
Introduction
Central Nervous System (CNS) tumors are the second most common cancers affecting children and adolescents after leukemia. Approximately half of these CNS tumors are childhood gliomas. Unlike in adult patients, the majority of childhood gliomas are low-grade gliomas (LGG), whereas high-grade gliomas (HGG) account for approximately 8-12% of all primary CNS tumors in children [1], with the most frequent types being anaplastic astrocytoma (WHO grade III) and glioblastoma (WHO grade IV) [2].
Treatment strategies have been similar for children and adolescents with HGG compared to adults. However, outcomes remain dismal with long-term survival rates around 10% [2, 3]. Recent knowledge in the biology, molecular and genetic characteristics of these tumors implies that pediatric HGG comprise one or most probably several distinct entities that might be treated differently based on their genetic and epigenetic features [4].
There are only scarce data available about HGG in children younger than three years of age due to their very low incidence in this age group. Reported data suggest that very young children with HGG might have a better prognosis compared to older children or adults [3, 5-7]. This might be – at least partly - explained by differences in molecular and biologic characteristics [8].
Here, we aim to review the current knowledge of very young children with HGG (here –in agreement with most pediatric neuro-oncology working groups- defined as patients younger than three years) regarding their epidemiology, genetic and epigenetic characteristics as well as their clinical management and treatment outcomes.
Epidemiology
Very young children differ from older children and adolescents regarding incidence and location of different histological entities of CNS tumors [1, 9]. Around 10% of primary CNS tumors occur during the first year of life with almost half of them during the first six months. About 18% of these tumors appear before the age of two years [10].
In a recent publication from the Central Brain Tumor Registry of the United States (CBTRUS), the highest overall incidence of childhood CNS tumors was in infants (below one year of age; 6.22 per 100,000 children) followed by ages 1 – 4 years (5.53 per 100,000 children) [1]. Earlier data from Surveillance, Epidemiology, and End Results (SEER) data-base for the period between 1973 and 2006 report on a much lower overall incidence of CNS tumors in infants (3.1 per 100,000) [11].
In a meta-analysis of data from 16 studies, the most frequent histological entity in pediatric CNS tumors was astrocytoma (37.6%) [12]. According to data from 60 countries published by the International Agency for Research on Cancer (IARC), annual incidence rate for patients with astrocytoma ranged between 0.7 – 2.2 per 100,000 children [13].
Regarding infant CNS tumors, Bishop et al. examined the SEER registry and found that gliomas had the highest incidence among infants with a rate of 1.38 per 100,000 [11]. Similarly, CBTRUS reports gliomas as the most common histological entity in infants (37.2% of tumors) and in children aged 1 – 4 years (58.1% of tumors). The majority of these gliomas were LGG (62.5% and 61.2%, respectively) and the highest incidence for HGG (26%) was observed in children aged 5 – 9 years [1]. In contrast, a German case series from 1984 to 2000 reported HGG as the most frequently occurring in very young children (17.7%) and in adolescents aged 15 – 17 years (21.7%) [12].
The annual incidence rates for CNS tumors in children as well as the frequency of different histologic diagnoses are largely variable among individual studies [14, 15]. In the pediatric oncology group (POG) experience, HGG were the 4th most common malignant brain tumor in very young children following medulloblastoma, ependymoma and primitive neuro-ectodermal tumors [16]. Interestingly, two thirds of these HGG occurred in infants younger than 6 months at diagnosis [16]. Similarly, the children’s cancer group (CCG) experience of HGG shows same tendency for clustering early in life with 21 out of 32 patients aged less than one year at diagnosis [6]. On the other hand, the French BBSFOP and United Kingdom UKCCSG 9204 studies didn’t show similar trend, where patients younger than 1 year comprised only 7 out of 21 patients and 6 out of 19 patients, respectively [5, 17]. In addition to true ethnic, geographical and socio-economic differences, this variability between reports is likely due to differences in patients’ numbers, variable age cut-offs, as well as the pathological classification system followed in each study. Table 1 summarizes the incidence of HGG in childhood in different age groups as reported by several studies and registries. However, the incidence of individual diagnoses might change in the future, as the pathological classification is continuously edited and modified to be complemented by novel molecular tests and characteristics.
HGG in very young children typically occur in hemispheric locations and less frequently in posterior fossa or other midline locations [18, 19]. This is evident in the POG, CCG and UKCCSG 9204 studies where 83%, > 50% and 90% of HGG were located in cerebral hemispheres, respectively [6, 16, 17].
Table 1: Incidence of childhood HGG in different age groups as reported by selected studies
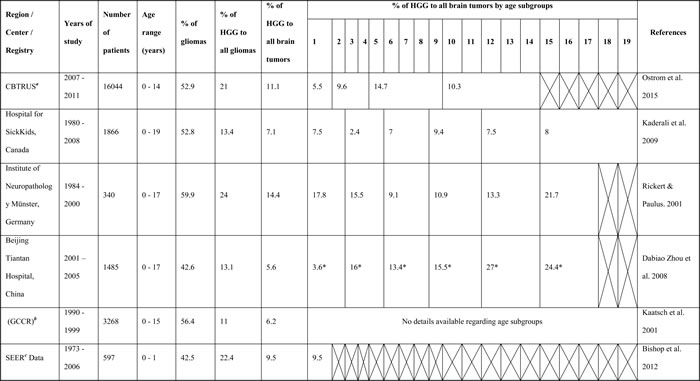
a CBTRUS: Central Brain Tumor Registry of the United States
* % of all astrocytic tumors; no data for HGG tumors alone.
b GCCR: German Childhood Cancer Registry
c SEER: Surveillance, Epidemiology, and End Results HGG: high-grade glioma
Pathological Diagnostics
Histology-based diagnosis is still the most reliable, fastest and cost-effective diagnostic approach in brain tumor classification, even though the most recent 2016 WHO classification of Tumors of the Central Nervous Systems[20] introduces multi-layered, i.e. histological and molecularly based definitions of brain tumors entities. In conventional histological and immunohistochemical staining, HGG in very young children do not show differences compared to pediatric or adult HGG (Figure 1). Routine diagnostic tools of formalin-fixed and paraffin-embedded tumor samples of diffusely infiltrating HGG should at least include a hematoxylin and eosin (H&E) staining and a reticulin silver staining as well as immunohistochemical examination with antibodies against glial fibrillary acidic protein (GFAP), p53 and Ki67 (Mib-1). Map2, Olig-2 and ATRX can be of use, while IDH-1 (R132H) immunohistochemistry does not play a role in diagnosis of HGG in very young children. Diffuse midline gliomas harboring histone H3 K27M mutations correspond to WHO grade IV, even though typical high-grade features (e.g. microvascular proliferation, necrosis, increased proliferative and mitotic activity) may be absent. Glial tumors arising in midline CNS structures should therefore always be tested for histone H3 mutations, possible also by a mutation-specific H3 (K27M) antibody. For HGG entities of much lower incidences (pediatric type anaplastic oligodendroglioma (WHO grade III), anaplastic (pilocytic) astrocytoma (analogue WHO grade III), pleomorphic xanthoastrocytoma with anaplastic features (WHO grade III) and anaplastic ganglioglioma (WHO grade III), additional immunohistochemical and molecular examination is often necessary. As a putative diagnostic pitfall especially in infant cases, the presence of desmoplastic infantile astrocytoma and ganglioglioma (DIA/DIG), corresponding to WHO grade I, should be taken into consideration, since these entities can present as large tumor masses with sometimes a poorly differentiated tumor cell component, possibly mimicking high-grade features.
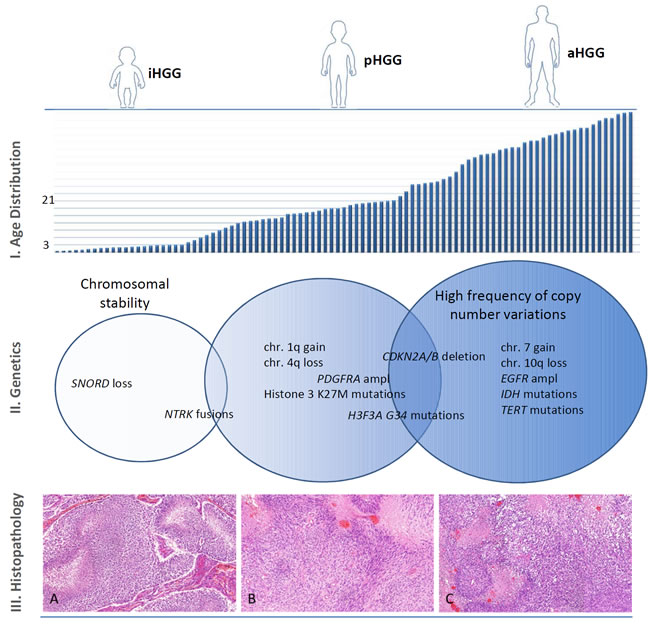
Figure 1: Age distribution, genetic abnormalities and histopathological findings of HGG. Diagrammatic illustration demonstrating the age distribution, genetic abnormalities and histopathologic findings of HGG in very young children, older children and adults. I. Age Distribution: Bar graph showing increasing incidence of high-grade glioma depending on age at diagnosis. Of notice, the age cutoff definition for HGG in very young children varies between different groups (from 2 - 5 years), but most pediatric neuro-oncology working groups define it at three years of age. Similarly, definition of age cutoff for pediatric HGG patients varies between different groups and ranges from 16 to 21 years. II. Genetics: HGG in very young children tend to display more stable genome and few identifiable mutations, with SNORD loss and NTRK fusion genes being the most common molecular abnormalities. HGG in older children are characterized by increasing frequency of copy number aberrations, like PDGFRA amplification and CDKN2A/CDKN2B deletions and/or histone 3 (K27M or G34R/V) mutations. Adult HGG frequently display copy number variations like gain of chromosome 7 and loss of chromosome 10q as well as EGFR amplification, IDH and TERT mutations. III. Histopathology: H&E stained tumor specimens of glioblastoma in a. a very young child, b. an older child and c. an adult patient. All three examples share histological high-grade features with cell-rich tumor areas, microvascular proliferation and pseudo-palisading necrosis as well as a high mitotic activity. A histology-based discrimination of these tumor samples allowing conclusion regarding the patient’s age is impossible.
Genetic and Epigenetic Features
Recent studies on the molecular features of pediatric HGG highlighting different genetic and epigenetic alterations strongly suggest a clear distinction from their adult counterparts [21-23] and they defined different subgroups in terms of cell of origin, clinical characteristics and prognosis [4]. Due to their rarity, HGG in very young children are still understudied and molecular insights are limited.
In this section we discuss the few available data on molecular characterization of HGG in very young children within the context of main molecular findings in pediatric HGG (Figure 1).
Structural abnormalities: Copy-number alterations and focal aberrations
Pediatric HGG exhibit broad chromosomal abnormalities at a much lower frequency compared to adult HGG, with chromosome 1q gains being the most common aberration while 7q gain and 10q loss are less commonly encountered [24,25].
Paugh et al. observed recurrent 1q gains in 29% of cases and focal platelet-derived growth factor receptor α (PDGFRA) amplifications in 12% of tumors in their report on 78 pediatric HGGs [25]. Other studies reported higher frequency of PDGFRA amplifications up to 39% of pediatric HGG [26, 27]. Unlike adult HGG, PDGFRA amplifications in pediatric HGG had no prognostic significance [27]. However, a recent study by Koschmann et al. reported worse outcome in correlation with PDGFRA mutations and not amplifications [28]. Other amplifications such as epidermal growth factor receptor (EGFR), MYC, MYCN and MDM4 are encountered in 1–4 % of tumors. The most frequent copy-number losses are 10q (38%), 13q (34%) and 14q (29%) [25].
In a recent study investigating the molecular features of 35 HGG from very young children, cytogenetic alterations were in general less frequent compared with HGG from older pediatric and adult patients but included some alterations which are commonly seen in HGG (gains of 1q and 7q or losses of 10q, 13q and 14q) [8]. Gain of 1q was seen in 22.7%, loss of 6q in 18.2%, and 10q loss in 9.1% of cases. Losses of 13q and 14q as well as focal amplifications of PDGFRA or EGFR were absent, while cyclin dependent kinase inhibitor -2A (CDKN2A) deletions were seen in two cases. Genomic alterations were more frequently seen in children older than nine months, which might imply an association between older age at diagnosis and accumulation of genetic alterations [8]. These findings are supported by results of Paugh et al. who included 11 very young children in their study cohort of pediatric HGG. In those very young children, 1q gain was not detected and 10q loss was observed in one case [25].
Loss of SNORD genes coding for small nucleolar RNAs (snoRNAs) has been previously reported in several types of cancer [29-32]. Recently, it was detected in 27.3% of HGG in very young children [8]. This finding raises some questions regarding the role of snoRNAs in gliomagenesis in HGG in young children and their potential uses as diagnostic markers or as novel therapeutic targets.
One of the common findings in pediatric HGG is the expression of fusion genes, yet with most of them being non-recurrent fusions. A study by Wu et al. identified recurrent fusion genes involving the kinase domain of the neurotrophin tyrosine receptor kinase gene (NTRK) in 4% of brainstem and 10% of non-brainstem HGG, being observed in 40% (4/10) of HGG in very young children [26]. Recently, recurrent NTRK fusion genes were reported in adult HGG as well as pediatric LGG [33, 34], and later shown to induce HGG in mice by injection of astrocytes transduced with retroviral vectors carrying NTRK fusion genes [26]. The authors suggested a significant oncogenic effect of NTRK fusion genes in very young children based on their high frequency in that age group, lack of other significant mutations, as well as rapid tumor induction in their animal model [26].
Somatic histone mutations
Detection of recurrent somatic histone H3 mutations is one of the major discoveries emphasizing the unique nature of pediatric HGG [35, 36]. Specific mutations in H3F3A coding for H3.3 and HIST1H3B/C coding for H3.1 were recently recognized in pediatric HGG as the first reported histone mutations associated with human malignancies [35, 36].
Initial reports identified two distinct single-nucleotide variant mutations, methionine replacing lysine 27 (K27M) frequently involving H3F3A and to a lesser extent HIST1H3B, and arginine (or less frequently valine) replacing glycine 34 (G34R/V) in H3F3A [35, 36]. These mutations were found in more than one third of cases of pediatric HGG in a mutually exclusive pattern. When other gliomas of different grades and across all ages were examined for these mutations, they were identified exclusively in HGG with the vast majority of them occurring in children and adolescents [23, 35, 36]. The anatomical tumor locations and age distribution of patients for each group of histone mutations differed largely. All G34R/V mutated tumors were found in non-midline cortical locations (hemispheric) and mainly affect young adults and adolescents. K27-mutated cases were found in midline tumors (thalamus, cerebellar vermis, brainstem, and spine) and affect younger children (~ 10 years of age) [35-37].
In contrast, Gielen et al. found H3F3A K27M mutations only in two cases of their cohort of very young children with HGG (6%). The authors suggested that the difference in frequency might be attributed to the anatomical distribution of tumors in their cohort. Only five cases – including those two with H3F3A K27M mutations – were midline tumors, while the majority of patients (30/35, 86%) had supratentorial hemispheric tumors [8]. However, the authors did not report H3F3A G34R/V mutations in any of the thirty hemispheric tumors. Thus, anatomical distribution alone might not adequately explain the different frequency of these mutations among very young children with HGG.
Schwartzentruber et al. found marked overlap of TP53 mutation (more than half of cases) with other mutations in H3F3A, the alpha thalassemia/mental retardation syndrome X-Linked (ATRX) gene, and the death-domain-associated protein (DAXX) gene [35]. Loss of ATRX protein expression in their samples was strongly associated with alternative lengthening of telomere (ALT), particularly in cases with simultaneous ATRX, H3F3A and TP53 mutations. This finding is in agreement with an earlier study of Heaphy et al. who reported a strong association of ATRX protein loss and ALT [38]. Conversely, Liu et al. failed to prove such an association in both adult and pediatric HGG [39]. As for HGG in very young children, Gielen et al. found loss of ATRX protein expression in only six cases (17%) and a similar frequency of ALT but with only minimal overlap, suggesting that other mechanisms for telomere lengthening maybe present in HGG of very young children [8]. However, 40% of their cases had either loss of ATRX protein expression or H3F3A mutation or p53 accumulation, pointing to the importance of the H3F3A/ATRX/DAXX pathway in a subgroup of HGG in very young children [8].
DNA methylation profiling
Genome-wide studies investigating global DNA methylation in HGG identified different subgroups in adult and pediatric HGG [23], of which three subgroups included most pediatric patients. One subgroup, receptor tyrosine kinase 1 (RTK-I), was characterized by predominant PDGFRA amplification and included patients from a wide age range. The other two subgroups correspond to the two H3F3A mutations (K27M and G34V/R). Among adolescents and young adult HGG, one of the identified subgroups was strictly related to IDH1 mutations [23].
Korshunov et al. investigated the DNA methylation profiles of more than 200 pediatric HGG. They found almost 20% of histologically confirmed HGG showing methylation patterns similar to LGG (LGG-like) and pleomorphic xanthoastrocytoma (PXA-like). The majority of those cases were of cortical location (around 75%), had better overall outcome (5-year OS > 90% for LGG-like and ~ 60% for PXA-like cases) and frequently harbored BRAF-V600E mutations (48% in PXA-like and 31% in LGG-like). Of interest, very young children with HGG comprised 54% of LGG-like cases and 11% of PXA-like cases in their retrospective study [40].
In summary, HGG in very young children tend to display a more stable genome with less frequent CNAs and fewer identifiable mutations.
Clinical Presentation and Diagnosis
The clinical presentation of a newly diagnosed child with a brain tumor depends on tumor location, and on the age rather than the underlying histology [41]. Different brain tumors share common initial symptoms of increased intracranial tension plus or minus focal deficits and other localizing symptoms according to their anatomical location [42]. Notably, anatomical distribution of brain tumors varies between very young children and their older counterparts with prevalent supratentorial locations in very young children [43, 44].
The onset of symptoms in very young children with a brain tumor is often delayed due to the elasticity and expandability of infants’ skulls that allow tumors to grow significantly before manifesting with increased intracranial pressure [45, 46]. The symptoms they develop in such young age are often non-specific and are usually attributed to other etiologies leading to further delay in diagnosis. These symptoms include macrocephaly, nausea and vomiting, irritability, lethargy and failure to thrive [41, 47-49].
For any child with suspected intracranial tumor, a magnetic resonance imaging (MRI) is the diagnostic standard. It helps identifying tumor location, defining its relation to surrounding structures and more importantly guiding further interventions like surgical resection and radiation field planning [50]. Non-contrast CT scan is particularly useful in emergency situations when acute hydrocephalus or CNS vascular accidents are suspected [50].
HGG tend to appear as ill-defined, irregularly shaped tumors with heterogeneous textures and nodular enhancement. This presentation often prevents a measurability of tumor size especially after surgery when postoperative changes blur the tumor margins additionally. They are usually accompanied by significant brain edema best evidenced on Fluid attenuation inversion recovery (FLAIR) and T2 sequences. They frequently display hypo-intense signals on T1 images and hyper-intense signals on T2 images [51]. Only rarely the T2-signal of the tumor is heavily decreased due to a very high cellularity comparable to embryonal tumors. In this case the imaging differential diagnosis to e.g. ependymomas may be difficult. Most HGG show intense contrast enhancement, however, the degree of contrast enhancement is not always consistent with histopathologic grading [51]. Figures 2 and 3 show some radiological findings of HGG in very young children.
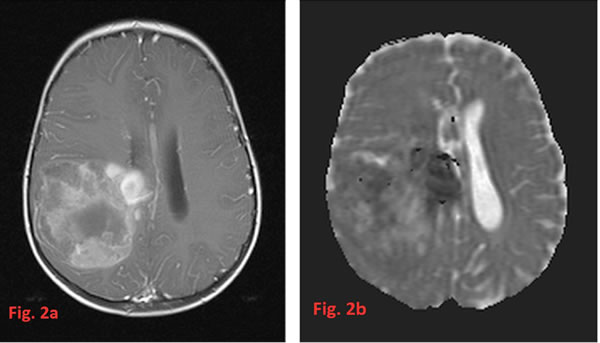
Figure 2: In a right frontoparietal large glioblastoma; enhancement on a T1-weighted MRI after contrast (a) is very inhomogeneous. The corresponding ADC-map (b) also shows a variability of restricted diffusion with the most restricted areas above the roof of the right lateral ventricle. On T1-weighted images before contrast (not shown) also met-hemoglobin as residue of subacute bleeding is present.
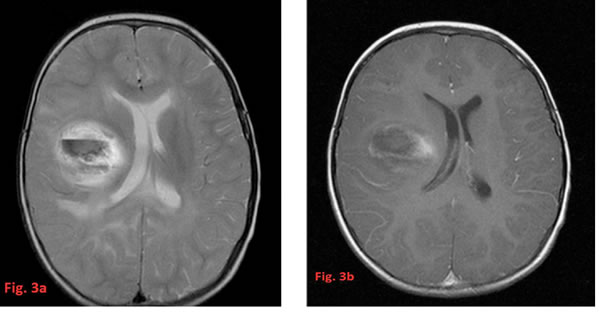
Figure 3: T2-weighted axial image (a) showing a central hemorrhagic part (with a fluid level) of a thalamic glioblastoma together with an increased T2-signal in the posterior corpus callosum, the right frontal region and the left frontobasal parts (not shown) of the brain as a result of multifocal growth. The T1-weighted image at the corresponding level after contrast (b) shows only slight enhancement surrounding the central deoxyhemoglobin core of this tumor part and very ill defined borders.
New imaging techniques have been lately developed for better prediction of the underlying tumor grade, including magnetic resonance spectroscopy (MRS) that works through analyzing the metabolic profile of tumor which reflects its grade [51-53]. Similarly, perfusion-weighted MRI can detect increased tumor blood flow caused by angiogenesis in high grade tumors [51, 54]. Diffusion-tensor MRI calculates an apparent diffusion coefficient (ADC) reflecting tumor cellularity and hence tumor grade [55]. All these modalities still need further validation of their predictability and accuracy. None of them can replace histopathologic examination of tumor tissue in order to reach a definite final diagnosis [50].
Around 3% of pediatric HGG are associated with dissemination into cerebrospinal fluid (CSF) at presentation [56]. Spread usually occurs with tumors related to ventricles or meninges where they can seed directly into the subarachnoid space [57]. The incidence of dissemination in very young children with HGG is quite variable among different studies; while the POG experience shows a much higher incidence with 4 out of 18 children having leptomeningeal dissemination at presentation, both the French BBSFOP and United Kingdom UKCCSG 9204 studies had no cases with CSF-dissemination. However, the very small numbers of patients in each of these studies preclude an accurate estimation of the real incidence of the disease dissemination in this very young patient subpopulation. Although leptomeningeal dissemination and distant spread are relatively uncommon in HGG patients [2, 42], it is usually recommended to perform a spinal MRI and CSF examination at time of diagnosis and whenever there is a suspicion for a relapse and/or dissemination [42, 56, 58]. Of interest, no survival advantage has been reported for non-disseminated gliomas compared to those with CSF dissemination [16, 57].
Treatment of High-Grade Gliomas in Pediatric Population
Over the past decades, several clinical trials have been conducted on pediatric HGG patients aiming for improving treatment outcome of this devastating disease, but unfortunately, little progress has been achieved [2, 3, 59-64]. Fewer trials have been conducted on very young children with HGG [5, 6, 16, 17], and despite their relatively better outcome compared to older children, no much progress has been achieved over the past decade since the last published trials [5, 17].
Surgery
As a general rule, whenever safe and feasible, gross total resection (GTR) of the tumor should be attempted in every child with HGG. However, in many occasions a safe complete tumor resection cannot be achieved [50]. This is particularly true for midline and infratentorial tumors that comprise –unfortunately - a major proportion of pediatric HGG cases [1]. The majority of HGG in very young children are supratentorial hemispheric tumors, and hence expected to be more amenable to GTR, yet this is not achievable in most patients. Despite marked decline in surgical mortality rates for very young children with malignant brain tumors, surgical morbidity is still a major issue [65]. Most cases present with enormous tumor sizes (tumor brains)[66] and are frequently associated with massive hydrocephalus, both of which cause marked distortion of brain vasculature, thus increasing the risk of intra and post-operative hemorrhage [65, 67]. In addition, given their relatively small blood volume, very young children are more vulnerable to hypovolemic shock and cardiac arrest due to blood loss during surgery [65, 67]. In most series, GTR was achievable in approximately one third of cases, and was reported in 6 out of 17 evaluable patients in the POG study [16], 7 out of 21 patients in the French series [5], 14 out of 32 patients in the CCG trial [6], and 3 out of 19 patients in the UKCCSG 9204 study [17].
In a recently published, relatively small single-center experience from Japan, neoadjuvant multi-agent chemotherapy was utilized for infants and very young children with brain tumors to facilitate tumor resection by reducing the vascularity of the tumor [68]. Eight out of 9 patients had GTR following neoadjuvant chemotherapy and only 3 of them required blood transfusion with no surgical mortality reported [68].
Generally, extent of resection is considered as a powerful prognostic factor that impacts survival outcome in HGG patients. Adults with completely resected HGG tumors appear to have a survival advantage [69]. Although patient numbers may be smaller, pediatric HGG patients appear also to benefit from GTR of the tumor with an improvement of survival [70-73]. In a report from the study-945 by the Children’s Cancer Group (CCG), pediatric HGG children with GTR (≥ 90%) had better 5-year progression-free survival (PFS) compared to those with lesser extent of resection (35±7% and 17±4%, respectively) [74].
However, this finding cannot be extrapolated to the very small population of very young children with HGG. Whereas some analyses showed evidence for a better survival after GTR of the tumor [16, 75], other studies could not confirm this survival advantage [5, 6]. These inconsistent findings might be -at least partly- explained by small patient numbers in each of these individual studies. Table 2 illustrates different studies and clinical trials for very young children with HGG.As residual tumor/disease appears to be associated with less favorable prognosis, a second surgery might be considered during the course of the treatment. In addition, Jeibmann et al. reported on a case of infant HGG that showed a differentiation towards low grade histology upon chemotherapy [76]. Thus, residual tumor after induction chemotherapy might be subjected to a second look surgery - even if only a partial resection or a biopsy can be performed - with the aims to increase the probability of survival and/or to avoid any second line treatment, e.g. radiotherapy, since this residual may no longer represent a HGG-like histology.
Table 2: Summary of the different studies in infants and very young children with primary high-grade gliomas
Reference |
No. / age of pts |
Treatment regimen |
Extent of resection |
Radiotherapy |
Treatment results |
Neuropsychologic sequelae |
Duffner et al. 1993, USA Multicenter trial (POG) |
< 3 years of age n=18 |
2 cycles Cyc/VCR followed by 1 cycle Cis/Eto, to be repeated for 1 or two years until radiotherapy can be performed at age > 3 years |
Better prognosis after gross total tumor resection (6/18) |
14/18 received radiotherapy according to the protocol |
1 y PFS 54% ; OS 83% 2 y PFS 54% ; OS 65% 3 y PFS 43% ; OS 50% |
No difference in cognitive functions evaluation between base-line and after 1 year of chemotherapy |
Geyer et al. 1995, USA Multicenter trial (CCG) |
< 2 years of age n=32 (20 AA, 3 AOA, 8 HGG, 1 GS) |
“Eight drugs in 1 day”: VCR, BCNU, Procarbazine, Hydroxyurea, Cis, ARA-C, DTIC, Pred. Potential RT after 10 cycles and/or PD |
No better prognosis after gross total (14/32) and subtotal (9/32) tumor resection |
4/32 in total with 2 at relapse |
3 y PFS : Total 31%, AA 44%, HGG 0% |
Not evaluated |
Dufour et al. 2006, France Multicenter trial (BBSFOP) |
<5 years of age n=21 (4 AO, 5 AOA, 7 AA, 5 HGG) |
Alternating cycles with Carbo/Procarbazine, Eto/Cis, VCR/Cyc |
No better survival after gross total tumor resection (7/21) |
6/21 at relapse/ progression |
5 y PFS 35.3%, 5 y OS 58.8% No significant survival differences between grade III and IV tumors |
For all evaluable survivors (n=7); the mean full scale intellectual quotient was 81.6 (range 55–104). |
Sanders et al. 2007, USA Single Center Study |
<3 years of age n=15 (9 AA, 5 HGG, 2 malignant glioma) |
Different chemotherapy protocolsa with 6 patients receiving scheduled radiotherapy during treatment |
Better OS, but no better EFS after gross total and near total tumor resection (11/16). |
12/15 in total with 6 at relapse/ progression |
5 y EFS / OS Total 28.6%, 66.3%%, AA 33.3%, 77.8%, HGG 40% 80% |
Evaluable survivors (n=9) with some neurocognitive impairment, overall cognitive ability ranging from average to significantly delayed |
Thorarinsdottir et al. 2007, USA Single Center Study |
<4 years of age n=5 (1 AO, 1 AGG, 2 AA). Brainstem glioma, n=1 (AA) |
3 cycles of induction Cis/Cyc/VCR/Eto followed by 3 cycles HDC with Carbo / Thiotepa and ASCR |
2/5 with 1 gross total and 1 near total tumor resection |
3/5 after high dose chemo-therapy |
1 dead; PFS & OS 17 & 22 months. 1 alive with progressive disease; PFS & OS: 3 & 10 months. 3 alive without progression; PFS/OS 8, 33 & 59 months. |
All evaluable survivors (n=4) with a normal cognitive development status |
Grundy et al. 2010, UK Multicenter trial UKCCSG CNS9204, |
<3 years of age n=18 (7 AA, 2 AO, 1 AAB, 5HGG, 3 unknown / unclassified) |
Alternating courses of: Course 1: VCR / Carbo Course 2: VCR / MTX Course 3: VCR / Cyc Course 4: Cisplatin |
3/18 total resection with only 1 long-term survivor, 11/18 partial tumor resection |
5/18 at relapse/ progression |
1-Y EFS 52.6%, OS 57.9% 3-Y EFS 24.1%, OS 40.5% 5-Y EFS 18.1%, OS 34.7% |
Not mentioned |
Mason et al. 1998, USA multicenter Study (Head Start 1) |
<6 years of age n=9 4 / 9 were < 3 years of age |
5 cycles of induction VCR / VP16 / Cis / Cyc followed by consolidation Carbo / thiotepa / VP16 with aBMT in 2 patients only |
2 GTR, 5 STR. For all diagnoses, GTR had better prognosis in survival time from diagnosis but not OS after aBMT |
5 / 9 due to progression after induction chemotherapy |
1-Y EFS 11%, OS 56% 2-Y EFS 11%, OS 22% |
Mean scores for un-irradiated children were within average range for academic achievement; verbal learning; visual memory, social-emotional, and behavioral functioning. |
Razzouk et al. 1995, USA multicenter Phase II Study |
< 4 years of age n=4 |
2 induction cycles of Thiotepa (TT) followed by alternative cycles of (Cyc/VCR), (Cispat/VP16) and (TT) |
Not detailed |
2 / 4 due to progression after induction chemotherapy |
b None had an objective response to induction with TT; 2 PD, and 2 SD |
Not mentioned |
Abbreviations: AA = anaplastic astrocytoma WHO III; AGG = anaplastic ganglioglioma WHO III; AO = anaplastic oligodendroglioma WHO III; AOA = anaplastic oligoastrocytoma WHO III; AAB = anaplastic astroblastoma WHO III; ARA-C = cytosine arabinoside; carbo = Carboplatin; CCG = Children’s Cancer Group; Cis = cisplatinum; Cyc = cyclophosphamide; DTIC = dacarbazine; EFS = event-free survival; Eto = etoposide; HGG = glioblastoma multiforme WHO IV; GS = gliosarcoma WHO IV; HGG = high-grade glioma; OS = overall survival; PFS = progression-free survival; POG = Pediatric Oncology Group; Pred= prednisone; RT = radiotherapy; VCR = vincristine
a Different chemotherapy regimens reported by Sanders et al. (2007): MOPP (n=1; Nitrogen Mustard/VCR/Procarbacine/Pred); CNS2 (n=1; Cis/Eto); Baby POG (n=4; VCR/Cyc alternating with Cis/Eto); CNS6 (n=1, upfront window with 2 cycles Thiotepa followed by Cis/Eto alternating with VCR/Cyc); CNS11 (n=2; VCR/Cyc alternating with Cis/Eto); CNS14 (n=5, Cyc/Carbo/Eto); BB98 (n=2; 2 cycles VCR/Cyc/intrathecal Mafosfamide alternating with oral Eto)
b Unacceptable high rate of Progressive Disease early termination of trial
Radiotherapy
In 1979, Walker et al. showed that post-surgical radiotherapy of glioblastoma significantly improved median survival time from 4-5 to 9-12 months [77]. However, vulnerability of the brain to cancer therapies – particularly radiation therapy - poses a challenge when therapeutic options are being considered. Children aged < 3 years are more vulnerable to the radiotherapy-induced sequelae when compared to older children [16, 78, 79]. These sequelae include neuropsychological and cognitive problems, endocrinopathy, vasculopathy with stroke, and secondary malignancies [78-82].
In order to minimize these damaging sequelae, chemotherapy-based therapies were utilized to defer or delay radiotherapy until the age of 3 years [5, 6, 16, 17]. Pediatric oncology group (POG) and CCG studies were designed to offer radiation therapy –at a reduced dose - for all patients at the end of chemotherapy, however, many patients in their cohorts did not receive any radiation therapy and were treated by chemotherapy alone [6, 16]. In four out of 18 infants with HGG treated in the Baby POG trial, radiation therapy was denied by their families and – interestingly - they were all alive at the end of the study and none of them experienced any recurrence for several years later [10]. In the CCG study, only four out of 32 patients received radiotherapy with two of them getting irradiation following disease relapse. Out of the four irradiated patients, only one – none relapsing – patient remained alive by the end of study [6].
Both the French BBSFOP and United Kingdom UKCCSG 9204 studies were restricting irradiation to the time of relapse or tumor progression only. This restriction did not seem to compromise the overall survival of studied cohorts [5, 17]. In the French cohort, out of 21 HGG patients below 5 years of age, 12 patients were still alive at the end of study, of whom 10 patients received no radiotherapy [5]. The UKCCSG 9204 study reported a cumulative radiotherapy rate of 29% at 5 years for HGG patients within their cohort, supporting the assumption that chemotherapy alone appears to be an adequate first-line treatment for very young children with HGG [17]. On the other hand, the experience from the St. Jude Children’s Research Hospital for 16 very young children with HGG suggested that radiotherapy was essential with 12/16 patients receiving irradiation whether as a part of their initial treatment or at time of disease progression/relapse [75]. However, it should be noted that this experience was based on a single center retrospective study and not a prospectively controlled trial as in other studies (Table 2).
Chemotherapy
Chemotherapy was established as an effective and essential element for the treatment of pediatric HGG when the CCG phase III clinical trial (CCG-943) showed for the first time that adjuvant radio-therapy plus chemotherapy (prednisone, chloroethyl-cyclohexyl nitrosourea [CCNU], and vincristine) significantly improved event-free survival (EFS) in children and adolescents with HGG compared to children and adolescents treated by radio-therapy alone (5-year EFS of 46% and 18%, respectively) [60]. Since then, multimodal treatment regimens for pediatric HGG patients with adjuvant chemotherapy was generally accepted. Design and treatment results of large trials for the treatment of children and adolescents with HGG are listed in Table 3.
Starting in the 1980s, different chemotherapy regimens were examined in the early “baby brain studies” for all types of brain tumors in very young children. Several chemotherapeutic agents were tried in different combinations that could be given for long periods to replace or postpone radiotherapy [5, 16, 83, 84]. Many of these studies suggested the effectiveness of chemotherapy alone to sustain long-term survival in very young children with HGG [5, 6, 16, 17]; for details see Table 2.
Table 3: Design and results of large international multicenter trials for the treatment of children and adolescents with high-grade gliomas
Clinical Trial |
Reference |
Chemotherapy and/or targeted therapy |
Radio-therapy |
Key results |
Special Remarks |
CCG-943 |
Sposto et al. 1989 |
VCR weekly concomitant to RT, thereafter 8 cycles in 6-week intervals with Pred, CCNU, VCR (pCV) vs. RT alone |
54 Gy |
46% 5y EFS (chemo+radio) vs. 18% (RT alone); significant only for HGG patients not for AA. |
Retrospective neuropathological review showing many LGG |
CCG-945 |
Finlay et al. 1995; Fouladi et al. 2003 |
pCV chemotherapy plus radiotherapy (standard treatment) vs. 2 cycles experimental treatment with “8-in-1” chemotherapy (VCR, CCNU, procarbazine, hydroxyurea, cisplatinum, cytarabine, dacarbazine, methylprednisolone) upfront RT, no concomitant chemotherapy to RT, 8 additional courses “8-in-1” after RT |
54 Gy |
33% 5y PFS, 36% 5y OS. No difference in survival between standard and experimental treatment. Corrected for centrally confirmed HGG: 5y PFS 19%, OS 22% |
29% of patients with LGG after consensus neuropathology review |
CCG-9933 |
MacDonald et al. 2005 |
Pre-irradiation chemotherapy with either 4 courses of Cyc/Eto or of carbo/Eto or of Ifos/Eto. All these treatment regimens were followed by RT with concomitant VCR and maintenance treatment with VCR/CCNU |
54 Gy |
8% 5y-EFS, 24% 5y-OS. No significance in survival between the different upfront chemotherapy groups. |
Randomized phase II pre-irradiation window study for non-completely resected HGG |
POG-9135 |
Finlay and Zacharoulis 2005 |
Pre-irradiation Cis/BCNU vs. pre-irradiation VCR/CyC |
54 Gy |
20% 5y-PFS for Cis/BCNU, 5% PFS for VCR/Cyc |
Randomized phase III pre-irradiation window study |
POG-9431 |
Chintagum-pala et al. 2006 |
Pre-irradiation chemotherapy with either 2 courses of procarbazine or of topotecan. This treatment was followed by RT with concomitant VCR as well as maintenance treatment with VCR / CCNU |
54 Gy |
10% 3y-EFS, 15% 3y-OS. No significant differences in survival between the different upfront chemotherapy groups. |
Randomized phase II pre-irradiation window study for non-controlled trial with historical control |
HIT-HGG A |
Wolff et al. 2000, 2002 |
21 day courses of oral Eto and trofosfamide with a subsequent rest for one week in parallel to RT and after RT for up to one year |
54-59 Gy |
4.3% 4y-EFS, 21.7% 4y-OS for non-brainstem HGG. 0.05% 4y-OS for DIPG |
Non-controlled trial with historical control |
HIT-HGG B |
Wolff et al. 2006 |
One cycle of Cis, and Eto at the beginning and one of Cis, Eto, and Ifos at the end of RT followed by maintenance treatment with daily interferon-γ and 3 weekly Cyc |
54-59 Gy |
18,3% 2y-OS for non-brainstem HGG 0% 2 y OS for DIPG |
Non-controlled trial with historical control |
Bronischer et al. 2009 |
Escalating daily doses of erlotinib in parallel and after RT for a planned maximum of 3 years. |
54-59 Gy |
2y-OS 485, 2y-PFS 35%; HGG: 1y-OS 67%, 1y-PFS 33%; AA: 1y-OS 86%, 1y-PFS 75%. |
Clinical phase I trial |
|
Geyer et al. 2010 |
Escalating daily doses of gefitinib in parallel and after RT for up to one year. |
55.8 Gy |
For non-brainstem HGG: 1y-OS 28.8%, 1y-PFS 15.4%. For BSG: 1y-OS 48%, 1y-PFS 16.1%. |
Clinical phase I trial |
|
HIT-HGG C |
Wolff et al. 2008, 2010 |
One cycle of Cis, Eto, and VCR at the beginning of RT followed by weekly VCR and then one cycle of Cis, Eto, and Ifos at the end of RT, thereafter repeated every 4 weeks up to week 29 as first maintenance. After week 29 valproic acid as second maintenance until progression. |
54-59 Gy |
For DIPG and non-brainstem HGG: 56% 1y-OS / 30% 2y-OS / 19% 5y-OS; 27% 1y-EFS / 16% 2y-EFS / 13% 5y-EfS. BSG: Median OS 1.13 y, median EFS 0.40y. |
Non-controlled trial with historical control |
HIT-HGG D pilot |
Wolff et al. 2011 |
Upfront 2 courses of HD MTX, then RT with 1 cycle of Cis, Eto, VCR at the beginning, weekly VCR during RT and 1 cycle of Cis, Eto, Ifos. at the end, Maintenance with VCR / CCNU / pred q 6 weeks for up to 8 cycles. |
54-59 Gy |
For DIPG and non-brainstem HGG: 77% 1y-OS / 40% 2y-OS / 13% 5y-OS; 43% 1y-EFS / 20% 2y-EFS / 13% 5y-EfS. |
Non controlled pilot trial with historical control |
ACNS 0126 |
Cohen et al. 2011a, b |
Radio-chemotherapy with daily TMZ followed by TMZ maintenance for 5 days every 28 d for 10 cycles |
54-59 Gy |
Non-brainstem HGG: 3y-OS 22%, 3y-EFS 11%. HGG: 3y-EFS 7%; AAIII: 3y-EFS 13%. |
Non-controlled trial with historical controls |
Abbreviations: AA = anaplastic astrocytoma WHO III; BSG = brainstem glioma; Carbo = carboplatin; Cis = cisplatinum; Cyc = cyclophosphamide; EFS = event-free survival; Eto = etoposide; HGG = glioblastoma multiforme WHO IV; HGG = high-grade glioma; OS = overall survival; PFS = progression-free survival; Pred= prednisone; RT = radiotherapy; VCR = vincristin
Response of post-operative measurable disease to initial chemotherapy varied between different studies. In the Baby POG study, 10/18 very young children with HGG had less than GTR and six of these 10 patients showed partial response after two cycles of chemotherapy [16]. The CCG trial of the 8-in-1 chemotherapy that included 32 infants with HGG did not show similar response rate. Only five out of 21 patients (24%) with measurable post-operative residual tumor showed partial response to 2 cycles of chemotherapy [6]. The BBSFOP study included 13 out of 21 patients with post-operative residual tumor [5]. Only 2 out of 13 patients showed partial responses to chemotherapy. In their analysis, age seemed to have a prognostic impact – though statistically non-significant – with 6 out of 11 children less than 2 years were long-term survivors compared with 2 out of the 10 patients older than 2 years [5].
Similar to studies of older children with HGG, variability in histopathologic classification of tumors was also evident among many studies of HGG in very young children. Six of 26 patients with malignant astrocytoma in the UKCCSG/SIOP CNS-9204 trial were found to have a different diagnosis following central pathological review [17] while 18% of infant HGG patients in the CCG-945 trial were reclassified after central review [6,10]. Similarly, 4 out of 16 patients (25%) in the St. Jude experience had revised diagnoses after pathologic review [75]. In contrast, 20 /21 patients in the BBSFOP study had same initial diagnosis after central review by a panel of four neuropathologists. Of notice, they reported one third of their cases as having oligodendrogliomas, a much higher percentage than those reported by other trials [5].
High-dose chemotherapy and autologous stem cell rescue
The potential role of high-dose chemotherapy (HDCT) followed by autologous stem cell rescue (ASCR) was previously investigated for very young children with CNS tumors including HGG [85-87]. Initially, it was mainly utilized for recurrent and progressive brain tumors [88, 89]. The Head Start 1 protocol investigated the efficacy of HDCT followed by ASCR for very young children with newly diagnosed brain tumors [90]. Sixty-two patients below 6 years of age – with 37 patients of them below 3 years of age - were enrolled including 9 patients with non-brainstem HGG. Only 37 patients were non-progressing after induction chemotherapy and hence received myeloablative chemotherapy followed by ASCR. Of those 37 patients, only 15 patients were long-term survivors without evidence of progression. Only 2 patients with HGG received HDC and ASCR and they were both alive and disease-free at end of study. The 3-year EFS and OS for all patients were 25% and 40%, respectively with no difference in outcome between the group of patients < 3 years of age and those > 3 years of age at diagnosis [90].
Tandem HDCT/ASCR treatment was investigated in children younger than 4 years old with newly diagnosed brain tumors [91]. Fifteen patients – including 5 patients with HGG – received induction chemotherapy followed by 3 cycles of HDCT (thiotepa / carboplatin) with ASCR. Their results were relatively better with 2-year PFS and OS of 52.2% and 72.1%, respectively. Of notice, 7 out of the 15 patients received selective radiotherapy after ASCR, however, the PFS of those who received irradiation did not differ significantly from those who did not. Ten out of the 15 patients –including 3 patients with HGG – were still alive and free of disease at end of study [91].
Conclusion
HGG in very young children are relatively rare tumors that are challenging to treat, yet they are characterized by a better prognosis compared to HGG in older children. Whether this better outcome when compared to older patients might be due to difference in biology of the disease, needs further investigations. The relation between age at diagnosis and accumulation of genetic alterations needs further exploration to help determine the best age cut-off that distinguish this particular group of tumors from their counterparts in older children. These tumors in very young children appear to be sensitive to chemotherapy and sometimes might be cured without irradiation. However, no advances of treatment strategies have been achieved in the past few years. More collaborative efforts are needed to decipher the molecular and genetic landscape of these rare tumors, to identify new potential therapeutic targets, and hence to move forward in the management of this disease in these very young patients.
ACKNOWLEDGMENTS
We would like to thank the CANSEARCH Foundation for continuous support.
Dr Moatasem El-Ayadi received a postdoctoral fellowship granted by the Egyptian ministry of Higher Education and Scientific Research.
The HIT-HGG trial office (Prof. Christof M. Kramm et al.) and the reference institutions (Neuropathology, Prof. Dr. Torsten Pietsch / Dr. Gerrit H. Gielen / Neuro-Radiology: Prof. Dr. Monika Warmuth-Metz et al..) are supported by the German Children’s Cancer Foundation (Deutsche Kinderkrebsstiftung).
ConflictS of Interest
There is no conflict of interest.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Ostrom QT, De Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, Stearns DS, Wolff JE, Wolinsky Y, Letterio JJ, Barnholtz-Sloan JS. Alex’s Lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007-2011. Neuro Oncol. 2015; 16: x1–35. doi: 10.1093/neuonc/nou327.
2. Broniscer A, Gajjar A. Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. Oncologist. 2004; 9: 197–206.
3. Cohen KJ, Pollack IF, Zhou T, Buxton A, Holmes EJ, Burger PC, Brat DJ, Rosenblum MK, Hamilton RL, Lavey RS, Heideman RL. Temozolomide in the treatment of high-grade gliomas in children: A report from the Children’s Oncology Group. Neuro Oncol. 2011; 13: 317–23. doi: 10.1093/neuonc/noq191.
4. Jones C, Karajannis MA, Jones DTW, Kieran MW, Monje M, Baker SJ, Becher OJ, Cho YJ, Gupta N, Hawkins C, Hargrave D, Haas-Kogan DA, Jabado N, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol. 2017; 9:153-161. doi: 10.1093/neuonc/now101.
5. Dufour C, Grill J, Lellouch-Tubiana A, Puget S, Chastagner P, Frappaz D, Doz F, Pichon F, Plantaz D, Gentet JC, Raquin MA, Kalifa C. High-grade glioma in children under 5 years of age: A chemotherapy only approach with the BBSFOP protocol. Eur J Cancer. 2006; 42: 2939–45. doi: 10.1016/j.ejca.2006.06.021.
6. Geyer JR, Finlay JL, Boyett JM, Wisoff J, Yates A, Mao L, Packer RJ. Survival of infants with malignant astrocytomas: A report from the Childrens Cancer Group. Cancer. 1995; 75: 1045–50.
7. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med. 2005; 352: 987–96. doi: 10.1056/NEJMoa043330.
8. Gielen GH, Gessi M, Buttarelli FR, Baldi C, Hammes J, zur Muehlen A, Doerner E, Denkhaus D, Warmuth-Metz M, Giangaspero F, Lauriola L, von Bueren AO, Kramm CM, et al. Genetic Analysis of Diffuse High-Grade Astrocytomas in Infancy Defines a Novel Molecular Entity. Brain Pathol. 2014; 25: 409–17. doi: 10.1111/bpa.12210.
9. Rickert CH, Probst-Cousin S, Gullotta F. Primary intracranial neoplasms of infancy and early childhood. Childs Nerv Syst. 1997; 13: 507–13.
10. Duffner PK, Horowitz ME, Krischer JP, Burger PC, Cohen ME, Sanford RA, Friedman HS, Kun LE. The treatment of malignant brain tumors in infants and very young children: an update of the Pediatric Oncology Group experience. Neuro Oncol. 1999; 1: 152.
11. Bishop AJ, McDonald MW, Chang AL, Esiashvili N. Infant brain tumors: Incidence, survival, and the role of radiation based on surveillance, epidemiology, and end results (SEER) data. Int J Radiat Oncol Biol Phys. 2012; 82: 341–7. doi: 10.1016/j.ijrobp.2010.08.020.
12. Rickert CH, Paulus W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Child’s Nerv Syst. 2001; 17: 503–11. doi: 10.1007/s003810100496.
13. Parkin D, Kramárová E, Draper GJ, Masuyer E, Michaelis J, Neglia J, Qureshi S, Stiller CA. International Incidence of Childhood Cancer, Vol. II - WHO - OMS -. Lyon: IARC; 1998.
14. Bauchet L, Rigau V, Mathieu-Daudé H, Fabbro-Peray P, Palenzuela G, Figarella-Branger D, Moritz J, Puget S, Bauchet F, Pallusseau L, Duffau H, Coubes P, Trétarre B, et al. Clinical epidemiology for childhood primary central nervous system tumors. J Neurooncol. 2009; 92: 87–98. doi: 10.1007/s11060-008-9740-0.
15. Kaatsch P. Epidemiology of childhood cancer. Cancer Treat Rev. 2010; 36: 277–85. doi: 10.1016/j.ctrv.2010.02.003.
16. Duffner PK, Horowitz ME, Krischer JP, Friedman HS, Burger PC, Cohen ME, Sanford RA, Mulhern RK, James HE, Freeman CR. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993; 328: 1725–31. doi: 10.1056/NEJM199306173282401.
17. Grundy RG, Wilne SH, Robinson KJ, Ironside JW, Cox T, Chong WK, Michalski A, Campbell RHA, Bailey CC, Thorp N, Pizer B, Punt J, Walker DA, et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: Results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer. 2010; 46: 120–33. doi: 10.1016/j.ejca.2009.09.013.
18. Isaacs H Jr. Perinatal brain tumors: a review of 250 cases. Pediatr Neurol. 2002; 27: 249–61. doi: 10.1016/S0887-8994(02)00472-1.
19. Packer RJ, MacDonald T, Vezina G. Central Nervous System Tumors. Pediatr Clin North Am. 2008; 55: 121–45. doi: 10.1016/j.pcl.2007.10.010.
20. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016; 131: 803–20. doi: 10.1007/s00401-016-1545-1.
21. Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nat Rev Cancer. 2014; 14: 1–11. doi: 10.1038/nrc3811.
22. Sturm D, Bender S, Jones DTW, Lichter P, Grill J, Becher O, Hawkins C, Majewski J, Jones C, Costello JF, Iavarone A, Aldape K, Brennan CW, et al. Paediatric and adult glioblastoma: multiform (epi)genomic culprits emerge. Nat Rev Cancer. 2014; 14: 92–107. doi: 10.1038/nrc3655.
23. Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012; 22: 425–37. doi: 10.1016/j.ccr.2012.08.024.
24. Bax DA, Mackay A, Little SE, Carvalho D, Viana-Pereira M, Tamber N, Grigoriadis AE, Ashworth A, Reis RM, Ellison DW, Al-Sarraj S, Hargrave D, Jones C. A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res. 2011; 16: 3368–77. doi: 10.1158/1078-0432.CCR-10-0438.A.
25. Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol. 2010; 28: 3061–8. doi: 10.1200/JCO.2009.26.7252.
26. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, Easton J, Edmonson M, Ma X, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014; 46: 444–50. doi: 10.1038/ng.2938.The.
27. Phillips JJ, Aranda D, Ellison DW, Judkins AR, Croul SE, Brat DJ, Ligon KL, Horbinski C, Venneti S, Zadeh G, Santi M, Zhou S, Appin CL, et al. PDGFRA Amplification is Common in Pediatric and Adult High-Grade Astrocytomas and Identifies a Poor Prognostic Group in IDH1 Mutant Glioblastoma. Brain Pathol. 2013; 23: 565–73. doi: 10.1111/bpa.12043.
28. Koschmann C, Zamler D, MacKay A, Robinson D, Wu YM, Doherty R, Marini B, Tran D, Garton H, Muraszko K, Robertson P, Leonard M, Zhao L, et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget. 2016; 7: 65696–706. doi: 10.18632/oncotarget.11602.
29. Mourtada-Maarabouni M, Pickard MR, Hedge VL, Farzaneh F, Williams GT. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009; 28: 195–208. doi: 10.1038/onc.2008.373.
30. Liao J, Yu L, Mei Y, Guarnera M, Shen J, Li R, Liu Z, Jiang F. Small nucleolar RNA signatures as biomarkers for non-small-cell lung cancer. Mol Cancer. 2010; 9: 198. doi: 10.1186/1476-4598-9-198.
31. Dong XY, Guo P, Boyd J, Sun X, Li Q, Zhou W, Dong JT. Implication of snoRNA U50 in human breast cancer. J Genet. 2010; 36: 447–54. doi: 10.1016/S1673-8527(08)60134-4.
32. Dong X, Rodriguez C, Guo P, Sun X. SnoRNA U50 is a candidate tumor-suppressor gene at 6q14. 3 with a mutation associated with clinically significant prostate cancer. Hum Mol. 2008; 7:1031-42.
33. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, Danussi C, Dolgalev I, Porrati P, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013; 45: 1141–9. doi: 10.1038/ng.2734.
34. Jones DTW, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DAK, Fontebasso AM, Stütz AM, Hutter S, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013; 45: 927–32. doi: 10.1038/ng.2682.
35. Schwartzentruber J, Korshunov A, Liu X, Jones DTW, Pfaff E, Jacob K. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012; 482:226-31.
36. Wu G, Broniscer A, Mceachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Gajjar A, Dyer MA, Mullighan CG, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012; 44: 251–3. doi: 10.1038/ng.1102.Somatic.
37. Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012; 124: 439–47. doi: 10.1007/s00401-012-0998-0.
38. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011; 333: 425. doi: 10.1126/science.1207313.
39. Liu XY, Gerges N, Korshunov A, Sabha N, Khuong-Quang DA, Fontebasso AM, Fleming A, Hadjadj D, Schwartzentruber J, Majewski J, Dong Z, Siegel P, Albrecht S, et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012; 124: 615–25. doi: 10.1007/s00401-012-1031-3.
40. Korshunov A, Ryzhova M, Hovestadt V, Bender S, Sturm D, Capper D, Meyer J, Schrimpf D, Kool M, Northcott PA, Zheludkova O, Milde T, Witt O, et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015; 129: 669–78. doi: 10.1007/s00401-015-1405-4.
41. Wilne S, Collier J, Kennedy C, Koller K, Grundy R, Walker D. Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol. 2007; 8: 685–95. doi: 10.1016/S1470-2045(07)70207-3.
42. Fangusaro J. Pediatric high-grade gliomas and diffuse intrinsic pontine gliomas. J Child Neurol. 2009; 24: 1409–17. doi: 10.1177/0883073809338960.
43. Rickert CH. Epidemiological features of brain tumors in the first 3 years of life. Childs Nerv Syst. 1998; 14: 547–50.
44. Giuffre R. Biological aspects of brain tumors in infancy and childhood. Childs Nerv Syst. 1989; 5: 55–9.
45. Wakai S, Arai T, Nagai M. Congenital brain tumors. Surg Neurol. 1984; 21: 597–609.
46. Reed UC, Rosemberg S, Gherpelli JL, Matushita H, de Almeida GG, Diament AJ. Brain tumors in the first two years of life: a review of forty cases. Pediatr Neurosurg. 1993; 19: 180–5.
47. Dobrovoljac M, Hengartner H, Boltshauser E, Grotzer MA. Delay in the diagnosis of paediatric brain tumours. Eur J Pediatr. 2002; 161: 663–7. doi: 10.1007/s00431-002-1088-4.
48. Reddy AT, Wellons JC. Pediatric high-grade gliomas. Cancer J. 2003; 9: 107–12.
49. Reulecke BC, Erker CG, Fiedler BJ, Niederstadt TU, Kurlemann G. Brain tumors in children: initial symptoms and their influence on the time span between symptom onset and diagnosis. J Child Neurol. 2008; 23: 178–83. doi: 10.1177/0883073807308692.
50. Fangusaro J. Pediatric High Grade Glioma: a Review and Update on Tumor Clinical Characteristics and Biology. Front Oncol. 2012; 2: 1–10. doi: 10.3389/fonc.2012.00105.
51. Panigrahy A, Blüml S. Neuroimaging of pediatric brain tumors: from basic to advanced magnetic resonance imaging (MRI). J Child Neurol. 2009; 24: 1343–65. doi: 10.1177/0883073809342129.
52. Lemort M, Canizares-Perez AC, Van der Stappen A, Kampouridis S. Progress in magnetic resonance imaging of brain tumours. Curr Opin Oncol. 2007; 19: 616–22. doi: 10.1097/CCO.0b013e3282f076b2.
53. Morita N, Harada M, Otsuka H, Melhem ER, Nishitani H. Clinical Application of MR Spectroscopy and Imaging of Brain Tumor. Magn Reson Med Sci. 2010; 9: 167–75.
54. Lev MH, Ozsunar Y, Henson JW, Rasheed AA, Barest GD, Harsh GR, Fitzek MM, Chiocca EA, Rabinov JD, Csavoy AN, Rosen BR, Hochberg FH, Schaefer PW, et al. Glial tumor grading and outcome prediction using dynamic spin-echo MR susceptibility mapping compared with conventional contrast-enhanced MR: confounding effect of elevated rCBV of oligodendrogliomas [corrected]. AJNR Am J Neuroradiol. 2004; 25: 214–21.
55. Gauvain KM, McKinstry RC, Mukherjee P, Perry A, Neil JJ, Kaufman BA, Hayashi RJ. Evaluating pediatric brain tumor cellularity with diffusion-tensor imaging. AJR Am J Roentgenol. 2001; 177: 449–54. doi: 10.2214/ajr.177.2.1770449.
56. Benesch M, Wagner S, Berthold F, Wolff JE. Primary dissemination of high-grade gliomas in children: experiences from four studies of the Pediatric Oncology and Hematology Society of the German Language Group (GPOH). J Neurooncol. 2005; 72: 179–83. doi: 10.1007/s11060-004-3546-5.
57. Irani DN. Cerebrospinal fluid in clinical practice. Saunders. 2009. p. 317.
58. Grabb PA, Albright AL, Pang D. Dissemination of supratentorial malignant gliomas via the cerebrospinal fluid in children. Neurosurgery. 1992; 30: 64–71.
59. Leonard A, Wolff JE. Etoposide improves survival in high-grade glioma: A meta-analysis. Anticancer Res. 2013; 33: 3307–16. doi: 10.1093/neuonc/nos102.
60. Sposto R, Ertel IJ, Jenkin RD, Boesel CP, Venes JL, Ortega JA, Evans AE, Wara W, Hammond D. The effectiveness of chemotherapy for treatment of high grade astrocytoma in children: results of a randomized trial. A report from the Childrens Cancer Study Group. J Neurooncol. 1989; 7: 165–77.
61. Wolff JE, Kortmann RD, Wolff B, Pietsch T, Peters O, Schmid HJ, Rutkowski S, Warmuth-Metz M, Kramm C. High dose methotrexate for pediatric high grade glioma - Results of the HIT-GBM-D Pilot study. J Neurooncol. 2011; 102: 433–42. doi: 10.1007/s11060-010-0334-2.
62. Wolff JE, Mölenkamp G, Westphal S, Pietsch T, Gnekow A, Kortmann RD, Kuehl J. Oral trofosfamide and etoposide in pediatric patients with glioblastoma multiforme. Cancer. 2000; 89: 2131–7.
63. Wolff JE, Driever PH, Erdlenbruch B, Kortmann RD, Rutkowski S, Pietsch T, Parker C, Metz MW, Gnekow A, Kramm CM. Intensive chemotherapy improves survival in pediatric high-grade glioma after gross total resection: Results of the HIT-gBM-c protocol. Cancer. 2010; 116: 705–12. doi: 10.1002/cncr.24730.
64. Wolff JE, Wagner S, Reinert C, Gnekow A, Kortmann RD, Kühl J, Van Gool SW. Maintenance treatment with interferon-gamma and low-dose cyclophosphamide for pediatric high-grade glioma. J Neurooncol. 2006; 79: 315–21. doi: 10.1007/s11060-006-9147-8.
65. Albright AL, Pollack IF, Adelson PD. Principles and practice of pediatric neurosurgery. New York: Thieme; 1999.
66. Giuffré R. Biological aspects of brain tumors in infancy and childhood. Child’s Nerv Syst. 1989; 5: 55–9. doi: 10.1007/BF00571110.
67. Raimondi AJ, Tomita T. Brain Tumors during the First Year of Life. Pediatr Neurosurg. 1983; 10: 193–207. doi: 10.1159/000120114.
68. Iwama J, Ogiwara H, Kiyotani C, Terashima K, Matsuoka K, Iwafuchi H, Morota N. Neoadjuvant chemotherapy for brain tumors in infants and young children. J Neurosurg Pediatr. 2015; 15: 488–92. doi: 10.3171/2014.11.PEDS14334.
69. Stummer W, van den Bent MJ, Westphal M. Cytoreductive surgery of glioblastoma as the key to successful adjuvant therapies: new arguments in an old discussion. Acta Neurochir (Wien). 2011; 153: 1211–8. doi: 10.1007/s00701-011-1001-x.
70. Albright AL, Sposto R, Holmes E, Zeltzer PM, Finlay JL, Wisoff JH, Berger MS, Packer RJ, Pollack IF. Correlation of neurosurgical subspecialization with outcomes in children with malignant brain tumors. Neurosurgery. 2000; 47: 879-85-7.
71. Heideman RL, Kuttesch J, Gajjar AJ, Walter AW, Jenkins JJ, Li Y, Sanford RA, Kun LE. Supratentorial malignant gliomas in childhood: a single institution perspective. Cancer. 1997; 80: 497–504.
72. Kramm CM, Wagner S, Van Gool S, Schmid H, Sträter R, Gnekow A, Rutkowski S, Wolff JE. Improved survival after gross total resection of malignant gliomas in pediatric patients from the HIT-GBM studies. Anticancer Res. 2006; 26: 3773–9.
73. Wisoff JH, Boyett JM, Berger MS, Brant C, Li H, Yates AJ, McGuire-Cullen P, Turski PA, Sutton LN, Allen JC, Packer RJ, Finlay JL. Current neurosurgical management and the impact of the extent of resection in the treatment of malignant gliomas of childhood: a report of the Children’s Cancer Group trial no. CCG-945. J Neurosurg. 1998; 89: 52–9. doi: 10.3171/jns.1998.89.1.0052.
74. Finlay J, Boyett JM, Yates AJ, Wisoff JH, Milstein JM, Geyer JR, Bertolone SJ, McGuire P, Cherlow JM, Tefft M. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J Clin Oncol. 1995; 13: 112–23.
75. Sanders RP, Kocak M, Burger PC, Merchant TE, Gajjar A, Broniscer A. High-grade astrocytoma in very young children. Pediatr Blood Cancer. 2007; 49: 888–93. doi: 10.1002/pbc.21272.
76. Jeibmann A, Hasselblatt M, Pfister S, Sträter R, Brentrup A, Holling M, Niederstadt T, Paulus W, Frühwald MC. From glioblastoma to gangliocytoma: an unforeseen but welcome shift in biological behavior. J Neurosurg Pediatr. 2009; 4: 475–8. doi: 10.3171/2009.6.PEDS097.
77. Walker MD, Strike TA, Sheline GE. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int J Radiat Oncol Biol Phys. 1979; 5: 1725–31.
78. Ellenberg L, McComb JG, Siegel SE, Stowe S. Factors affecting intellectual outcome in pediatric brain tumor patients. Neurosurgery. 1987; 21: 638–44.
79. Spunberg JJ, Chang CH, Goldman M, Auricchio E, Bell JJ. Quality of long-term survival following irradiation for intracranial tumors in children under the age of two. Int J Radiat Oncol Biol Phys. 1981; 7: 727–36.
80. Shalet SM, Gibson B, Swindell R, Pearson D. Effect of spinal irradiation on growth. Arch Dis Child. 1987; 62: 461–4.
81. Armstrong GT, Liu Q, Yasui Y, Huang S, Ness KK, Leisenring W, Hudson MM, Donaldson SS, King AA, Stovall M, Krull KR, Robison LL, Packer RJ. Long-Term Outcomes Among Adult Survivors of Childhood Central Nervous System Malignancies in the Childhood Cancer Survivor Study. J Natl Cancer Inst. 2009; 101: 946–58. doi: 10.1093/jnci/djp148.
82. Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004; 5: 399–408. doi: 10.1016/S1470-2045(04)01507-4.
83. Ater JL, van Eys J, Woo SY, Moore B, Copeland DR, Bruner J. MOPP chemotherapy without irradiation as primary postsurgical therapy for brain tumors in infants and young children. J Neurooncol. 1997; 32: 243–52.
84. White L, Kellie S, Gray E, Toogood I, Waters K, Lockwood L, Macfarlane S, Johnston H. Postoperative chemotherapy in children less than 4 years of age with malignant brain tumors: promising initial response to a VETOPEC-based regimen. A Study of the Australian and New Zealand Children’s Cancer Study Group (ANZCCSG). J Pediatr Hematol Oncol. 1998; 20: 125–30.
85. Dunkel J, Finlay JL. High-dose chemotherapy with autologous stem cell rescue for brain tumors. Crit Rev Oncol Hematol. 2002; 41: 197–204.
86. Finlay J. The role of high-dose chemotherapy and stem cell rescue in the treatment of malignant brain tumors: a reappraisal. Pediatr Transplant. 1999; 3: 87–95.
87. Wolff J, Finlay JL. High-Dose Chemotherapy in Childhood Brain Tumors. Oncol Res Treat. 2004; 27: 239–45. doi: 10.1159/000077973.
88. Dupuis-Girod S, Hartmann O, Benhamou E, Doz F, Mechinaud F, Bouffet E, Coze C, Kalifa C. Will high dose chemotherapy followed by autologous bone marrow transplantation supplant cranio-spinal irradiation in young children treated for medulloblastoma? J Neurooncol. 1996; 27: 87–98.
89. Guruangan S, Dunkel IJ, Goldman S, Garvin JH, Rosenblum M, Boyett JM, Gardner S, Merchant TE, Gollamudi S, Finlay JL. Myeloablative chemotherapy with autologous bone marrow rescue in young children with recurrent malignant brain tumors. J Clin Oncol. 1998; 16: 2486–93.
90. Mason WP, Grovas A, Halpern S, Dunkel IJ, Garvin J, Heller G, Rosenblum M, Gardner S, Lyden D, Sands S, Puccetti D, Lindsley K, Merchant TE, et al. Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol. 1998; 16: 210–21.
91. Thorarinsdottir HK, Rood B, Kamani N, Lafond D, Perez-Albuerne E, Loechelt B, Packer RJ, MacDonald TJ. Outcome for children <4 years of age with malignant central nervous system tumors treated with high-dose chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer. 2007; 48: 278–84. doi: 10.1002/pbc.20781.