
INTRODUCTION
The underlying genetic cause of colorectal cancer (CRC) can only be identified for 5-10% of cases despite approximately 20% of all CRC cases thought to be due to inherited genetic factors [1], highlighting that the genetic cause for the majority of the heritable CRC is still unknown [2]. Germline mutations in the DNA mismatch repair (MMR) genes [3] and the APC [4] gene were discovered over 20 years ago, accounting for 2-5% of CRC overall. Since then, linkage studies have led to some progress in identifying additional highly penetrant genes including MUTYH [5], STK11 [6], BMPR1A [7], SMAD4, and PTEN [8], when combined might explain a further 1% of CRC. Genome-wide association-based studies (GWAS) have identified common germline alleles, but all have been weakly associated with CRC risk and collectively are likely to explain only a few percent of the missing heritability for CRC [9]. Therefore, while a number of established hereditary CRC genes exist, the cause of the majority of inherited CRC remains explained.
Up to half of the CRC cases with a very strong family history of CRC (fulfilling the Amsterdam criteria I) have microsatellite stable (MSS) tumours and do not carry an inherited MMR gene mutation [10]. For almost all of these families, no mutation can be identified which has important negative clinical implications for family members. These families have been named “Familial Colorectal Cancer Type X” (FCCTX) [11]. FCCTX is probably not a single disorder, rather, it is more likely to be a heterogeneous group of CRCs including: CRC cases with a chance aggregation of CRC in their relatives (lifetime risk of CRC is 5% in the general population); CRC cases with an undiagnosed syndrome e.g. undetected Lynch syndrome or MUTYH-associated polyposis [12]; but for the majority of CRC cases it will be yet-to-be-discovered genetic mutations.
Next-generation sequencing (NGS) (whole genome or exome) has facilitated further discovery of cancer susceptibility genes including RECQL, FANCM, FANCC, XRCC2, POT1, and BAP1 for breast and melanoma [13], and POLE, POLD1, and NTHL1 for CRC [14, 15]. Highly penetrant cancer mutations, such as those observed in the APC or MMR gene mutations in CRC and BRCA1/2 genes in breast cancer, are rare and usually population-specific. Therefore, screening for mutations in these and other cancer susceptibility genes using NGS requires large sample sizes, which makes this strategy prohibitively expensive.
The cost of whole genome sequencing has dropped from about $10 million in 2007 to a reasonable $3,000-$4,000 per genome nowadays [16]. However, it is widely agreed that the cost of variant interpretation is not going down anytime soon. While sequencing projects, such as NHLBI GO Exome Sequencing Project (ESP) and 1000 Genomes Project, have generated an enormous amount of data on common and rare variants, the interpretation of the significance of these variants in the etiology of hereditary syndromes is limited. Genome or exome analyses of well-defined case-control studies to identify rare, highly penetrant mutations associated with hereditary syndromes are still prohibitively expensive and frequently rely on the sequencing of cases only to reduce the cost.
Common variants and very rare variants have not explained heritability of complex diseases and the research paradigm has shifted towards the role of large sets of rare variants with moderate effect sizes [17, 18]. While very rare variants do not explain the entirety of missing heritability of complex diseases, they may help to elucidate new mechanisms of the development of a disease. Moreover, rare, highly penetrant mutations have great importance for genetic counseling, disease screening, and primary prevention of hereditary cancer. Recent identification of POLE and POLD1 genes with mutations predisposing to hereditary CRC [15] are the perfect example of rare, highly penetrant genes that have expanded our understanding of CRC pathogenesis by implicating inherited impairment of DNA base-excision repair in CRC predisposition.
The aim of the study was to apply a DNA pooling NGS strategy to screen 40 established or candidate CRC susceptibility genes in order to identify rare, likely pathogenic variants across a study of 1,046 familial CRC cases and 1,006 controls from the Colon Cancer Family Registry (CCFR). The DNA pooling strategy presented here is at least five times less expensive than traditional NGS approaches and could be applied to other familial diseases.
RESULTS
Analysis of very rare variants
The characteristics of the familial CRC cases according to their recruitment category (Tiers 1 to 6) and controls are shown in Table 1 where 89% of the cases were whites. A total of 9,985 unique non-synonymous, stop-gain, stop-loss, and frameshift variants in 40 genes were identified across all cases and controls. Subsequent variant filtering based on allele frequency and predicted functional impact identified a total of 264 rare, likely deleterious variants in 38 genes (no variants met selection criteria in PTEN or STK11) in 287 out of 1046 CRC cases, not found in the tested controls and observed at a very low frequency or absent in reference population datasets (MAF <0.001). Among all variants, six were stop-gain, three frameshifts, and 255 were non-synonymous variants. Out of 264 rare variants, 20 were found in more than one pool (Supplementary Table 1). The frequency of likely deleterious variants identified across the six Tiers ranged from 8.2% (Tier 5) to 15.6% (Tier 2) (Figure 1). A total of 24 MMR variants were identified across all cases, including those with MSS CRCs, with Tier 2 cases having the highest proportion of MMR gene variants identified. A total of 6 POLE and 2 POLD1 likely deleterious variants were identified across all CRC cases tested, all of which had tumors that were MSS. Considering only those variants that met the more stringent ACMG criteria of pathogenicity [19], reduced the number of variants from 264 to 21 variants for both established and candidate CRC genes, the majority of which were in Tier 2 cases (4/21) (Table 2).
Table 1: Characteristics of the study sample
FCCTX-like cases |
Lynch-like cases |
Cases, n (%) |
Controls, n (%) |
Total |
|||||
|---|---|---|---|---|---|---|---|---|---|
Tier |
Tier 1 |
Tier 3 |
Tier 4 |
Tier 5 |
Tier 2 |
Tier 6 |
|||
Tier criteria |
AC-I |
AC-I |
AC-II |
CRC<60 |
AC-I/II |
CRC ≥1 FDR/SDR |
|||
Tier MSI status |
MSS |
MSS |
MSS |
MSS |
MSI-H |
MSI-H |
|||
Total |
139 (13.3%) |
202 (19.3%) |
36 (3.4%) |
501 (47.9%) |
64 (6.1%) |
104 (10%) |
1046 |
1006 |
2052 |
Sample origin |
|||||||||
Fred Hutchinson Cancer Research Center |
6 |
22 |
5 |
67 |
6 |
15 |
121 (12%) |
504 (50%) |
625 |
University of Hawaii |
5 |
13 |
- |
36 |
- |
6 |
60 (5%) |
110 (11%) |
170 |
Cancer Care Ontario |
26 |
84 |
10 |
96 |
13 |
27 |
256 (25%) |
26 (3%) |
282 |
USC Consortium |
15 |
32 |
10 |
52 |
25 |
17 |
151 (14%) |
0 |
151 |
University of Melbourne |
58 |
34 |
5 |
155 |
16 |
20 |
288 (28%) |
256 (25%) |
544 |
Mayo Clinic |
29 |
17 |
6 |
95 |
4 |
19 |
170 (16%) |
110 (11%) |
280 |
Age (mean (range)) |
55 (26–87) |
63 (23-92) |
58 (39-76) |
53 (29-94) |
55 (20-87) |
58 (29-84) |
56 (20-94) |
61 (43-84) |
|
Sex |
|||||||||
Male |
61 |
105 |
15 |
251 |
31 |
49 |
512 (49%) |
395 (39%) |
907 |
Female |
78 |
97 |
21 |
249 |
33 |
55 |
533 (51%) |
611 (61%) |
1144 |
Unknown |
0 |
0 |
0 |
1 |
0 |
0 |
1 (0%) |
0 |
1 |
Race |
|||||||||
White |
127 |
180 |
36 |
436 |
59 |
96 |
934 (89%) |
863 (85%) |
1797 |
Black |
1 |
1 |
0 |
9 |
0 |
0 |
11 (1%) |
20 (2%) |
31 |
Asian |
6 |
17 |
0 |
45 |
2 |
7 |
77 (7%) |
97 (10%) |
174 |
American Indian |
1 |
1 |
0 |
2 |
0 |
0 |
4 (1%) |
2 (1%) |
6 |
Other or unknown |
4 |
3 |
0 |
9 |
3 |
1 |
20 (2%) |
24 (2%) |
44 |
CRC site |
|||||||||
Large intestine |
102 |
140 |
23 |
374 |
59 |
98 |
796 (76%) |
- |
796 |
Rectum |
37 |
57 |
13 |
123 |
5 |
6 |
241 (23%) |
- |
241 |
Appendix |
0 |
4 |
0 |
4 |
0 |
0 |
8 (1%) |
- |
8 |
MSI |
|||||||||
MSS |
112 |
174 |
33 |
417 |
- |
- |
736 (70%) |
- |
736 |
MSI-Low |
27 |
28 |
3 |
84 |
- |
- |
142 (14%) |
- |
142 |
MSI-High |
- |
- |
- |
- |
64 |
104 |
168 (16%) |
- |
168 |
AC – Amsterdam criteria, MSI – microsatellite instability, MSS – microsatellite stable, MSI-H – highly microsatellite unstable, FDR – first degree relative, SDR – second degree relative.
*AC-I (no age) describes Tier 3 cases that fulfill all AC-I criteria except “At least 1 of the cancers diagnosed before age 50”.
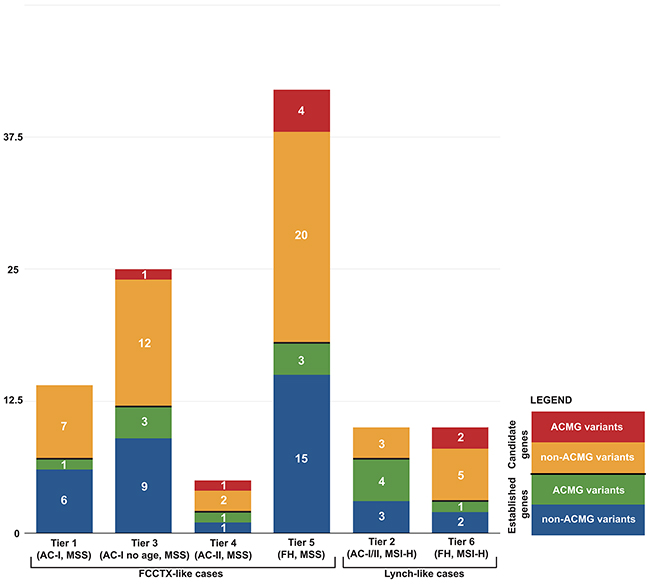
Figure 1: Distribution of 106 germline variants from 40 established and candidate CRC genes by case tiers.
Table 2: Distribution of 99 identified germline variants from 40 CRC genes by case tiers
FCCTX-like cases |
Lynch-like cases |
||||||
|---|---|---|---|---|---|---|---|
Tier |
Tier 1 |
Tier 3 |
Tier 4 |
Tier 5 |
Tier 2 |
Tier 6 |
|
Criteria |
AC-I |
AC-I (no age)** |
AC-II |
CRC<60 |
AC-I/II |
CRC |
|
MSI status |
MSS |
MSS |
MSS |
MSS |
MSI-H |
MSI-H |
|
|
Established CRC genes |
All variants |
7 (5.0%) |
12 (5.9%) |
2 (5.6%) |
18 (3.6%) |
7 (10.9%) |
3 (2.9%) |
ACMG pathogenic variants |
1 (0.7%) |
3 (1.5%) |
1 (2.8%) |
3 (0.6%) |
4 (6.3%) |
1 (1.0%) |
|
Genes with variants |
APC, MLH1, MSH2*, POLE, TGFBR2 |
MLH3*, MSH2, MSH6*, POLD1, POLE |
MUTYH, POLE* |
APC*, AXIN2, MLH1, MLH3, MSH2*, MSH6, MUTYH, POLD1, POLE* |
MLH1*, MSH2*, MSH6* |
MLH1, MSH2, MSH6* |
|
|
Candidate CRC genes |
All variants |
7 (5.0%) |
13 (6.4%) |
3 (8.3%) |
24 (4.8%) |
3 (4.7%) |
7 (6.7%) |
ACMG pathogenic variants |
- |
1 (0.5%) |
1 (2.8%) |
4 (0.8%) |
- |
2 (1.9%) |
|
Genes with variants |
ALPK2, CDH1, LAMA2, MSH3, NOS1, PREX1 |
ALPK2, BLM, LAMA2, MSH3, NOS1, PALB2, POLQ, PTCH1, SH2B3* |
LAMA2, MSH3, SH2B3* |
ALPK2, BAP1, BLM, CDH1, HELQ, LAMA2, LRIG1, MSH3, NOS1*, POLQ*, PREX1, PTCH1, SH2B3* |
HELQ, PALB2, PTCH1 |
CHEK2*, ENG, LAMA2, LRIG1, MSH3, NOS1, POLQ* |
|
All ACMG pathogenic variants |
1/139 (0.7%) |
4/202 (2.0%) |
2/36 (5.6%) |
7/501 (1.4%) |
4/64 (6.3%) |
3/104 (2.9%) |
|
All variants |
14/139 (10.0%) |
25/202 (12.4%) |
5/36 (13.9%) |
42/501 (8.4%) |
10/64 (15.6%) |
10/104 (9.6%) |
|
*Genes with ACMG classified mutations.
**AC-I (no age) describes Tier 3 cases that fulfill all AC-I criteria except “At least 1 of the cancers diagnosed before age 50”.
AC – Amsterdam criteria, MSI – microsatellite instability, MSS – microsatellite stable, MSI-H – highly microsatellite unstable, FDR – first degree relative, SDR – second degree relative, ACMG – American College of Medical Genetics.
A subset of 264 rare variants were selected for Sanger sequencing validation of those variants predicted to be deleterious by either SIFT, PolyPhen2, or MutationTaster, listed as pathogenic in ClinVar, or were stop-gain variants (n=108) in 348 cases from 116 pools. We found no variants in STK11, PTEN, GREM1, and WDR78; and we found only variants predicted to be benign in EPCAM, TP53, BRAP, ENTPD7, MET, and FZD7. Six variants failed in Sanger sequencing for various reasons including inability to design primers in repeat-prone loci, mispriming, and technical error. Two variants were not found on chromatograms, although high depth of targeted sequencing of the locus (84/295 and 196/548 reads) suggests a technical error of using wrong DNA sample for Sanger sequencing. Unfortunately, we did not have enough DNA to repeat sequencing. Considering these two variants as “unconfirmed”, we had sensitivity of >98% (99/101 variants). Thus, Sanger sequencing was successful for 99 variants from 106 pools (Table 3 and Supplementary Table 2). Eight variants were found in two cases and one variant was found in three cases. Out of 99 variants 24 (24%) were novel.
Table 3: The 40 established and candidate CRC susceptibility genes used for targeted sequencing and the distribution of 99 identified variants in 106 cases
Number of patients with identified variants |
||||||||
|---|---|---|---|---|---|---|---|---|
FCCTX-like cases |
Lynch-like cases |
Total |
||||||
Tier 1 |
Tier 3 |
Tier 4 |
Tier 5 |
Tier 2 |
Tier 6 |
|||
Criteria |
AC-I |
AC-I (No age)**** |
AC-II |
CRC<60 |
AC-I/II |
CRC |
||
MSI status |
MSS |
MSS |
MSS |
MSS |
MSI-H |
MSI-H |
||
Established CRC genes |
BMPR1A, EPCAM, PMS2, PTEN, SMAD4, STK11, TP53 |
- |
- |
- |
- |
- |
- |
0 |
AXIN2 |
- |
- |
- |
1 |
- |
- |
1 |
|
TGFBR2 |
1 |
- |
- |
- |
- |
- |
1 |
|
POLD1 |
- |
1 |
- |
1 |
- |
- |
2 |
|
MUTYH |
- |
- |
1 |
2 |
- |
- |
3 |
|
APC |
2 |
- |
- |
2* |
- |
- |
4 |
|
MLH1 |
1 |
- |
- |
1 |
2 |
1 |
5 |
|
POLE |
1 |
1 |
1 |
3 |
- |
- |
6 |
|
MLH3 |
- |
4 |
- |
3 |
- |
- |
7 |
|
MSH6** |
- |
2* |
- |
2 |
2* |
1* |
7 |
|
MSH2 |
2 |
4 |
- |
3 |
3 |
1 |
13 |
|
Candidate CRC genes |
BRAP, CTNNB1, ENTPD7, GREM1, MET, FZD7, WDR78 |
- |
- |
- |
- |
- |
- |
0 |
BAP1 |
- |
- |
- |
1 |
- |
- |
1 |
|
CHEK2 |
- |
- |
- |
- |
- |
1* |
1 |
|
ENG |
- |
- |
- |
- |
- |
1 |
1 |
|
BLM |
- |
1 |
- |
1 |
- |
- |
2 |
|
PREX1 |
1 |
- |
- |
1 |
- |
- |
2 |
|
HELQ |
- |
- |
1 |
1 |
- |
2 |
||
PALB2 |
- |
1 |
- |
- |
1 |
- |
2 |
|
POLQ** |
- |
1 |
- |
1 |
- |
1 |
3 |
|
CDH1 |
1 |
- |
- |
2 |
- |
- |
3 |
|
LRIG1 |
- |
- |
- |
3 |
- |
1 |
4 |
|
PTCH1 |
- |
1 |
- |
2 |
1 |
- |
4 |
|
ALPK2 |
1 |
2 |
- |
2 |
- |
- |
5 |
|
MSH3 |
1 |
2 |
1 |
1 |
- |
1 |
6 |
|
LAMA2 |
1 |
2 |
1* |
1* |
- |
1 |
6 |
|
SH2B3*** |
- |
1 |
1 |
5 |
- |
- |
7 |
|
NOS1** |
2 |
2 |
- |
3 |
- |
1 |
8 |
|
Total |
14 |
25 |
5 |
42 |
10 |
10 |
106 |
|
*Including one nonsense variant, all other variants are missense.
**One variant was found in two patients.
***One variant was found in two patients.
****AC-I (no age) describes Tier 3 cases that fulfill all AC-I criteria except “At least 1 of the cancers diagnosed before age 50”.
Variants within the MMR genes
While 34% (90/264) of the rare variants have never been reported before, almost all (33/36) MMR genes (MLH1, MSH2, MSH6, and PMS2) variants have been reported in dbSNP (Supplementary Table 1). However, only 8 out of 36 MMR variants had annotation in ClinVar database (6 pathogenic and 2 benign), other variants did not have sufficient evidence to determine their effect and were classified as variants of uncertain clinical significance (VUS) (Supplementary Table 1).
We found 10 MMR gene variants (3 MLH1, 4 MSH2, 3 MSH6) in Lynch-like cases (Tiers 2 and 6) and 14 MMR gene variants (2 MLH1, 8 MSH2, 4 MSH6) in FCCTX-like cases (Tiers 1, 3, 4, 5), which correspond to 6% and 1.6% of all cases respectively. Two variants in Lynch-like cases were nonsense, while all variants in FCCTX-like cases were missense. Median age at diagnosis for MMR variant carriers was different between Lynch-like (50.5 years, range 28 – 62) and FCCTX-like cases (57 years, range 36 – 73), although not statistically significant (p=0.144). In Lynch-like cases 50% (5/10) of the variants were pathogenic according to ACMG criteria in comparison to 21% (3/14) of variants in FCCTX-like cases. In 67% of Lynch-like cases MMR proteins had impaired expression in IHC, while all FCCTX-like cases had intact MMR protein expression.
Among five MLH1 variants, we found two known mutations, putatively pathogenic variants p.R100P, p.R226L and a novel p.A125E in three AC-I positive patients with MSI-High CRC diagnosed before 50 years (Tier 2). We found 12 MSH2 variants in 13 patients; 70% of the patients had multiple cancers (Supplementary Figure 1). The MSH2 p.G692V variant, currently considered a VUS in ClinVar, was identified in a patient with metachronous CRC at 29 and 44 years demonstrating MSI-H and loss of MSH2/MSH6 protein expression, providing further support for variant pathogenicity. Six MSH6 variants were found in seven patients. MSH6 p.K295I, p.S541R, and p.T767S were found in cases with CRC diagnosed before 50 years of age. The MSH2 p.H46Q classified as VUS by ClinVar was found in two cases, both with normal protein expression of MSH2, and we identified two cases carrying the nonsense MSH6 mutation p.R911* and a further case carrying the p.R298* mutation. Some variants classified as VUS were found in patients with young age metachronous CRC and/or endometrial cancer (MLH1 p.A125E, MSH2 p.H466R, and MSH6 p.T767S). Of note, over 20% (5/24) of sequenced MMR variants were identified in East Asians and Native American, although 89% of the all cases from this study were white.
Other established CRC genes
We found a novel nonsense APC p.C1410* variant in male with metachronous MSS CRC at 28, 48, and 49 years. APC variants p.T1160K and p.A1358T (both VUS in ClinVar) were found in patients with MSS CRC at 43 and 51 years respectively (Supplementary Figure 2). All APC patients had polyps except p.T1160K carrier. Very rare (MAF<0.0003) p.R594W variant in AXIN2 was found in male with MSI-Low CRC at 45 years. We found one BMPR1A variant p.R406C in two patients with MSI-High CRC below age 53 and below age 47 years old (individual genotypes were not available). Three MUTYH heterozygous variants predicted to be deleterious and highly conserved (all VUS in ClinVar) were found in patients with CRC before age 50 years but no polyps. Of the two POLD1 variants identified (neither in ClinVar), p.Q411H (melanoma at 28 years and MSS CRC at 58 years) and p.Q684H (MSS CRC at 55 years), only the former resided within the exonuclease domain. In POLE exonuclease domain, we found one novel (p.D301G) and one previously reported (p.R231C) variants. Novel (p.N143D) and previously reported (p.H144R) variants were identified close to the exonuclease domain of POLE. All six MLH3 variants (p.A1394T, p.N1147I, p.L1111F, p.D1049N, p.L880V, and p.F168S) were in cases with MSS CRC before age 60 (Supplementary Figures 3 and 4). MLH3 p.F168S was found in two females with CRCs at 55 and 65 years from families that met AC-I without age restriction. TGFBR2 p.G169R variant was found in male with MSS CRC at 31 years from AC-I positive family.
Candidate CRC genes
We found p.R389C variant in BAP1, a well-established tumor suppressor gene, in the case with CRC, squamous cell carcinoma (SCC), and basal cell carcinoma (BCC) at age 66. BLM variants (p.S897C and p.Y1044C) were found in patients with metachronous CRCs, Japanese male (CRC at 48 and 72 years) and white female (CRC at 72 and 79 years). Three CDH1 variants (p.R335Q, p.L630V, and p.A817V) listed as VUS in ClinVar were found in cases with CRC before age 60 and no history of gastric cancer. While CHEK2 is considered a low-risk gene for CRC, we found previously reported truncating CHEK2 mutation p.R95* in a 52 year old woman diagnosed with MLH1/PMS2 deficient MSI-High CRC. In MSH3 only p.A1064T was found in a female with MSI-High CRC at 62 years, other variants (p.D143N, p.L432W, p.I440M, p.V682L, and p.M892V) were in cases with MSS and MSI-Low CRC between 51 and 74 years. However, all MSH3 variants were heterozygous. Two of the tumor suppressor PTCH1 variants (p.R1391W and p.T1106M) were found in cases with MSS CRC before 50 years. ENG exon 12 variant p.T550M carrier had polyps at 53 years and CRC at 70 years.
Variants in LRIG1, PREX1, NOS1, and SH2B3 have been recently found to be associated with CRC in a large GWAS [20]. Variants in LRIG1, a known tumor suppressor downregulated in CRC [21], were identified in cases with CRC before age 50 (adjacent p.V805I, p.R738W, and p.R723C). In PREX1, another known tumor suppressor, we found two variants (p.R1243W and p.V569M) in cases with MSS CRC at 55 and 50 years. Seven NOS1 variants were found in cases with CRC between 36 and 72 years (Supplementary Figures 5 and 6). All five variants in the tumor suppressor SH2B3 were found in cases with MSS CRC. A highly conserved SH2B3 p.E395K was identified in three cases with MSS CRC at 47, 54, and 62 years. Another conserved SH2B3 variant p.N271T was found in a female with CRC and breast IDC at 43 years. Among others, SH2B3 p.I568T was found in a case with synchronous MSS CRCs at 45 years and p.P512T was found in a case with MSS CRC at 50 years.
In POLQ, a DNA polymerase involved in DNA repair with helicase activity, we found two variants predicted to be deleterious: p.P291L (C-terminal helicase domain) in two cases with CRC at 24 and 55 years and p.Y2420C (polymerase domain) in a case with CRC at 50 years. LAMA2 is a methylation target in CRC [22] with mutations predisposing to congenital muscular dystrophy type 1A (MCD1A). Nonsense variants were found in a female with MSS CRC at 73 years and genital malignancy at 68 years (p.Y1334*) and a male with MSI-Low CRC at 53 years and prostate adenocarcinoma at 57 years (p.R2578*). We found novel PALB2 variant p.H1076Y in a Chinese male with MSS CRC at 55 years.
DISCUSSION
In this study, we present a DNA pooling targeted NGS analysis of CRC-related and candidate genes in a large cohort of familial CRC patients Over a third of 264 identified variants were novel. Variants classified as pathogenic by ACMG (Table 2) represent the clinically actionable mutations; however, ACMG classification is partly based on prior publications and recently established CRC genes or candidate CRC genes have insufficient functional data yet. Variants classified as VUS by ACMG criteria include candidates suggestive of being pathogenic, such as POLE p.H144R (MSS CRC at 48 years), BLM p.Y1044C (MSS CRC at 48 and 72 years), and MLH1 p.R100P (MSI-H CRC at 28 years). All VUS were predicted to be deleterious by several bioinformatics tools and the majority are highly conserved, which suggests that these variants may include genuine CRC mutations. ACMG criterion PS4, the prevalence of the variant in affected individuals is significantly increased compared with the prevalence in controls, is particularly problematic when very rare or private mutations are studied.
Despite previous screening of the CRC-affected individuals using dHPLC, Sanger sequencing and MLPA, variants in the core MMR genes (MLH1, MSH2, and MSH6), comprised 22.6% (24/106) of the cases identified to carry one of the rare, predicted pathogenic variants identified in our study. This included carriers who had developed MSI-High CRCs (Lynch-like cases) but also those who developed MSS CRCs from within the FCCTX-like cases. MMR gene mutation carriers who develop MSS CRCs has been reported previously, particularly for MSH6 mutation carriers with missense mutations. We observed MSS CRCs in not only MSH6 missense variant carriers but also for carriers of missense variants in the MSH2 and MLH1 genes. While further validation of the pathogenicity of these MMR gene missense variants is needed, the observation from our study that six MMR gene missense variants classified as VUS by ClinVar were identified in individuals who developed MSS CRCs may warrant further consideration by organizations working towards implementing population-based screening programs for Lynch syndrome that are based on screening CRCs via MMR immunohistochemistry for evidence of tumor mismatch repair deficiency before subsequent germline MMR gene testing. Some of the identified variants are worth separate discussion. Only three germline mutations have been reported so far in AXIN2 [23]. We report another putative pathogenic variant AXIN2 p.R594W found in a patient with CRC at 45 years, however, we did not have information on existence of oligodontia in this carrier, a feature previously associated with AXIN2 germline mutations. All identified MUTYH variants were heterozygous and found in the cases with CRC before 50 without polyps. Prior reports have suggested an increased risk of CRC in MUTYH heterozygotes [24], so it appears that some MUTYH variants are more penetrant than others. While TGFBR2 variant p.G169R was found in a young patient (MSS CRC at 31) from AC-I positive family, it is not conserved and predicted to be benign by PolyPhen2. While two POLD1 and four POLE variants were found in exonuclease domain, POLE p.V2152M and p.R1077C in patients with young onset (<50 years) metachronous CRC were located outside of this domain. It is noteworthy that we found two mucinous adenocarcinomas of colon associated with POLE variants. MLH3 involvement in hereditary CRC is still controversial [25], which may be explained by late age of onset of MLH3 associated CRC. All six MLH3 variants in our study were found in cases with MSS CRC diagnosed between 50 and 65 years, and three of the variants were found in cases that met AC-I criteria without age restriction. As expected, variants in APC, POLE, POLD1 were found in FCCTX-like cases only; however, MLH3 variants were also found in MSS CRC cases only, as well as variants in CDH1, ALPK2, and SH2B3 candidate genes (Table 3). CHEK2 and ENG variants were found in Lynch-like cases only. Several genes had variants in both FCCTX-like and Lynch-like cases including all MMR genes (MLH1, MSH2, MSH6, and MSH3), HELQ, POLQ, LRIG1, PTCH1, LAMA2, and NOS1. Some MMR variants found in FCCTX-like MSS cases may still be pathogenic, since MMR mutations have been reported in MSS CRC patients, especially those with MSH6 mutations. However, in some cases better characterization of the cases is needed. For example MSH6 p.R911* mutation was found in cases 61 and 62 (Supplementary Table 2); while case 61 had MSI-High CRC, case 62 did not have MSI tested and misclassified as MSS ending up among Tier 3 cases.
BAP1 is a well-established tumor suppressor gene [26], and its downregulation is associated with decreased CRC survival [27]. A conserved variant BAP1 p.R389C was found in a white male with CRC, squamous cell carcinoma, and basal cell carcinoma at age 66 years. This allele, detected by ExAC in only one European, is located in the same C-terminal hydrolase domain that harbors a mutation predisposing to melanoma, thyroid cancer, and mesothelioma [28–30]. Our finding raises a question regarding the role of BAP1 germline mutations predisposing to squamous and basal cell carcinomas. It is noteworthy that two LAMA2 variants p.R2578* and p.I136M were detected in cases with both colon and prostate adenocarcinomas. CDH1 is known to be associated with hereditary diffuse gastric cancer (HDGC) and other cancer types; however, none of the CDH1 variant carriers had history of gastric cancer. CHEK2 mutation p.R95* has been described in breast cancer patients [31]; here, we report it in a patient with CRC at 52. ENG was found to cause Familial Juvenile Polyposis (FJP) with mutations in exons 11 and 12 reported in patients with late onset of CRC (60 and 68 years) and early age polyps (3 and 5 years) [32]. We found ENG exon 12 variant p.T550M in a case with polyps at 53 years and MSI-High CRC at 70 years. It is possible that ENG mutations are more common because early age polyps may remain undetected until the age colonoscopy surveillance starts. Our findings of variants in LRIG1, PREX1, NOS1, and SH2B3 further implicate these genes in hereditary CRC following the recent large GWAS [20]. However, additional segregation and functional studies are needed to confirm these results.
BLM is a known tumor suppressor associated with higher CRC risk in heterozygotes, in addition to Bloom syndrome in homozygotes [33]. Recent study showed that heterozygote BLM mutations are associated with early onset CRC [34]. We found highly conserved BLM variants p.Y1044C and p.S897C (C-terminal helicase domain) in patients with metachronous CRC. Variants in the C-terminal helicase domain were found in both BLM and POLQ. These findings suggest that C-terminal helicase domain mutations in POLQ and BLM may be involved in predisposition to hereditary CRC. It is noteworthy that identified HELQ variants were found outside of the helicase domain in carriers of candidate mutations in other genes.
DNA pooling has been previously used for genetic and genomic analysis including attempts to use DNA pooling for GWAS with mixed results, because pipetting errors interfered with estimated allele frequency. In NGS analysis pooling is frequently used as multiplexing with barcoded DNA samples pooled together to reduce the cost of sequencing. Pooling of non-barcoded samples is the further step to make sequencing even less expensive. It has been demonstrated that rare variants can be effectively identified in large populations using pooled NGS [35, 36]. Several guidelines and optimization algorithms for the analysis of rare variants in pooled NGS samples have been reported [37–39]. Previously, in our analysis of pooled exome sequencing, we showed that high depth of sequencing is important for identification of rare variants [40]. Therefore, our strategy is based on pooling DNA samples prior to preparation of the sequencing libraries followed by high depth sequencing and genotype validation, which makes our strategy more accurate and cost-efficient. A key factor in our strategy is that pooled sequencing is ideal for detection of very rare variants where the mere presence of the variant, and not its allele frequency, is important.
There are a number of limitations of the pooled sequencing strategy for analysis of very rare variants. Possibility that variants/mutations in genes not tested in this study account for the CRC in some of these cases cannot be excluded. For example, other candidate CRC genes including RPS20 [41], SEMA4A [42] and NTHL1 [14] have been published recently. Use of a specific percent of minor allele reads representing one heterozygote in a pool may lead to insufficient sensitivity of the analysis and some valid variants may be excluded. On the other hand, we used relatively relaxed frequency criteria (from 10% to 50% in a pool of three DNAs) to increase sensitivity and still >98% of the variants chosen for Sanger sequencing were validated. In addition, a number of genes, such as EPCAM and GREM1, have been reported to have copy number variations (CNVs) that have not been investigated in this study. The identified variants were predicted to be deleterious by at least one commonly used in silico variant effect prediction tool, however, it has been shown that in silico tools and their algorithms for missense variant effect prediction are only 65-80% accurate when examining known disease causing missense variants, therefore, further studies are needed before assigning pathogenicity to the missense variants identified in this study.
In conclusion, we performed a large targeted sequencing study using a DNA pooling strategy on 1046 CRC-affected cases selected for a positive family history of CRC and inclusive of both MSS and MSI-High subgroups of CRC. Our variant filtering criteria identified rare, predicted pathogenic variants in 106 cases representing 10% of all the cases tested. The cases with MMR gene variants comprised almost a quarter of the identified carriers, with other prominent genes identified in the established CRC genes group (MLH3 and POLE) and the candidate CRC genes group (NOS1, SH2B3, LAMA2 and MSH3) requiring further validation studies at both the gene and variant level. The DNA pooling NGS strategy applied in this study for identifying rare variants in hereditary CRC was a cost-effective approach for this large case-control targeted sequencing study and could be applied to other cancer types or complex diseases with a hereditary component, and may further facilitate studies aimed at identifying rare genetic risk factors in populations that are underrepresented in resequencing projects, such as Middle East or Slavic ethnicities. The results from our study support the concept that familial CRC is highly heterogeneous with regards to underlying genetic etiology with additional high-risk genes yet to be identified. Additional, large case-control studies supported by studies on functional effect and variant segregation are needed to generate the evidence needed to translate gene and rare variant discovery into improvements in clinical practice and actionability.
MATERIALS AND METHODS
Study population
The selection of CRC cases for this study was primarily based on CRC-affected individuals with a family history of CRC such as those who would be referred to Family Cancer Clinic and/or for genetic testing for hereditary CRC syndrome including individuals with both MSS and microsatellite unstable (MSI-H) tumors in order to provide findings of broad clinical relevance. We selected 1,046 familial CRC cases and 1,006 unrelated healthy controls from the Colon Cancer Family Registry Cohort (CCFR) [43]. Germline mutation testing for MMR gene mutations had been performed previously using different methods, including denaturing HPLC (dHPLC) screening prior to Sanger sequencing and MLPA during Phase II testing (2001-2006) while Phase III testing (2007-2012) involved Sanger sequencing and MLPA only; MUTYH gene testing involved genotyping and Sanger sequencing [43–45]. CRC cases were selected from six prioritized groups based on family structure of affected relatives, age at CRC diagnosis and tumor MMR status (Table 1). Therefore, the sample included FCCTX-like cases (MSS CRC patients from Tiers 1, 3, 4, and 5) and Lynch-like cases (MSI-H CRC patients from Tiers 2 and 6): Tier 1 included CRC patients with MSS tumors fulfilling Amsterdam criteria I (AC-I), also known as Familial Colorectal Cancer Type X [11], Tier 2 included AC-I or AC-II CRC patients with MSI-High tumors and no known MMR gene mutation or methylation of the MLH1 gene promoter (Lynch-like), Tier 3 included CRC patients with MSS tumors from AC-I positive families without fulfilling the criteria of age (CRC <50 years), Tier 4 included AC-II CRC patients with MSS tumors, Tier 5 included CRC patients with MSS tumors from families with a proband <60 years old at CRC diagnosis and had ≥1 FDR or SDR with CRC, Tier 6 included CRC patients with an MSI-High tumor and no known MMR gene mutation or methylation of the MLH1 gene promoter (Lynch-like) from families where proband had ≥1 FDR or SDR with CRC (although not fulfilling the AC-I or AC-II criteria). Controls were spouses or unrelated healthy individuals without cancer and selected to be older than cases at time of study (mean age 61 years) to account for variable CRC penetrance. Male to female ratio was 1:1 in cases and 1:1.5 in controls.
Targeted sequencing and bioinformatics analysis
We performed targeted sequencing of pooled DNA samples as a cost-efficient way to screen a large case-control sample set. Our strategy included four steps: 1) targeted sequencing of pooled cases and controls, 2) identification of case-only variants, 3) Sanger sequencing of case-only variants to identify individual genotypes and validate targeted sequencing (Figure 2). Blood-derived DNA from 1,046 cases and 1,006 controls were pooled into 480 pools in equimolar proportions (~3 cases/pool and ~8 controls/pool) for targeted sequencing of 40 established and candidate CRC genes (Table 2). Each sample was sequenced once. Genes were selected based on the following criteria: 1) established hereditary CRC genes (n=17), or 2) novel candidate CRC susceptibility genes (n=23) identified from the literature and those identified from whole exome sequencing study of familial CRC cases lead by the first author (Raskin et al., unpublished data). Custom libraries for each DNA pool were prepared using Qiagen GeneRead DNAseq Customized Targeted Panel covering exons and exon/intron boundaries and sequenced on the Illumina HiSeq 2500 to a mean depth of >1000X per case pool (>330X per case in each pool) and >700X per control pool (~90X per control in each pool) at Vanderbilt Technologies for Advanced Genomics (VANTAGE). See Supplementary Methods and Supplementary Table 3 for more details.
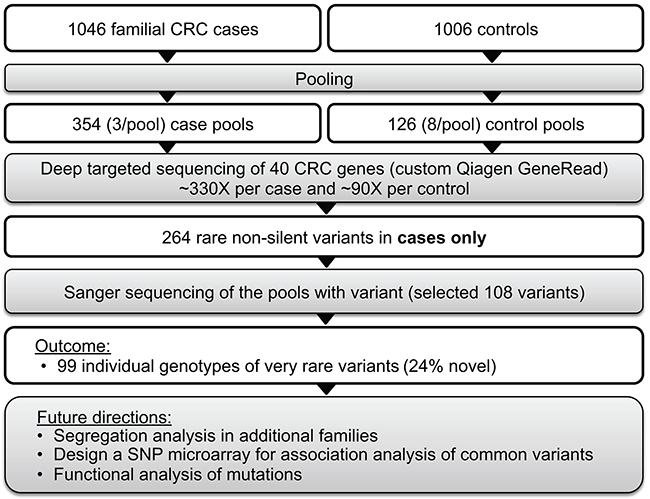
Figure 2: Targeted sequencing of pooled samples for identification of rare variants of large effect.
Analysis of the raw sequencing data was performed at Vanderbilt Technologies for Advanced Genomics Analysis and Research Design (VANGARD) including a multi-stage quality control protocol developed previously [46, 47]. No quality concerns were observed. Alignments were performed using BWA against human genome reference hg19. We marked duplicates using Picard, then performed local realignment and local recalibration using the Genome Analysis Toolkit (GATK). Single nucleotide variants (SNVs) and indels were inferred using GATK’s Unified Genotyper. Results were further filtered based on GATK’s best practice. Annotations of SNV and indel were performed using ANNOVAR. Additional annotations were obtained through wANNOVAR [48], and Oncotator [49]. Variant reference databases including 1000 Genomes and ExAC were used as a source of variant allele frequency in addition to the controls tested. We selected all variants that had ≥2% reads with alternative allele to exclude false positives. Fractions of the alternative allele were calculated per pool for further analysis (alternative allele reads/total reads). A minor allele percentage in a case pool reads between 10% and 50% and ≥50 reads were used as a threshold to select variants. Likely deleterious variants were defined as variants (nonsense, frame-shift, splice-site variants) likely to result in protein truncation or disrupt a consensus splice site (i.e. +/- 1, 2) and non-synonymous variants predicted to be pathogenic according to MutationTaster, PolyPhen-2, and SIFT in silico tools from ANNOVAR. Rare likely deleterious variants identified in only the CRC cases from targeted sequencing were tested by Sanger sequencing to exclude a false positive variant and to confirm which CRC case in the DNA pool was the carrier (Sanger sequencing primers available on request).
American College of Medical Genetics and Genomics (ACMG) recommended an updated standards and guidelines for interpretation of sequence variants as benign or pathogenic based on the following types of data: population, computational and predictive, functional, segregation, de novo status, and presence in other databases [19]. ACMG guidelines were used for interpretation of the clinical significance of the variants.
Abbreviations
CRC – colorectal cancer, NGS – next-generation sequencing, MSI – microsatellite instable, MSS – microsatellite stable, MMR – mismatch repair, FCCTX – familial colorectal cancer type X, CCFR - Colon Cancer Family Registry, ACMG – American College of Medical Genetics.
Author contributions
Leon Raskin (study concept and design, data acquisition, data analysis and interpretation, drafting of the manuscript, obtained funding, study supervision); Yan Guo (data analysis); Liping Du (statistical analysis); Mark Clendenning (data acquisition); Christophe Rosty (data acquisition); CCFR (data acquisition, critical revision of the manuscript, obtained funding); Noralane Lindor (data acquisition, critical revision of the manuscript); Stephen Gruber (study design); Daniel Buchanan (data acquisition, data interpretation, drafting of the manuscript).
ACKNOWLEDGMENTS
The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute any SEER program or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government, SEER or the CCFR.
CONFLICTS OF INTEREST
The authors have no conflict of interest to declare.
FUNDING
This work was supported by the NIH NCI Pathway to Independence Award K99/R00CA158141. This work was also supported in part by grant UM1 CA167551 from the National Cancer Institute and through cooperative agreements with the following Colon CFR centers: Australasian Colorectal Cancer Family Registry (U01 CA074778 and U01/U24 CA097735), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (U01/U24 CA074800), Ontario Familial Colorectal Cancer Registry (U01/U24 CA074783), Seattle Colorectal Cancer Family Registry (U01/U24 CA074794), University of Hawaii Colorectal Cancer Family Registry (U01/U24 CA074806), USC Consortium Colorectal Cancer Family Registry U01/U24 CA074799), with additional support through grant R01 CA104132 (to L LeMarchand). Additional support was provided from the Surveillance, Epidemiology and End Results (SEER) Program of the National Cancer Institute to Fred Hutchinson Cancer Research Center (Control Nos. N01-CN-67009 and N01-PC-35142, and Contract No. HHSN2612013000121), the Hawai‘i Department of Health (Control Nos. N01-PC-67001 and N01-PC-35137, and Contract No. HHSN26120100037C, and the California Department of Public Health (contracts HHSN261201000140C awarded to the Cancer Prevention Institute of California and HHSN261201000035C awarded to the University of Southern California.
REFERENCES
1. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343:78-85.
2. Aaltonen L, Johns L, Jarvinen H, Mecklin JP, Houlston R. Explaining the familial colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res. 2007; 13:356-361.
3. Vasen HF. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J Clin Oncol. 2000; 18:81S-92S.
4. Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001; 10:721-733.
5. Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C -->T:A mutations in colorectal tumors. Nat Genet. 2002; 30:227.
6. Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, Krush AJ, Yardley JH, Luk GD. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987; 316:1511-1514.
7. Haidle JL, Howe JR. (1993). Juvenile Polyposis Syndrome. In: Pagon RA, Bird TD, Dolan CR and Stephens K, eds. Gene Reviews. (Seattle, WA: University of Washington, Seattle).
8. Mallory SB. Cowden syndrome (multiple hamartoma syndrome). Dermatol Clin. 1995; 13:27-31.
9. Dunlop MG, Tenesa A, Farrington SM, Ballereau S, Brewster DH, Koessler T, Pharoah P, Schafmayer C, Hampe J, Volzke H, Chang-Claude J, Hoffmeister M, Brenner H, et al. Cumulative impact of common genetic variants and other risk factors on colorectal cancer risk in 42 103 individuals. Gut. 2012.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003; 348:919-932.
11. Lindor NM, Rabe K, Petersen GM, Haile R, Casey G, Baron J, Gallinger S, Bapat B, Aronson M, Hopper J, Jass J, LeMarchand L, Grove J, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA. 2005; 293:1979-1985.
12. Jenkins MA, Croitoru ME, Monga N, Cleary SP, Cotterchio M, Hopper JL, Gallinger S. Risk of colorectal cancer in monoallelic and biallelic carriers of MYH mutations: a population-based case-family study. Cancer Epidemiol Biomarkers Prev. 2006; 15:312-314.
13. Sokolenko AP, Suspitsin EN, Kuligina E, Bizin IV, Frishman D, Imyanitov EN. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015; 369:274-288.
14. Weren RD, Ligtenberg MJ, Kets CM, de Voer RM, Verwiel ET, Spruijt L, van Zelst-Stams WA, Jongmans MC, Gilissen C, Hehir-Kwa JY, Hoischen A, Shendure J, Boyle EA, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015; 47:668-671.
15. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013; 45:136-144.
16. Christensen KD, Dukhovny D, Siebert U, Green RC. Assessing the costs and cost-effectiveness of genomic sequencing. J Pers Med. 2015; 5:470-486.
17. Mancuso N, Rohland N, Rand KA, Tandon A, Allen A, Quinque D, Mallick S, Li H, Stram A, Sheng X, Kote-Jarai Z, Easton DF, Eeles RA, et al. The contribution of rare variation to prostate cancer heritability. Nat Genet. 2016; 48:30-35.
18. Rand KA, Rohland N, Tandon A, Stram A, Sheng X, Do R, Pasaniuc B, Allen A, Quinque D, Mallick S, Le Marchand L, Kaggwa S, Lubwama A, et al. Whole-exome sequencing of over 4100 men of African ancestry and prostate cancer risk. Hum Mol Genet. 2016; 25:371-381.
19. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405-424.
20. Schumacher FR, Schmit SL, Jiao S, Edlund CK, Wang H, Zhang B, Hsu L, Huang SC, Fischer CP, Harju JF, Idos GE, Lejbkowicz F, Manion FJ, et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun. 2015; 6:7138.
21. Wang S, Li N, Yousefi M, Nakauka-Ddamba A, Li F, Parada K, Rao S, Minuesa G, Katz Y, Gregory BD, Kharas MG, Yu Z, Lengner CJ. Transformation of the intestinal epithelium by the MSI2 RNA-binding protein. Nat Commun. 2015; 6:6517.
22. Lee S, Oh T, Chung H, Rha S, Kim C, Moon Y, Hoehn BD, Jeong D, Lee S, Kim N, Park C, Yoo M, An S. Identification of GABRA1 and LAMA2 as new DNA methylation markers in colorectal cancer. Int J Oncol. 2012; 40:889-898.
23. Mazzoni SM, Fearon ER. AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 2014; 355:1-8.
24. Jones N, Vogt S, Nielsen M, Christian D, Wark PA, Eccles D, Edwards E, Evans DG, Maher ER, Vasen HF, Hes FJ, Aretz S, Sampson JR. Increased colorectal cancer incidence in obligate carriers of heterozygous mutations in MUTYH. Gastroenterology. 2009; 137:489-494, 494e1; quiz 725-486.
25. Wu Y, Berends MJ, Sijmons RH, Mensink RG, Verlind E, Kooi KA, van der Sluis T, Kempinga C, van dDer Zee AG, Hollema H, Buys CH, Kleibeuker JH, Hofstra RM. A role for MLH3 in hereditary nonpolyposis colorectal cancer. Nat Genet. 2001; 29:137-138.
26. Kadariya Y, Cheung M, Xu J, Pei J, Sementino E, Menges CW, Cai KQ, Rauscher FJ, Klein-Szanto AJ, Testa JR. Bap1 is a bona fide tumor suppressor: genetic evidence from mouse models carrying heterozygous germline Bap1 mutations. Cancer Res. 2016.
27. Tang J, Xi S, Wang G, Wang B, Yan S, Wu Y, Sang Y, Wu W, Zhang R, Kang T. Prognostic significance of BRCA1-associated protein 1 in colorectal cancer. Med Oncol. 2013; 30:541.
28. McDonnell KJ, Gallanis GT, Heller KA, Melas M, Idos GE, Culver JO, Martin SE, Peng DH, Gruber SB. A novel BAP1 mutation is associated with melanocytic neoplasms and thyroid cancer. Cancer Genet. 2016; 209:75-81.
29. Ohar JA, Cheung M, Talarchek J, Howard SE, Howard TD, Hesdorffer M, Peng H, Rauscher FJ, Testa JR. Germline BAP1 mutational landscape of asbestos-exposed malignant mesothelioma patients with family history of cancer. Cancer Res. 2016; 76:206-215.
30. Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011; 43:1018-1021.
31. Chrisanthar R, Knappskog S, Lokkevik E, Anker G, Ostenstad B, Lundgren S, Berge EO, Risberg T, Mjaaland I, Maehle L, Engebretsen LF, Lillehaug JR, Lonning PE. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One. 2008; 3:e3062.
32. Sweet K, Willis J, Zhou XP, Gallione C, Sawada T, Alhopuro P, Khoo SK, Patocs A, Martin C, Bridgeman S, Heinz J, Pilarski R, Lehtonen R, et al. Molecular classification of patients with unexplained hamartomatous and hyperplastic polyposis. JAMA. 2005; 294:2465-2473.
33. Gruber SB, Ellis NA, Scott KK, Almog R, Kolachana P, Bonner JD, Kirchhoff T, Tomsho LP, Nafa K, Pierce H, Low M, Satagopan J, Rennert H, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002; 297:2013.
34. de Voer RM, Hahn MM, Mensenkamp AR, Hoischen A, Gilissen C, Henkes A, Spruijt L, van Zelst-Stams WA, Kets CM, Verwiel ET, Nagtegaal ID, Schackert HK, van Kessel AG, et al. Deleterious germline BLM mutations and the risk for early-onset colorectal cancer. Sci Rep. 2015; 5:14060.
35. Futschik A, Schlotterer C. The next generation of molecular markers from massively parallel sequencing of pooled DNA samples. Genetics. 2010; 186:207-218.
36. Jin SC, Pastor P, Cooper B, Cervantes S, Benitez BA, Razquin C, Goate A, Cruchaga C, Ibero-American Alzheimer Disease Genetics Group Researchers. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimers Res Ther. 2012; 4:34.
37. Anand S, Mangano E, Barizzone N, Bordoni R, Sorosina M, Clarelli F, Corrado L, Martinelli Boneschi F, D’Alfonso S, De Bellis G. Next generation sequencing of pooled samples: guideline for variants’ filtering. Sci Rep. 2016; 6:33735.
38. Evangelista D, Zuccaro A, Lancinskas A, Zilinskas J, Guarracino MR. A web-oriented software for the optimization of pooled experiments in NGS for detection of rare mutations. BMC Res Notes. 2016; 9:111.
39. Zilinskas J, Lancinskas A, Guarracino MR. Application of multi-objective optimization to pooled experiments of next generation sequencing for detection of rare mutations. PLoS One. 2014; 9:e104992.
40. Guo Y, Samuels DC, Li J, Clark T, Li CI, Shyr Y. Evaluation of allele frequency estimation using pooled sequencing data simulation. ScientificWorldJournal. 2013; 2013:895496.
41. Nieminen TT, O’Donohue MF, Wu Y, Lohi H, Scherer SW, Paterson AD, Ellonen P, Abdel-Rahman WM, Valo S, Mecklin JP, Jarvinen HJ, Gleizes PE, Peltomaki P. Germline mutation of RPS20, encoding a ribosomal protein, causes predisposition to hereditary nonpolyposis colorectal carcinoma without DNA mismatch repair deficiency. Gastroenterology. 2014; 147:595-598.e595.
42. Schulz E, Klampfl P, Holzapfel S, Janecke AR, Ulz P, Renner W, Kashofer K, Nojima S, Leitner A, Zebisch A, Wolfler A, Hofer S, Gerger A, et al. Germline variants in the SEMA4A gene predispose to familial colorectal cancer type X. Nat Commun. 2014; 5:5191.
43. Newcomb PA, Baron J, Cotterchio M, Gallinger S, Grove J, Haile R, Hall D, Hopper JL, Jass J, Le Marchand L, Limburg P, Lindor N, Potter JD, et al. Colon Cancer Family Registry: an international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev. 2007; 16:2331-2343.
44. Rosty C, Clendenning M, Walsh MD, Eriksen SV, Southey MC, Winship IM, Macrae FA, Boussioutas A, Poplawski NK, Parry S, Arnold J, Young JP, Casey G, et al. Germline mutations in PMS2 and MLH1 in individuals with solitary loss of PMS2 expression in colorectal carcinomas from the Colon Cancer Family Registry Cohort. BMJ Open. 2016; 6:e010293.
45. Win AK, Dowty JG, Cleary SP, Kim H, Buchanan DD, Young JP, Clendenning M, Rosty C, MacInnis RJ, Giles GG, Boussioutas A, Macrae FA, Parry S, et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology. 2014; 146:1208-1211.e1201-1205.
46. Guo Y, Zhao S, Sheng Q, Ye F, Li J, Lehmann B, Pietenpol J, Samuels DC, Shyr Y. Multi-perspective quality control of Illumina exome sequencing data using QC3. Genomics. 2014; 103:323-328.
47. Guo Y, Ye F, Sheng Q, Clark T, Samuels DC. Three-stage quality control strategies for DNA re-sequencing data. Brief Bioinform. 2013.
48. Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012; 49:433-436.
49. Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015; 36:E2423-2429.