, Ebisui E, Fleig A, Penner R, Mikoshiba K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins. Cell Calcium. 2010; 47:1-10.
21. Hendron E, Wang X, Zhou Y, Cai X, Goto J, Mikoshiba K, Baba Y, Kurosaki T, Wang Y, Gill DL. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium. 2014; 56:482-92.
22. Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985; 260:3440-50.