
Introduction
Pre-mature aging phenotypes were eminent in the klotho (Kl)-deficient mice, which have ~ 10 copies of a transgene integrated in the 5’ flanking region of the Kl gene disrupting its expression [1]. The klotho mice die around ~ 2 months of age after birth due to multiple aging-related organ failures [1]. Later, the role of KL in aging was confirmed by the reproduction of the same aging phenotypes in Kl knockout homozygous (Kl -/-) mice [2]. On the other hand, overexpression of KL extends lifespan by 20-30% [2, 3]. The protein products of Kl gene can be divided into two forms: membrane-integrated form of Kl and non-integrated form of Kl which includes secreted and soluble Kl (sKl). These two type of proteins are produced from the two transcripts that arise from a single kl gene due to alternative RNA splicing [4, 5]. It has been reported that sKl also can be produced by enzymatic cleavage of the extracellular domain of membrane Kl [5-7]. The membrane-integrated Kl is a single-pass transmembrane protein composed of a short intracellular domain and a long extracellular domain that resembles β-glucuronidase [8]. The membrane Kl functions as a co-receptor for fibroblast growth factor-23 (FGF23), while sKl is believed to have diverse systemic roles and function as a hormone [5, 9-11].
Kl is primarily expressed in brain and kidney, particularly at choroid plexus and distal tubules, respectively [5, 11]. Two other Kl-related genes have been identified based on the DNA sequence similarity; they are designated as β- and γ-klotho, respectively [12]. They are type I transmembrane proteins and behave as coreceptors of FGFs similar to the original Kl which is designated as αKl, but they are expressed in different tissues [13].
The KL expression level decreases proportionally with aging, but molecular mechanisms for the decrease are largely unknown [1, 3]. An increase of DNA methylation on the Kl CpG islands was found in aged rhesus monkeys [14]. Overexpression of Kl was able to rescue the aging phenotypes in Kl-deficient mice. Therefore, induction of Kl might be an effective therapeutic strategy for the onset of aging-related diseases. Several transcription factors and small molecules were reported as Kl inducers. Vitamin D receptor (VDR), Egr1 (early growth response protein 1), and peroxisome proliferator activation receptor γ (PPARγ) are the reported transcription factors for klotho transcription [15-17]. Klotho expression can be induced by rapamycin, 25-OH vitamin D, and statins - inhibitors for HMG-CoA reductase [17-20]. Because of the powerful anti-aging effect of klotho, it will be intriguing to explore the molecular mechanism of induction of klotho.
In an effort to find molecular mechanisms for Kl induction, we examined the small molecules and transcription factors that bind to the promoter/enhancer sequences of Kl gene. Candidate transcription factors were selected by analyses of the 2.0 kb upstream DNA sequences of human and mouse Kl genes using TRANSFAC®. Transactivation functions of the selected transcription factors and small molecules were investigated using a reporter plasmid in which firefly luciferase gene expression was under control of human KL promoter/enhancer [21].We found an activation mechanism for induction of Kl expression by a small chemical compound H. The induction by Compound H was accomplished through demethylation on the CpG islands of Kl gene and subsequent binding of transcription factors to the promoter. These results suggest that artificial DNA demethylation could be an activation mechanism for Kl expression.
Results
Expression levels of the candidate transcription factors
Human and mouse KL gene promoter/enhancer sequences up to -2.0 kb from the first ATG codon were analyzed with TRANSFAC® from Biobase to deduce candidate transcription factors (TFs) that could occupy the promoter sequences. Under the highest stringency, total 36 and 35 putative transcription factors were obtained through the analyses of human and mouse klotho gene promoter/enhancer region, respectively. TFs commonly identified for human and mice were selected for further analyses, which includes Kid3 (kidney transcription factor-3), Pax4 (paired box gene 4), ZF5 (zinc finger transcription factor-5), and CTCF (CCCTC-Binding factor) (Table 1). Expression levels of these TFs and conventional coactivators were analyzed using kidney cell lines such as HEK293 (human embryonic kidney cells), HRCE (human cortical cells that are mixture of proximal tubule and distal tubule), and HK-2 (epithelial cells from human kidney proximal tubule). All of the selected TFs except kid3 were expressed equally in the three kidney cell lines (Figure 1A). The expression level of kid3 was lower in HEK293 cells than that in other cell lines (Figure 1A). There are three kid TFs in the kid family, therefore we investigated expression levels of other kid TFs, kid1 and kid2. Among the kid TFs, kid3 was the only TF expressed lower in HEK293 cells (Figure 1B). Expression levels of KL mRNA and protein were higher in HK-2 cells than in other types of cells (Figure 1A, 1C). Consistent with the mRNA level, the protein level of Kid3 (ZFP354C) was lower in HEK-293 cells than in HK-2 cells, whereas there was no significant difference in the expression level of ZF5 (ZFP161) protein between the cell lines (Supplementary Figure 1A & B).
Table 1: Candidate transcription factors and their response elements in the KL promoter region.

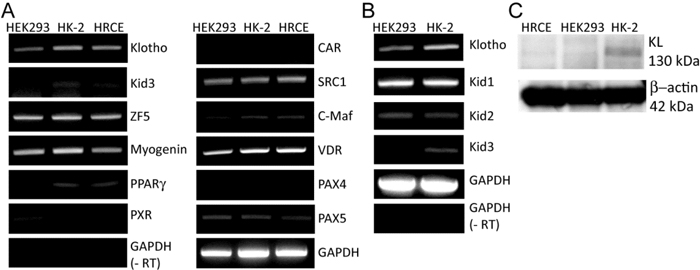
Figure 1: Expression levels of the candidate transcription factors (TFs). A. Expression levels of the identified candidate TFs were analyzed by semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR). Total RNA (1 µg) purified from the kidney cell lines, HEK293, HK-2, and HRCE, were used to synthesize the first cDNA, which was used as a template DNA for the PCR reactions. B. Expression levels of the Kid family TFs and klotho (KL) in HK-2 cells were compared with their expression levels in HEK293 cells by semi-quantitative RT-PCR. (-RT) indicates a negative control that was processed equally with the other experiment except reverse transcriptase. C. Whole cell lysates (10 µg) from the three cell lines were used for quantifying endogenous expression level of KL protein.
Screening of the small molecules
The selected TFs were functionally tested using a reporter plasmid (pHKP-luc) in which fire fly luciferase gene was expressed under the control of human KL promoter [21]. Marginal repression and activation of the reporter expression were induced by overexpression of kid3 and ZF5, respectively (Supplementary Figure 2). We believed that simple overexpression of a TF might not be strong enough to induce endogenous KL expression. Therefore, we focused on the small molecules that were reported to induce KL expression using the same reporter plasmids. Rapamycin, pravastatin, vitamin D3, and compound H were selected according to the previous reports [18-20, 22].These small molecules were not able to induce the reporter expression strongly except compound H. Compound H increased expression of the pHKP-luc reporter by 7-folds in HRCE cells (Figure 2A). Interestingly, this strong activation effect of compound H was not observed in HEK293 cells; compound H induced only 2-fold induction of the reporter expression in HEK293 cells with an irregular dose-response curve (Figure 2B). To define a response element for compound H, the pHKP-luc reporter plasmid was modified to have a shorter fragment of the promoter sequence (Figure 2C). Through transient transfection assays, it was confirmed that compound H treatment induced expression of all the reporter plasmids even one having the shortest response element, pSac2-luc, in HRCE cells (Figure 2D), but failed to induce obvious expression of any reporter plasmids in HEK293 cells (Figure 2E). Thus, the effect of compound H on the KL promoter activation varies with cell lines.
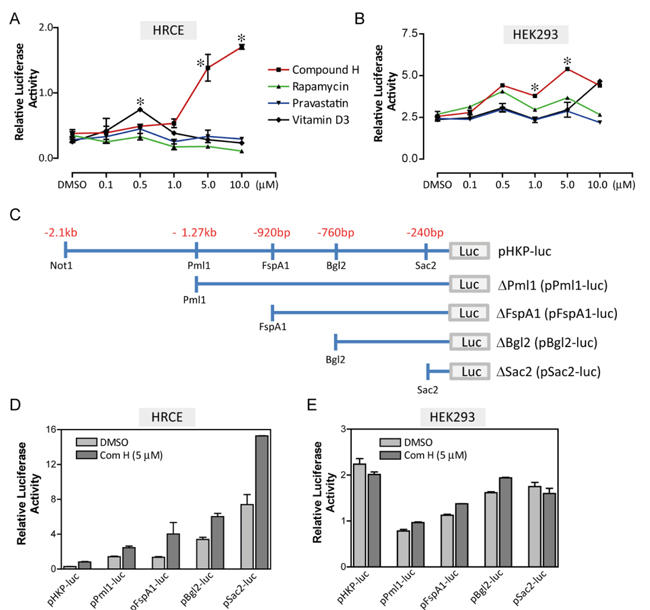
Figure 2: Screening of the small molecules. HRCE cells or HEK293 cells were transiently transfected with two reporter plasmids, one contained a KL promoter and the other contained thymidine kinase (tk) promoter as a control. Dose-response curves for the small molecules in HRCE cells A. and HEK293 cells B.. C. Fragmented KL promoter used for constructing the reporter plasmids. Effects of Compound H on luciferase activity of fragmented reporter plasmids were examined in HRCE cells D. and HEK293 cells E.. *indicates statistically significant (p < 0.01) induction of the reporter expression by the small molecules. N = 3 independent experiments.
Induction of endogenous KL expression by compound H
Next we examined whether compound H could induce endogenous KL expression. Total RNA was purified from mammalian cells treated with DMSO as a control or compound H for analyzing KL mRNA expression. It was uncovered through semi-quantitative PCR experiments that endogenous KL expression was not induced by compound H treatment in the human cell lines originated from kidney, such as HRCE and HEK293 cells (Figure 3A). A mouse cell line originated from distal convoluted tubule (DCT) in the kidney increased expression of Kl mRNA in response to compound H treatment (Figure 3B). The Kl mRNA expression was absent in a mouse vascular smooth muscle cell line (MOVAS), indicating that Kl is not expressed in smooth muscles (Figure 3B). Compound H did not affect Kl expression (Figure 3B). The Kl protein expression was also increased in the DCT cells treated with compound H in accordance with the Kl mRNA induction (Figure 3C). These results suggest that endogenous Kl expression in mammalian cells can be induced by compound H. However, the Kl induction was limited to the distal convoluted cells.
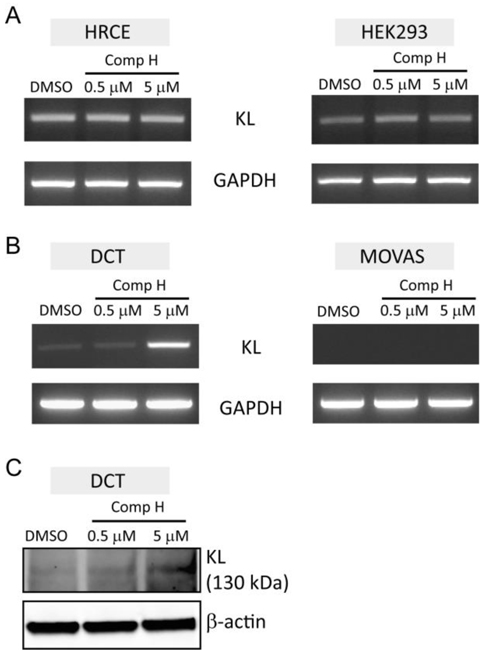
Figure 3: Induction of endogenous KL expression by compound H. mRNA purified from the cells treated with DMSO or compound H was used for the semi-quantitative PCR. A. KL and GAPDH expression levels in the human cell lines, HRCE cells and HEK293 cells, following treatment with compound H for 6 hr. B. Expression levels of Kl and GAPDH in the mouse cell lines, DCT and MOVAS, following treatment with compound H for 6 hr. C. Expression levels of Kl protein in DCT cells treated with compound H were measured by western blot.
Compound H increased binding of Kid3 and Pax4 to the KL promoter
We investigated whether the candidate TFs are involved in compound H-induced signaling to activate KL expression. Specifically, we examined whether binding of the TFs toward KL promoter could be changed by compound H. To this end, we employed a DNA pull-down assay. Magnetic beads coated with streptavidin protein were labeled with the biotin-conjugated DNA fragment encompassing 200 bp upstream from the first ATG codon of human KL gene. The 200 bp DNA contains all the response elements for the TFs. The beads were incubated with nuclear extracts purified from DCT cells treated with DMSO or 5 µM compound H. Binding proteins were recovered using a magnet and were subject to analysis with SDS-PAGE and western blot. The binding of the two TFs, Kid3 and Pax4, to the 200 bp DNA was increased by compound H, whereas binding affinity of CTCF and ZF was not increased (Figure 4A). The total cellular protein expression levels of the TFs, such as ZF5, Pax4, Kid3 and CTCF, were not changed by compound H treatment (Figure 4B).
These results indicate that the KL induction mechanism of compound H may not be through the increase of the TFs, but through the increased binding of these selected TFs (Kid3, Pax4) to the KL promoter.
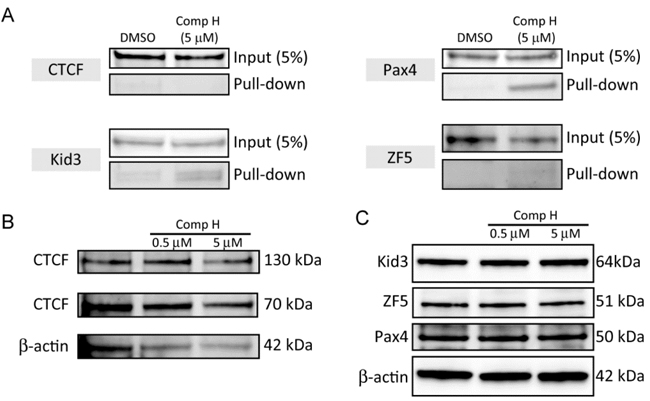
Figure 4: Compound H increased binding of Kid3 and Pax4 to the KL promoter. A. Nuclear extracts (1 mg) of DCT cells treated with DMSO or compound H (5 µM) for 24 hr were incubated with the 200 bp DNA containing the KL promoter fragment. And the binding proteins were subject to analyses of binding of the TFs to the DNA by western blot B.. Whole cell extracts (10 µg) from the DCT cells treated with DMSO or compound H for 24 hr were loaded onto each well for western blot analyses of protein expression levels of transcription factors. The 70 kDa CTCF is the major fragment of full-length (130 kDa) CTCF.
Two transcription factors, Pax4 and Kid3, mediated the effects of compound H
Because binding of Kid3 and Pax4 to the KL promoter was increased by compound H treatment, we next investigated whether Kid3 or Pax4 are functionally involved in compound H-induced activation of Kl expression. To this end, DCT cells were transfected with siRNA for Kid3 or Pax4 to suppress expression of these transcription factors. A scrambled sequence of siRNA was used as a control siRNA. The Pax4 expression level was suppressed in the siPax4-transfected cells (Figure 5A, 5B) which attenuated the induction of secreted Kl (sKl) by compound H (Figure 5A, 5C). Kid3 suppression by siKid3 was hard to detect (Figure 5D, 5E): many transfected cells were floated and dead (data not shown). In a low concentrated transfection of the Kid3 siRNA, we were able to detect that Kid3 suppression significantly potentiated compound H-induced induction of sKl expression (Figure 5D, 5F). These results suggest that Pax4 and Kid3 are involved in induction of Kl expression positively and negatively, respectively, by compound H.
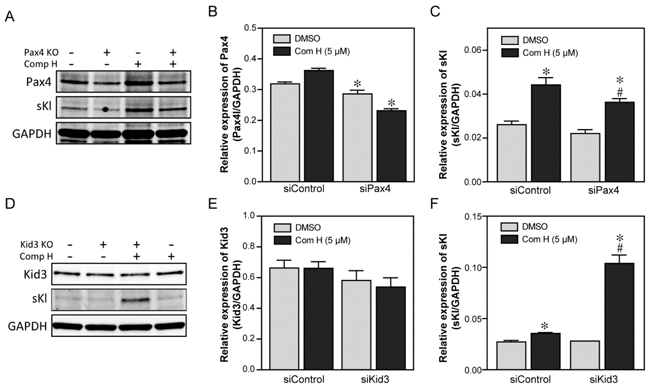
Figure 5: Two transcription factors, Pax4 and Kid3, mediated the effects of compound H. Effects of the two transcription factors, Pax4 and Kid3, on the compound H-induced induction of Kl were analyzed following siRNA-mediated suppression. DCT cells were transfected with siRNA for Pax4 or Kid3, and then treated with DMSO or compound H. Whole cell extracts were collected for protein expression analyses with specific antibodies. A. Representative western blot results for Pax4 and secreted Kl (sKl) expression in the Pax4-supressed DCT cells. B. Quantification of Pax4 expression. *indicates statistically significant ( p < 0.05) suppression of Pax4 by siPax4 transfection (vs. siControl). C. Quantification of the sKl expression. *indicates statistically significant (p < 0.01) induction of sKl by compound H treatment (vs. DMSO). # indicates statistically significant (p < 0.01) decrease of the compound H-induced sKl expression in siPax4-transfected cells compared with that in control siRNA-transfected cells treated with compound H. D. Representative western blot results of Kid3 and sKl expression in Kid3-suppressed DCT cells. E. Quantification of the Kid3 expression. F. Quantification of sKl expression. *indicates statistically significant ( p < 0.05) induction of sKl by compound H treatment.# indicates a statistically significant (p < 0.01) increase of the compound H-induced sKl expression in the siKid3-transfected cells compared with that in the control siRNA-transfected cells treated with compound H. N = 3 independent experiments.
Effect of compound H on DNA demethylation
It has been known that the Kl expression level decreases with aging probably due to an increase of DNA methylation on the CpG islands of Kl. Therefore, demethylation could be a mechanism for induction of Kl expression. We examined the methylation levels on the CpG islands of Kl following treatment with compound H. The DNA methylation status was quantitatively measured using a kit, with which methylated DNA was selectively captured by Mecp2, a protein exclusively binds to methylated DNA, and an anti-Mecp2 antibody-labeled beads. Eluted methylated DNA was used to amplify CpG islands of Kl with specific primers for the known methylated CpG region. For this experiment, we designed a primer set for amplifying between +168 and +282 from the first ATG codon of mouse Kl. Methylation on the CpG island of Kl was decreased in the DCT cells treated with compound H for 24 hr, whereas treatment with a DNA methylase inhibitor ‘5-azacytidine’ failed to decrease methylation (Figure 6A). The demethylation effect of compound H on Kl was detected in DCT cells as early as 6 hours after treatment with compound H (Figure 6B). The methylation status on p16 (CDKN2A) gene was measured to determine whether the demethylation effect of compound H was exclusive to Kl. Similar to its effect on Kl, compound H treatment induced demethylation of CpG islands of p16 gene (Figure 6C). Methylation of p16 gene was also decreased by 5-azacytidine treatment which was not observed for the Kl gene (Figure 6A and 6C).
We expected that compound H might be an activator for DNA demethylases rather than a methylase inhibitor such as 5-azacytidine because compound H was able to induce the demethylation in such a short period of the incubation time (6hr). To confirm it, enzyme activity of DNA demethylases was measured using nuclear extracts purified from the DCT cells treated with compound H for 6 hr. DNA demethylase activity was measured by quantifying left over methylated DNA amounts with an anti-methyl cytosine antibody after incubation with the nuclear extracts. The DNA demethylase activity in the DCT cells treated with compound H was increased significantly in DCT cells (Figure 6D).
These results indicate that compound H induced Kl expression through demethylation on CpG islands of the Kl gene by enhancing the demethylase activity.
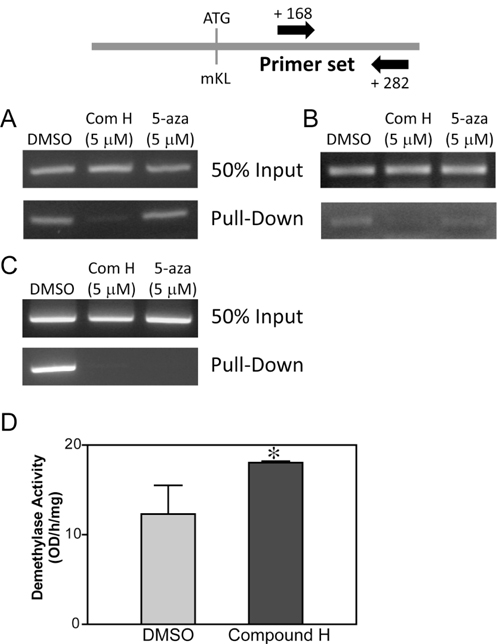
Figure 6: Effect of compound H on DNA demethylation. Genomic DNA was purified from DCT cells treated with compound H. After fragmentation with Mse 1, the genomic DNAs were incubated with Mecp2 protein to capture the methylated DNA fragments, which were used to amplify CpG islands on Kl or p16. PCR amplification of the CpG islands on the mouse Kl gene using purified methylated DNA from DCT cells were treated with compound H (Com H) or 5-azacytidine (5-aza) for 24 hr A. or 6 hr B.. C. CpG island on the p16 gene was amplified by PCR as described above. D. The demethylase enzyme activity was measured using nuclear extracts from DCT cells treated with DMSO or compound H for 24 hr. n = 3. *indicates statistically significant (p < 0.01) change of demethylase enzyme activity in the DCT cells treated with compound H (vs. DMSO).
Discussion
Klotho is an aging-suppressor gene [5]. Emerging evidence suggests that induction of klotho expression may be an effective therapeutic strategy for preventing or ameliorating aging-associated diseases [3, 23-27]. However, it remains a challenge to induce KL expression for the therapeutic purposes owing to insufficient knowledge of the induction mechanism. This study demonstrates a new molecular mechanism for induction of Kl expression, i.e., demethylation of Kl gene, by a small molecule called compound H. Compound H was reported to induce Kl expression but its induction mechanism has never been explored [20]. We found that compound H activated the expression of a reporter plasmid, which was composed of human KL gene promoter followed by luciferase gene [21]. The effect of compound H in induction of KL expression was much stronger than that of 1,25-dihydroxy vitamin D (calcitriol), an agonist ligand for vitamin D receptors and is known to induce KL expression [17, 19]. Calcitriol induces KL expression through vitamin D receptor response element existing in the KL promoter. Therefore, it is highly likely that calcitriol may be an endogenous inducer of KL expression because calcitriol expresses in the proximal tubule in the kidney that is physically close to the distal tubule where KL is expressed. We found that calcitriol activity was not as much strong as compound H in stimulating expression of the KL promoter-containing reporter in the transfected kidney cells. It is likely that the endogenous inducer, calcitriol, may not be able to activate KL expression if a prerequisite condition exists, i.e., DNA methylation. Therefore, we expect that KL expression could not be induced by a transcription factor alone under the methylation condition. Increased binding of Pax4 and Kid3 to the KL promoter by compound H treatment support our notion; DNA demethylation might be a favorable condition for the binding of these TFs (Figure 4B).
DNA methylation is a general mechanism that blocks a gene expression. Methylated DNA is hardly occupied by transcription factors, so that demethylation is required for proper binding of the TFs, such as Pax4 and Kid3, to the promoter. Indeed, we found that compound H treatment increased demethylation and enhanced binding of Pax4 and Kid3 to the Kl promoter (Figure 4). Downregulation of Kid3 potentiated compound H-induced sKl expression, whereas downregulation of Pax4 attenuated the activity of compound H (Figure 5). Although silencing of Kid3 potentiated the effect of compound H (Figure 5D-F), it may be too early to conclude that Kid3 is a repressor for Kl expression. Kid3 is a C2H2 (Krüppel-like) zinc finger protein which is involved in the development [28]. We speculate that Kid3 might be an integrator which resists against any signals that could change the integrity in the cells. We found that cells were detached from the culture dish surface and died when Kid3 expression was decreased.
An increase in DNA methylation on the CpG islands of Kl gene might be an important contributor for the age-related decrease in KL expression [14]. In the artificial absence of methylation by compound H, the transcription factors could bind to the promoter and activate gene expression. We found that the compound H-induced demethylation was accomplished through an active process by stimulating demethylases. Inhibition of methylation by a methylase inhibitor, 5-azacytidine, failed to induce Kl expression. As we expected, DNA demethylase activity was increased in the cells treated with compound H only for 6hr which is too short for methylase inhibitors to exert their activity. Enhanced demethylase activity by compound H also resulted in demethylation on p16 gene promoter. Thus, compound H-mediated DNA demethylation may not be gene-specific. Selective induction of KL expression should be considered when screening for an anti-aging therapeutic agent. We showed in a recent study that compound H also induced klotho expression, increased circulating klotho levels, and improved arterial stiffening in aged mice (unpublished data). Since compound H has not been tested for its safety, caution is needed for inducing KL in vivo because it may cause unwanted effects.
This is an initial study to explore whether and how compound H may induce kotho expression. The finding showed that the compound H activated DNA demethylase leading to demethylation and klotho expression. However, the mechanism of compound H-induced activation of DNA demethylase is not clear. A further study is warranted to investigate how compound H acts on demethylase.
Materials and Methods
Plasmid constructions
The pHKP-luc reporter plasmid was kindly provided by Dr. Kadir Turan [21]. This plasmid contains -2.0 kb upstream sequence from the translation start codon of human KL gene. Reporter plasmids containing a fragment of the human KL promoter were constructed by cutting the pHKP-luc plasmid with restriction enzymes and followed by ligation with klenow fragment. The restriction enzymes used for the constructions are listed in Figure 2C. Each reporter plasmid containing the fragment was named following the restriction enzyme used for cutting with Not1. DNA sequences of every plasmid used for the experiments were confirmed by DNA sequencing. siRNA for Kid3 and Pax4 were purchased from Santa Cruz Biotechnology.
Transient co-transfection assay
Mammalian cells (HRCE, HK-2, HEK293 and DCT) were used for the transfection assay. HRCE (human renal cortical epithelial) cells were purchased from Lonza. HK-2 (human kidney proximal tubule) cells and HEK293 (human embryonic kidney) cells were purchased from ATCC. A DCT (mouse distal convoluted tubule) cell line was kindly provided by Peter Friedman in University of Pittsburgh [29]. X-treme GENE transfection reagent from Roche Applied Science was used for transfection. For analyses of KL gene expression, plasmid reporters in which fragments of human KL gene promoter drive the fire fly luciferase gene expression were used. For transfection control, a reporter plasmid expressing renilla luciferase under control of thymidine kinase promoter was used. Luciferase assays were conducted using Dual-Luciferase® Reporter Assay System from Promega. Fire fly luciferase activity was divided by matched renilla luciferase activity to present relative luciferase activity. Transfected cells were treated with DMSO or small molecules for the indicated time period in each result after 24 h of transfection.
Reverse-transcription polymerase chain reaction (RT-PCR)
Semi-quantitative RT-PCR was conducted with purified RNA from mammalian cells [30, 31]. One µg of RNAs purified using RNeasy kit (Qiagen) was used for synthesis of the first strand cDNA with a random hexamer (Roche) and Superscript III first-strand synthesis kit (Invitrogen). PCR was conducted with high fidelity 2X PCR master mix (NEB).
DNA pull-down experiments
All of the processes were done following the protocol published by BL Jutras et al. with minor modifications [32]. Biotin-labeled 200-bases DNA encompassing human KL promoter/enhancer was hybridized with a 200-bases complementary DNA, with which streptavidin-coated Dynabeads from Life Technologies (Grand Island, NY) were labeled. Nuclear extracts prepared from DCT cells treated with DMSO or compound H for 24 h were incubated with the labeled magnetic beads at room temperature for 30 min. Binding proteins to the DNA were purified with a DynaMag™ (Life Technologies) and analyzed on SDS-PAGE followed by western blot.
DNA methylation analysis
Methylation on the CpG islands of the mouse Kl gene was analyzed with a kit ‘Promoter methylation PCR kit’ from the Affymetrix (Santa Clara, CA) by following the manufacturer’s protocol. Briefly, mouse DCT cells treated with DMSO, the compound H, or 5-azacytidin (a methyltransferase inhibitor) were collected to purifying genomic DNAs, which were cut with Mse 1 restriction enzyme to make fragments of the DNAs. For quantification of DNA methylation, the fragmented DNAs were incubated with Mecp2, a protein exclusively binds to methylated DNA, and then the Mecp2-bound DNAs were captured to the beads pre-labeled with an anti-Mecp2 antibody in a column. After heavy washings to remove unmethylated DNAs, methylated DNAs were eluted and used for amplifying CpG islands of KL gene with PCR. An increase of the amplified PCR products indicates an increase of methylation and vice versa.
DNA demethylase assay
The DNA demethylase assay kit was purchased from Epigentek. The process was conducted by following manufacturer’s protocol. Briefly, pre-methylated DNA substrates were incubated with nuclear extracts purified from the DCT cells treated with DMSO or compound H (5 M) for 24 hr. DNA demethylase activity was quantified by measuring the amounts of left over methylated DNA using a specific antibody for methylated cytosine. Low observance indicates there is a strong DNA demethylase activity.
Western blot
The western blot analysis was performed as described in our recent studies [33-37]. Ten µg protein from whole cell extracts were loaded onto each well of Criterion™ TGX™ precast gel from Bio-Rad. Protein amounts were quantified using the BCA protein assay kit from Thermo Scientific following manufacturer’s protocol. Migrated protein bands in SDS-PAGE were transferred onto nitrocellulose membrane (0.2 µm) from Bio-Rad using a Bio-Rad trans-blot turbo system. Expression levels of KL proteins were measured with an anti-mouse KL antibody from R & D systems or an anti-human KL antibody from Cosmo bio (Clone No. KM2076). An anti-actin antibody was purchased from Abcam. Fluorescence-labeled secondary antibodies were purchased from Invitrogen. Protein bands were developed and analyzed with the ChemiDoc™ MP imaging system from Bio-Rad.
Statistical analysis
The data were analyzed by one-way analysis of variance (ANOVA). The unpaired t-test was used for comparisons between two groups. Significance was set at a 95% confidence limit.
Author contributions
Z.S. developed the concepts and hypotheses and participated in critical revision of the manuscript. D.J. designed the study, conducted the experiments, analyzed the data, and participated in writing the manuscript. Y.X. participated in some experiments.
Conflicts of Interest
There is no competing financial interest.
Funding
This work was supported by NIH R01 AG049780, HL118558, DK093403, HL105302, HL116863, HL122166, and HL102074. This publication was made possible by NIH Grant Number 9P20GM104934-06 from the COBRE Program of the National Institute of General Medical Sciences.
References
1. Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997; 390:45-51.
2. Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003; 17:2393-2403.
3. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, Shimomura I, Takayama Y, Herz J, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005; 309:1829-1833.
4. Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998; 242:626-630.
5. Xu Y, Sun Z. Molecular basis of klotho: from gene to function in aging. Endocr Rev. 2015; 36:174-193.
6. Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci USA. 2007; 104:19796-19801.
7. Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, Kaether C. Klotho is a substrate for alpha-, beta- and gamma-secretase. FEBS Lett. 2009; 583:3221-3224.
8. Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998; 424:6-10.
9. Kuro-o M. Klotho as a regulator of fibroblast growth factor signaling and phosphate/calcium metabolism. Curr Opin Nephrol Hypertens. 2006; 15:437-441.
10. Huang CL. Regulation of ion channels by secreted Klotho: mechanisms and implications. Kidney Int. 2010; 77:855-860.
11. Wang Y, Sun Z. Current understanding of klotho. Ageing Res Rev. 2009; 8:43-51.
12. Ito S, Kinoshita S, Shiraishi N, Nakagawa S, Sekine S, Fujimori T, Nabeshima YI. Molecular cloning and expression analyses of mouse betaklotho, which encodes a novel Klotho family protein. Mech Dev. 2000; 98:115-119.
13. Kuro-o M. Klotho and the aging process. Korean J Intern Med. 2011; 26:113-122.
14. King GD, Rosene DL, Abraham CR. Promoter methylation and age-related downregulation of Klotho in rhesus monkey. Age (Dordr). 2012; 34:1405-1419.
15. Zhang H, Li Y, Fan Y, Wu J, Zhao B, Guan Y, Chien S, Wang N. Klotho is a target gene of PPAR-gamma. Kidney Int. 2008; 74:732-739.
16. Choi BH, Kim CG, Lim Y, Lee YH, Shin SY. Transcriptional activation of the human Klotho gene by epidermal growth factor in HEK293 cells; role of Egr-1. Gene. 2010; 450:121-127.
17. Haussler MR, Haussler CA, Whitfield GK, Hsieh JC, Thompson PD, Barthel TK, Bartik L, Egan JB, Wu Y, Kubicek JL, Lowmiller CL, Moffet EW, Forster RE, Jurutka PW. The nuclear vitamin D receptor controls the expression of genes encoding factors which feed the “Fountain of Youth” to mediate healthful aging. J Steroid Biochem Mol Biol. 2010; 121(1-2):88-97.
18. Tataranni T, Biondi G, Cariello M, Mangino M, Colucci G, Rutigliano M, Ditonno P, Schena FP, Gesualdo L, Grandaliano G. Rapamycin-induced hypophosphatemia and insulin resistance are associated with mTORC2 activation and Klotho expression. Am J Transplant. 2011; 11:1656-1664.
19. Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I, Haussler MR, Kerr Whitfield G. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011; 414:557-562.
20. King GD, Chen C, Huang MM, Zeldich E, Brazee PL, Schuman ER, Robin M, Cuny GD, Glicksman MA, Abraham CR. Identification of novel small molecules that elevate Klotho expression. Biochem J. 2012; 441:453-461.
21. Turan K, Ata P. Effects of intra- and extracellular factors on anti-aging klotho gene expression. Genet Mol Res. 2011; 10:2009-2023.
22. Yoon HE, Lim SW, Piao SG, Song JH, Kim J, Yang CW. Statin upregulates the expression of klotho, an anti-aging gene, in experimental cyclosporine nephropathy. Nephron Exp Nephrol. 2012; 120:e123-e133.
23. Hu MC, Moe OW. Klotho as a potential biomarker and therapy for acute kidney injury. Nat Rev Nephrol. 2012; 8:423-429.
24. Nabeshima Y. Klotho: a fundamental regulator of aging. Ageing Res Rev. 2002; 1:627-638.
25. Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009; 54:810-817.
26. Lin Y, Sun Z. In vivo pancreatic beta-cell-specific expression of antiaging gene klotho: a novel approach for preserving beta-cells in type 2 Diabetes. Diabetes. 2015; 64:1444-1458.
27. Lin Y, Sun Z. Antiaging Gene Klotho Attenuates Pancreatic beta-Cell Apoptosis in Type 1 Diabetes. Diabetes. 2015; 64:4298-4311.
28. Gao L, Sun C, Qiu HL, Liu H, Shao HJ, Wang J, Li WX. Cloning and characterization of a novel human zinc finger gene, hKid3, from a C2H2-ZNF enriched human embryonic cDNA library. Biochem Biophys Res Commun. 2004; 325:1145-1152.
29. Pizzonia JH, Gesek FA, Kennedy SM, Coutermarsh BA, Bacskai BJ, Friedman PA. Immunomagnetic separation, primary culture, and characterization of cortical thick ascending limb plus distal convoluted tubule cells from mouse kidney. In Vitro Cell Dev Biol. 1991; 27A:409-416.
30. Lin Y, Sun Z. Antiaging gene Klotho enhances glucose-induced insulin secretion by up-regulating plasma membrane levels of TRPV2 in MIN6 beta-cells. Endocrinology. 2012; 153:3029-3039.
31. Wang X, Wang Q, Sun Z. Normal IgG downregulates the intracellular superoxide level and attenuates migration and permeability in human aortic endothelial cells isolated from a hypertensive patient. Hypertension. 2012; 60:818-826.
32. Jutras BL, Verma A, Stevenson B. Identification of novel DNA-binding proteins using DNA-affinity chromatography/pull down. Curr Protoc Microbiol. 2012; Chapter 1:Unit1F. 1.
33. Gao D, Zuo Z, Tian J, Ali Q, Lin Y, Lei H, Sun Z. Activation of SIRT1 Attenuates Klotho Deficiency-Induced Arterial Stiffness and Hypertension by Enhancing AMP-Activated Protein Kinase Activity. Hypertension. 2016; 68:1191-1199.
34. Varshney R, Ali Q, Wu C, Sun Z. Monocrotaline-Induced Pulmonary Hypertension Involves Downregulation of Antiaging Protein Klotho and eNOS Activity. Hypertension. 2016; 68:1255-1263.
35. Fan J, Sun Z. The Antiaging Gene Klotho Regulates Proliferation and Differentiation of Adipose-Derived Stem Cells. Stem Cells. 2016; 34:1615-1625.
36. Chen J, Lin Y, Sun Z. Deficiency in the anti-aging gene Klotho promotes aortic valve fibrosis through AMPK alpha-mediated activation of RUNX2. Aging Cell. 2016; 15:853-860.
37. Lin Y, Chen J, Sun Z. Anti-aging gene klotho deficiency promoted high fat diet-induced arterial stiffening by inactivation of AMP-activated protein kinase. Hypertension. 2016; 67:564-573.