
INTRODUCTION
The recently described Salvador-Warts-Hippo pathway (SWH or Hippo pathway) is highly conserved throughout evolution [1] and is crucially involved in organ size regulation [2,3]. Hippo pathway consists of several negative growth regulators acting in a kinase cascade that ultimately inactivates Yorkie in Drosophila or YAP1 (Yes-associated protein 1) in mammalian; these two effector proteins are both transcriptional coactivators that positively regulate cell growth, survival and proliferation [4]. Therefore YAP1, which is localized in 11q22 genomic region, represents a critical downstream regulatory target of this signaling pathway [5].
Literature data so far described the involvement of YAP1 in carcinogenesis contradictorily. Initially, YAP1 was classified as a tumor suppressor gene (or at least as helper of tumor suppressors), as it was reported to exert pro-apoptotic functions. Following DNA damage, YAP1 functions as a co-activator of TP73-mediated apoptosis in TP53 null cells [6, 7], after phosphorylation of YAP1 at tyrosine 357 [8], following YAP1 dissociation from cytoplasmatic multiprotein complex with 14-3-3 and Akt [9] and as well as a result of RASSF1A activation [10]. As a consequence YAP1 translocates into the nucleus promoting the assembly of the active complex inducing the transcription of target genes [7]. YAP1 was also proposed to be a tumor suppressor in breast cancer, as the target of loss of heterzygosity in 11q22 genomic region [11].
On the contrary, YAP1 was also described to function as an oncogene by promoting increased organ size and cancer development. YAP1 resulted amplified in human hepatocellular carcinoma and cooperated with myc oncogene to induce tumor growth in nude mice [12]. In non-transformed mammary cells YAP1 ectopic overexpression induces alterations typical of a transformed phenotypes, namely anchorage-independent growth, EMT, growth factor independent proliferation, activation of AKT/ERK and inhibition of apoptosis [13] . In addition, in transgenic mouse models the liver-specific YAP1 overexpession induced a dramatic increase of liver organ size, eventually leading to cancer development [14, 15]. Moreover, recent data indicated that YAP1 activity correlates with high histological grade and metastasis in breast cancer [16]. Furthermore, the 11q22 genomic region was found amplified in individual cases of several human tumor types [12, 17-29] but the direct evidence of YAP1 amplification is described in very few of these cases [12, 21, 24, 26-28].
Notably, YAP1 point/small mutations have not been described so far and the reported 11q22 amplification events include multiple flanking genes in addition to YAP1. Some of these genes have been involved in cancer development, including a cluster of matrix metalloproteinase (MMP) genes [30], two members of the BIRC family of caspase inhibitors (BIRC2 and BIRC3) [31] and the progesterone receptor (PGR) [32]. As a consequence, there is no direct evidence about the oncogenic role of endogenous YAP1 in the context of cancer cells carrying the 11q22 amplification event.
In the present work we corroborate that YAP1 plays an important role in the tumorigenic phenotype of 11q22-amplified cancer cell lines, as it effectively supports multiple transformed properties. Moreover we detect YAP1 copy number amplification in clinical series of different human tumor types and identify the downstream genes and pathways that are critical as YAP1 effectors in carcinogenesis.
RESULTS
Identification of cancer cell lines and clinical specimen carrying 11q22 amplification and YAP1 overexpression.
Public and private genomic copy-number databases were interrogated for the copy number status of loci encompassing YAP1 (Supplemental Table 1). Notably, homozygous deletion encompassing YAP1 gene was a very rare event, in fact it was found only in 3/664 (0.5%) cancer cell lines and in 3/1629 (0.2%) cancer tissue samples. In contrast, YAP1 copy amplification event was found in a greater percentage of the same samples (Chi-square test p<0.0001). In fact, it was reported in 40/664 (6%) cancer cell lines, in 31/1629 (1.9%) cancer tissue samples, in 2/110 (1.8%) primary cancer cell cultures and in 1/20 (5%) xenograft tumors (Supplemental Table 1).
We focused our attention on tumor subtypes with little or no established involvement of YAP1 gene, and selected representative cancer cell lines, including Ca-Ski cell line (Cervical squamous cell carcinoma), RO82 cell line (Follicular thyroid carcinoma) and EKVX cell line (Non small-cell lung adenocarcinoma). Preliminary experiments were performed in order to verify YAP1 amplification in these established cancer cell lines and to evaluate the occurrence of YAP1 protein overexpression. Ca-Ski, RO82 and EKVX cell lines showed YAP1 high-copy number (Figure 1a). FISH analysis allowed to define that YAP1 copy amplification is contained in a homogeneously staining region in Ca-Ski cell line or as multiple interspersed copies (double minutes) in RO82 and in EKVX cell lines (Figure 1b). YAP1 amplification correlated with protein overexpression as detected by western blot analysis. In fact, Ca-Ski, RO82 and EKVX cell lines show high protein level of both total- and phospho(S127)- YAP1, compared to other cancer cell lines lacking 11q22 amplification (Figure 1c). Mutational analysis by exon amplification and sequencing was performed, but no small/point mutations were detected in Ca-Ski, RO82 and EKVX cancer cell lines (not shown). The major known genetic and epigenetic traits of the cell lines under study is reported in Supplemental Table2.
Literature data report that chromosome region 11q22, containing YAP1 gene, is amplified in a small percentage of samples from multiple human tumor types [12, 17-29]. We directly investigated the frequency of YAP1 copy number amplification in clinical series from different human tumor types and revealed YAP1 amplification in 4/25 (16%) cervical cancer samples, in 18/77 (23%) non small cell lung cancers (NSCLC), in 2/56 (3.6%), central nervous system tumors (CNS) and in 0/15 thyroid cancers (Figure 1d). The 77 NSCLC samples arrayed on a tissue microarray were assayed using double-colour FISH. Since cyclin D1 is present in the same chromosomal arm as YAP1 but in 11q13 and CCND1 is frequently amplified in lung cancer [33, 34], we used one probe specific for cyclin D1 gene and another probe specific for YAP1 gene or FISH analyses. Using such approach, we detected a large amplification which included both YAP1 and CCND1 genes in 16/77 (21%) NSCLC samples, while 2/77 samples (2.6%) carried an amplification restricted to YAP1 (Figure 1e). We investigated the YAP1 copy number status of the other clinical samples, which were available as individual paraffin blocks, by a quantitative, real-time PCR approach. YAP1 amplification was found in 4/25 (16%) cervical cancer samples, including high copy amplification in 3/4 and low copy amplification in 1/4. YAP1 protein expression was assayed by immunohistochemistry in these cervical cancer samples, revealing that YAP1 protein level was higher in samples carrying YAP1 copy amplification compared to samples with normal YAP1 copy number (p=0.021, Figure 1f).
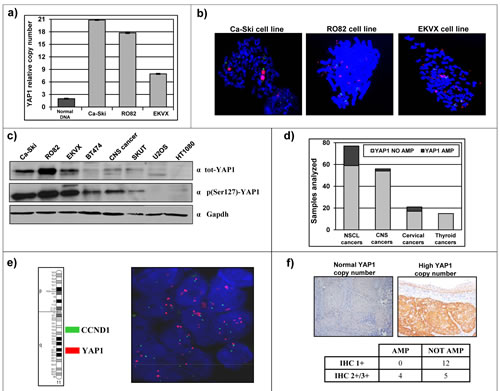
Figure 1: Identification of cancer cell lines and clinical cancer samples carrying 11q22 amplification and overexpression. a) Gene dosage analysis by qPCR shows YAP1 copy number amplification in Ca-Ski, RO82 and EKVX cell lines (grey bars). Normal DNA (2 copies of YAP1) was used as a control (black bar). b) FISH analysis defined the presence of the amplicon spanning YAP1 gene in Ca-Ski, RO82 and EKVX cell lines (Green = Control BAC clone; Red = YAP1-containing BAC clone). c) Total and phospho (Ser127)-YAP1 protein level in Ca-Ski, RO82 and EKVX cell lines (high protein levels) and in other cancer cell lines. GAPDH was used as loading control. d) Number of cases with normal YAP1 copy number (in grey) or YAP1 copy number amplification (in black) in NSCL non small cell lung cancers samples, CNS central nervous system cancers samples, cervical cancers samples and thyroid cancers samples. e) Representative FISH analysis of a NSCLC sample carrying YAP1 amplification and normal CCND1 (located in 11q13) copy number. (Green = CCND1-containing BAC clone; Red = YAP1-containing BAC clone). f) Representative immunohistochemistry analysis of YAP1 protein level in a cervical cancer sample with normal YAP1 copy number (left panel) and a sample carrying YAP1 copy amplification (right panel). Fisher’s exact test two-tailed p=0.0211.
YAP1 is efficiently downregulated in 11q22-amplified cancer cell lines.
Endogenous YAP1 expression was modulated in Ca-Ski, EKVX and RO82 cell lines by RNA interference strategies. YAP1 silencing was performed using both short-hairpin RNA (shRNA) lentiviral particles and small-interfering RNAs (siRNA). In stably infected sh-YAP1 bulk cell population, YAP1 residual expression of mRNA was on average 30% and the YAP1 residual protein expression was 15-30% (Figure 2a). In si-YAP1 short-term silenced cells, YAP1 mRNA was silenced with a residual expression of about 10-20% and the protein was efficiently downregulated with minimal expression between 48 and 96 hours (Figure 2b). The three individual siRNAs used effectively silenced YAP1 as much as an equal total amount of the pool of the three combined siRNAs (Supplemental Figure 1a). YAP1 protein level in parental and silenced cell lines was similarly quantified by multiple antibodies, raised against different YAP1 epitopes (Supplemental Figure 1b).
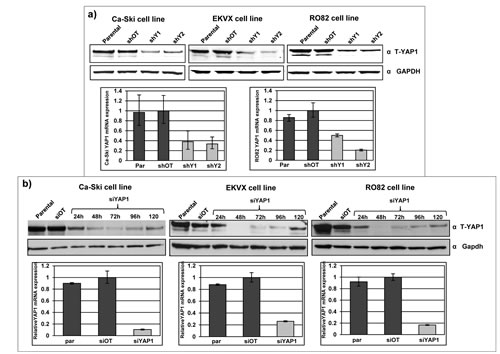
Figure 2: Effective YAP1 silencing in 11q22-amplified cancer cell lines. a) YAP1 protein expression in Ca-Ski, EKVX and RO82 bulk cell population after puromycin selection of off target cells (shOT) and YAP1 silenced cells (shY1 and shY2) was defined by western blot analysis using anti-total YAP1 antibody. GAPDH was used as loading control. Lower panel: YAP1 mRNA expression level in Ca-Ski and RO82 parental cells, off target cells (shOT) and YAP1 silenced cells (shY1 and shY2). YAP1 mRNA expression level was determined by real-time quantitative PCR (qPCR) analysis. b) YAP1 protein level in Ca-Ski, EKVX and RO82 parental cells, off target cells (siOT) and YAP1 silenced cells (siYAP1) was determined by western blot analysis after short-term, siRNA-mediated silencing. The panel shows a time-course analysis 24 to 120 hours after transfection with siYAP1. Lower panel: YAP1 mRNA expression level in Ca-Ski, EKVX and RO82 parental cells, off target cells (siOT) and of YAP1 silenced cells (siYAP1), 24 hours post transfection, as determined by qPCR analysis.
Identification of YAP1 target genes in 11q22-amplified cancer cell lines.
Literature data report that YAP1 gene is a transcriptional cofactor [35] but YAP1 target genes remain poorly characterized and therefore we performed gene expression profiling experiments with the aim to identify YAP1 target genes that may mediate an oncogenic stimulus in YAP1-amplified cell lines. Using class comparison analyses comparing global gene expression profiles of YAP1 proficient cells versus YAP1-silenced cells we identified 707 statistically significantly modulated genes at the uninominal p-value= 0.001 (multivariable global test p-value=0.002, Figure 3 and Supplemental Table 3). Among these target genes, 505 are down-regulated and 202 are up-regulated upon YAP1 silencing (Chi-square p-value < 0.0001), thus suggesting that YAP1 mainly acts as a transcriptional co-activator in the cancer cell lines under study. Using different tools of functional annotation, we found that the 707 genes identified are significantly enriched for gene ontologies related to cell proliferation and to cell movement molecular functions and suggesting that YAP1 overexpression positively sustains these two biological functions (Figure 3b and Supplemental Table 3). In order to pinpoint the YAP1 target genes specifically modulated in the context of YAP1 amplification in cancer cells, we filtered our YAP1-gene signature with the YAP1-gene signature from two previous studies reporting gene expression profiling in normal cellular contexts in which YAP1 gene is ectopically overexpressed [15, 36]. Among the 427 significantly regulated genes that could be compared across the three studies, we found that 88 are in common in our study and in at least one study from the literature and 86/88 are concordantly (up- or down-) co-regulated (9/9 genes concordantly regulated in all analyses) (Figure 3a). Indeed, these genes represent YAP1 targets modulated in multiple different normal and tumor cell contexts. Additional 339 genes are uniquely modulated in the 11q22-amplified cancer cell lines (Figure 3a and Supplemental Table 3). We functionally annotated the filtered 339 gene-list using Ingenuity functional annotation tool, revealing that they were mainly annotated for cell proliferation and for cell movement ontologies again suggesting that YAP1 overexpression positively sustains these two biological functions (Figure 3b and Supplemental Table 3), similar to the global 707 gene-list.
The validation of individual YAP1 target genes identified by gene expression analysis was performed using qPCR on independent silencing experiments. We analyzed the modulation of CTGF gene [37], which represents one of the few known YAP1 target genes, a group of genes involved in cell cycle regulation (CCNA2, CCNG1, CDK2, SKP2) [38-42]; the ITGA5 [43] and NRG1 genes involved in cell-cell interaction; the genes LATS2, NF2, STK3, STK4 and TEAD2, which are components of the Hippo pathway [44]; GADD45A gene which is involved in DNA repair [45]; VEGFA gene involved in angiogenesis [46] and ETS1 which is a transcription factor that acts as an oncogene via multiple pathways [47]. The genes modulated in agreement with the microarray data were 13 out of 16 in Ca-Ski and RO82 cell lines and 15 out of 16 in EKVX cell line (Figure 3c). Among them, the SKP2 protein was validated by western blotting on Ca-Ski, EKVX and RO82 cell lines revealing that protein levels are modulated in agreement with the microarray and qPCR results (Figure 3d).
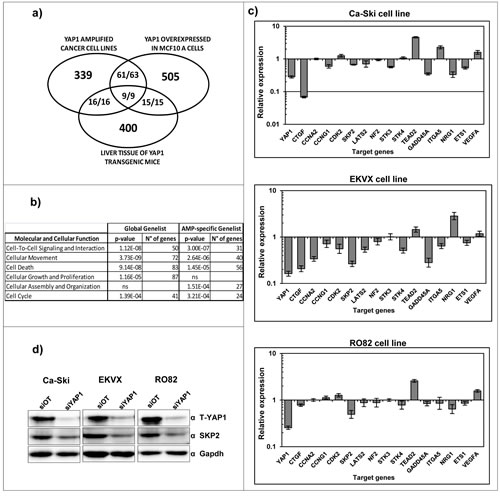
Figure 3: Identification of YAP1 target genes in 11q22-amplified cancer cell lines. a) Number of unique and common target genes identified in YAP1 amplified cell lines following downmodulation of endogenous YAP1 expression (Supplemental Table 2) and in normal cell contexts in which YAP1 gene is overexpressed. The fraction numerators indicate the number of genes which are concordantly co-regulated in the studies. b) Molecular function pathways significantly modulated following downmodulation of endogenous YAP1 expression. c) qPCR analysis showing the modulation of YAP1 target genes in Ca-Ski, EKVX and RO82 cell lines after YAP1 silencing using siRNAs. The siRNA off target was used as a calibrator. d) Total YAP1 and SKP2 protein levels were analyzed by western blotting analysis in Ca-Ski siOT and siYAP1 cells. GAPDH was used as loading control.
YAP1 downregulation induces a moderate reduction of cell proliferation and influences the cell cycle.
Uncontrolled, aberrant cell proliferation is a common characteristic of aggressive cancer cells [48]. Therefore, in order to evaluate whether YAP1 sustains the cell proliferation, as suggested by gene expression analyses, we investigated cell growth dynamics in 11q22-amplified cancer cell lines, using the bulk cell populations following sh-mediated stable silencing. As detected by both SRB assay and cell counting, YAP1 silencing induced a moderate reduction in cell proliferation in all cell lines analyzed, which is statistically significant in Ca-Ski cell line (Figure 4a). Concordantly, YAP1 silencing correlated with a reduction in viable cell counting without induction of cell death (Figure 4b). In addition, YAP1 silencing induced a modest reduction of the proliferative cell compartment of cells in S phase of the cell cycle, as demonstrated by BrdU incorporation assay (Figure 4c). Notably, we detected a significant increase of the protein levels of p21 and p27, which are implicated in the negative regulation of cell-cycle [49, 50], in YAP1 silenced cells compared to the off target cells in all YAP1-amplified cancer cell lines (Figure 4d).
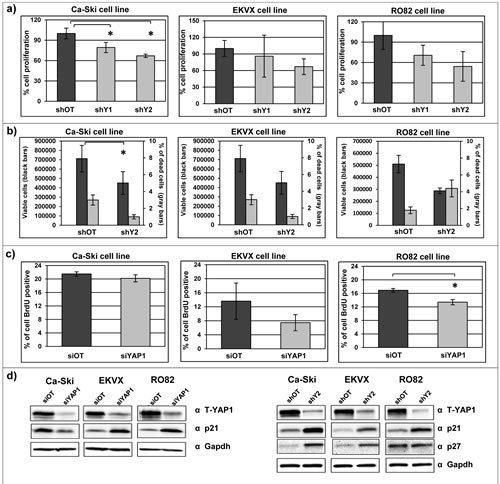
Figure 4: YAP1 downregulation induces a moderate reduction of cell proliferation and perturbation of the cell cycle. a) Sulforhodamine B assay was performed on Ca-Ski, EKVX and RO82 cell lines silenced for YAP1 using stable silencing system. The percentage of cell proliferation was evaluated 72 hours post seeding. Means+ SEM from three independent experiments. In Ca-Ski cell line paired t-test shOT versus shY1 p=0.0124, shOT versus shY2 p=0.0157. b) Cell counting and trypan blue staining was performed on Ca-Ski, EKVX and RO82 cell lines silenced for YAP1 using stable YAP1 silencing system (shY2). The count of viable cells (left y axis) and the percentage of the dead cells (right y axis) were determined 72 hours after cells seeding. Means+ SEM from three independent experiments. In Ca-Ski cell line unpaired t-test shOT versus shY2 p=0.0079. c) BrdU assay was performed on Ca-Ski, EKVX and RO82 cell lines. The histograms showed the BrdU positive cells in siOT and in siYAP1 cells. Mean percentage+ SEM from three independent experiments. In RO82 cell line paired t-test siOT versus siYAP1 p=0.0215. d) p21 and p27 protein expression levels were analyzed by western blotting on Ca-Ski, EKVX and RO82 YAP1 silencing cells (siYAP1 and shY2) and off target cells (siOT and shOT). GAPDH was used as loading control.
YAP1 downregulation strongly reduces the tumorigenic phenotype.
Normal cells typically are not able to grow and form cell colonies in semisolid media because under these conditions they undergo anoikis-mediated cell death. In contrast, aggressive cancer cells acquire the capability to grow without anchorage-dependence to the substrate, within a semi-solid medium such as agar [51]. In order to evaluate whether YAP1 positively affected the anchorage-independent growth we evaluated the colonies growth in off target cells and in YAP1 stably silenced cells. YAP1 silencing induced a strong and significant reduction in the number and size of colonies counted in all 11q22-amplified cancer cell lines analyzed, compared to the control cells (Figure 5a). Furthermore, subcutaneous injection of stably infected Ca-Ski sh-off target and Ca-Ski sh-YAP1 cells into nude mice showed a significant reduction in tumor volume in YAP1 silencing cells, providing a direct evidence that reduction of anchorage-independent growth correlated with significantly reduced in vivo cell expansion of cancer cells (Figure 5b).
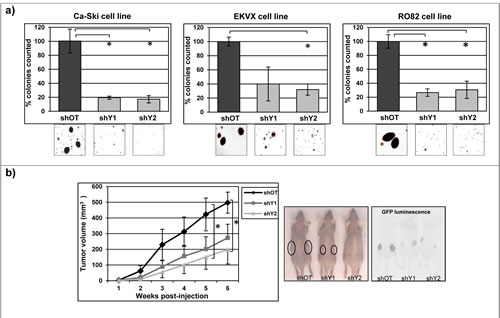
Figure 5: YAP1 downregulation strongly reduces tumorigenic phenotype. a) Soft agar assay was performed on Ca-Ski, EKVX and RO82 cell lines silenced for YAP1 using stable sh-silencing system. Colonies were counted from 60 mm dishes and one randomly chosen field was photographed (10x magnification). The values correspond to the mean percentage + SEM from three independent experiments. In Ca-Ski cell line unpaired t-test shOT versus shY1 and shY2 p=0.009, in EKVX cell line unpaired t-test shOT versus shY2 p=0.022 and in RO82 cell line unpaired t-test shOT versus shY1 p=0.003, shOT versus shY2 p=0.012. b) In vivo mice tumor growth was analyzed on Ca-Ski cell line off target cells (shOT) and YAP1 silenced cells (shY1 and shY2). The values correspond to the means + SEM from three independent experiments (12 mice per condition in total). Two-way ANOVA shOT versus shY1 and shY2 p=0.041 and p=0.027. A representative picture (left) and GFP luminescence (right) of tumors grown in mice 6 weeks after cells injection.
YAP1 silencing affects cell migration in Ca-Ski and in RO82 cell lines.
Active migration of tumor cells is a prerequisite for tumor cell invasion and for metastasis development [52] and our gene expression analyses indicated that YAP1 modulates several target genes that are reported to control cell-cell contact and cell migration. To investigate if migration activity is effectively affected by the modulation of YAP1 expression, cell tracking analysis experiments using video time-lapse microscopy were performed in Ca-Ski and RO82 cell lines, while EKVX parental cell line was uninformative as it is characterized by very limited migration ability (not shown). In Ca-Ski and RO82 cell lines the short-term YAP1 downregulation produced a moderate reduction of cell migration as measured by the distance covered by cells during 12 hours of observation, compared to the control off-target cells (Figure 6a). The phenomenon was qualitatively evident in RO82 cell line also by wound healing assay (Figure 6b), further supporting that the cell motility is reduced in YAP1 silenced cells. The same results were obtained using either siRNA (Figure 6a-b) or shRNA (data not shown) cells. Similarly, cell migration was reduced after short term YAP1 silencing in chemotaxis assay (Figure 6c).
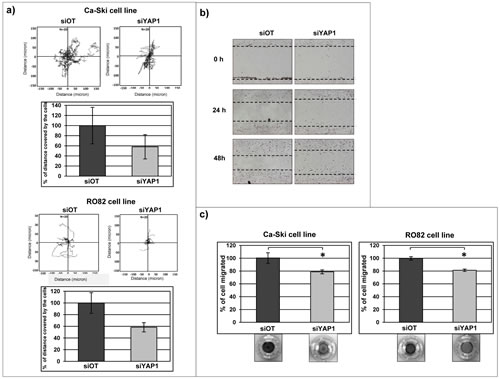
Figure 6: YAP1 silencing affects cell migration in Ca-Ski and in RO82 cell lines. a) Tracking experiment using time-lapse microscopy was performed on Ca-Ski and RO82 cells transfected with siRNA. The graphs show the distance covered by cells (micron) in 12 hours. The values in the histograms correspond to the percentage calculate on the means + SEM from three independent experiments. b) Wound healing assay was performed on Ca-Ski and RO82 cell lines depleted for YAP1 using siRNA silencing systems. The pictures show representative results. The T0 represents the cells 24 hours post-transfection (10x magnification). b) Chemotaxis experiment was performed on Ca-Ski and RO82 siOT and siYAP1 cells. The pictures show the cells migrated in low chamber of transwell stained with crystal violet. The values correspond to the means from one representative experiment out of three. In Ca-Ski cell line paired t-test siOT versus siYAP1 p=0.0487, in RO82 cell line paired t-test siOT versus siYAP1 p=0.0287.
YAP1 silencing increases DNA damage response in Ca-Ski cell line.
In Ca-Ski cell line, YAP1-silenced cells are more sensitive to genotoxic stress induced by cisplatin treatment, compared to control cells (Figure 7a). We detected a statistically significant increase in annexin V and 7AAD positivity in YAP1 silenced cells compared to the off target cells, indicating that in these cells the DNA damage induced a more prominent reduction of viability and increased cell death (Figure 7b). Concordantly, following DNA damage, we detected an increase in caspase 8 and PARP protein activation, which are essential mediators of the apoptosis process, in Ca-Ski YAP1 silenced cells compared to the control cells (Figure 7c). To a lesser extent, cisplatin induced an analogous effect also in RO82 and EKVX cell lines (Supplemental Figure 2).
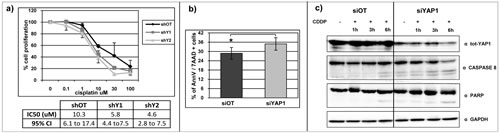
Figure 7: YAP1 silencing increases DNA damage response in Ca-Ski cell line. a) Percentage of cell proliferation reduction determined by SRB assay in Ca-Ski off target and YAP1-silenced cells after cisplatin treatment. The table shows the values of IC50 and the 95% confidence interval. b) Flow cytometer analysis was performed on Ca-Ski off target cells and YAP1 silenced cells with siRNA silencing cell system. The histograms showed the the mean + SEM percentage of single or double annexin V and 7AAD positivity cells after cisplatin treatment. Paired t-test siOT versus siYAP1 p=0.0107. c) Total YAP1, caspase 8 and PARP protein levels were analyzed by western blotting analysis in Ca-Ski siOT and siYAP1 cells 1, 3 and 6 hours after treatment with 100 uM cisplatin. GAPDH was used as loading control.
DISCUSSION
In this study we demonstrate that YAP1 effectively supports multiple transformed properties in 11q22-amplified cancer cell lines. In particular, in our experiments, we exploited RNA interference strategies to silence YAP1 in cancer cell lines (Ca-Ski, EKVX and RO82) that carried 11q22 copy number amplification and YAP1 overexpression and compared YAP1 silenced cells and control cells for gene expression profiling, cell proliferation tumorigenic potential, cell migration and response to genotoxic stress.
Literature data report apparently conflicting results on YAP1 role in carcinogenesis. In fact, YAP1 has been reported to exhibit both oncogenic properties [12-15] and tumor-suppressive functions [7, 9, 10] in distinct in vitro or in vivo models. Our results provide definitive evidence that endogenous YAP1 expression has oncogenic properties in cancer cells carrying YAP1 gene amplification.
Several studies reported that the 11q22 locus is amplified in human tumor samples [12, 17-29]. The majority of the data published until now detected 11q22 amplification in different tumor types without directly investigating the YAP1 copy alteration. We here provided direct evidence of YAP1 copy amplification in up to 23% of samples from multiple tumor types, providing compelling evidence that it represents a cancer associated alteration in a fraction of multiple tumor types. In particular, we detected YAP1 copy amplification in 4/25 (16%) of cervical cancer samples. Literature data reported 11q22 copy number amplification in cervical cancer samples [19, 25, 29], predominantly in patients with advanced stage of the disease [25]. These evidences, together with our data directly addressing YAP1 copy amplification, support that YAP1 is the target of the amplification at 11q22 locus in cervical carcinoma. Whether YAP1 copy amplification event represents a marker of cervical cancer progression and prognosis remains an intriguing but unexplored issue.
Genes mapping within amplicons are considered as candidate oncogenes. Amplification is an important mechanism for gene copy number gain, for protein overexpression and sustained oncogene activation in tumor cells. Most amplification events include a relatively large region of DNA that contains several genes that can be considered possible targets of the amplification event. The 11q22 amplicons include, in addition to YAP1, several genes which could be considered as possible candidate oncogenes such as BIRC2 (cIAP1) and BIRC3 (cIAP2) encoding apoptosis inhibitor proteins. Evidences reported that BIRC2 cooperates with YAP1 to accelerate tumorigenesis and promote mouse liver carcinogenesis [12]. In addition, several members of MMP gene family, known to be involved in extracellular matrix remodeling, in cell invasion and in metastasis development [53], have been proposed as oncogene targets of the 11q22 amplicon [54, 55]. Finally, progesterone receptor (PGR) is only 1 Mb proximal to YAP1 and its co-amplification may play a role in female cancers. Therefore, the combined oncogenic role of co-amplified genes within 11q22 cannot be ruled out and remains a complex and fascinating issue to be explored.
Interestingly, we found that five of the significantly modulated genes identified as targets of YAP1 are components of Hippo pathway (STK3/Mst2, STK4/Mst1, LATS2, NF2 and TEAD2). Specifically, TEAD2, which is a transcription factor that interacts with YAP1 to promote the expression of the Hippo responsive genes [56] results to be upregulated after YAP1 silencing. All the other modulated genes represent negative growth regulators acting in a kinase cascade that ultimately inactivates YAP1 and we revealed that they are downregulated upon YAP1 silencing. Therefore, considering that Hippo pathway is a cascade of phosphorylation events which inhibits YAP1, we can speculate that in YAP1-amplified cancer cell lines the transient YAP1 downregulation, by mimicking an over-activation of Hippo pathway, induces a feedback loop function on Hippo pathway components, that tends to counterbalance the YAP1 silencing.
In conclusion, our results demonstrate that in the YAP1-amplified cancer cell lines under study the YAP1 gene effectively sustains multiple transformed traits, indicating that YAP1 is a direct oncogenic target of the 11q22 amplicons. In addition, we directly detected that YAP1 amplification is actually present in a variable but significant fraction of carcinoma subtypes.
MATERIALS AND METHODS
Cancer cell lines.
Ca-Ski and EKVX cell lines were purchased from ATCC, RO82 cell line from Interlab Cell Line Collection, Genova, Italy (ICLC). Ca-Ski and EKVX were grown in RPMI 1640 (Sigma) supplemented with 10% heat-inactivated FBS (Lonza), RO82 cell line was grown in DMEM (Sigma), Ham’s F12 (Lonza), MCDB 105 (Sigma) (2:1:1) supplemented with 10% heat-inactivated FBS (Lonza), in a humidified incubator at 37°C and 5% CO2 (Thermo Electron Corporation). Cell identity was monitored by microsatellite typing and absence of mycoplasma contamination was checked regularly.
For stable YAP1 silencing experiments, short-hairpin RNA (shRNA) expression vectors were used targeting YAP1 (shY1 SH-012200-02-10, shY2 SH-012200-03-10, Thermo Scientific) or an off-target sequence (shOT, s-004000-02, Thermo Scientific) as negative control. Cells were infected with MOI 5-10 in the presence of Polybrene (4 ug/mL), cells were selected using puromycin 72 hours post infection and bulk cell population was collected and screened for YAP1 expression.
For transient YAP1 silencing, cells were transfected using the Dharmafect Transfection Reagent (Thermo Scientific) and small-interfering RNAs (siRNA) targeting YAP1 (siYAP1) (AM16708A-107951, AM16708A-107952, AM16708A-114602, Ambion) and an off-target sequence as negative control (siOT, AM4611 Ambion).
In order to establish Ca-Ski xenograft, cells (106) were injected subcutaneously into the each flank of six-week-old immunocompromised athymic nude mice (Harlan). Tumor size was monitored weekly. Tumor volume was calculated using the formula 1/2r3. Animal experimentation was approved by Institutional IRB and performed according to National laws. The data showed the means and the s.e.m of three independent experiments, each composed of 4 mice per experimental condition. Two classes’ comparisons with multiple measurement points have been performed by two-way analysis of variance.
Tumor samples.
Paraffin sections of 21 human cervical cancer samples, 15 human thyroid cancer samples, 56 human CSN tumor samples and of 77 human non small cell lung cancer samples were obtained from the Departments of Pathology of collaborating centers (Treviso General Hospital, Italy; “Fondazione Pascale” of Naples, Italy; “Fondazione Istituto Nazionale Tumori” of Milan, Italy).
Protein expression.
For western blotting, 40 ug of protein lysates were used as described [57]. Antibodies used and conditions are listed in Supplemental Table 4. Protein expression was analyzed using Odyssey infrared imaging system (Li-Cor) and the data were normalized on siOT and on shOT. Immunohistochemistry was performed using anti-YAP1 antibody diluted 1:25 (Cell Signaling, 4912) using Ultra vision detection system (LabVision), upon heat-induced epitope retrieval. Endogenous peroxidase was blocked with 0.3% hydrogen peroxide in methanol for 30 minutes. Intensity scoring of tumor cells was performed by pathologist based on a 4-tiered scale. The comparisons between two classes were performed by Fisher two tailed extract test by grouping negative/1+ cases and 2+/3+ cases.
Anchorage-independent growth.
Cells (105) were resuspended in 0.35% agar complete medium and seeded on 0.5% bottom agar medium in 60 mm dish. After two weeks, plates were stained with iodonitrotetrazolium violet (1mg/ml, Sigma) and clones were counted at the microscope (Olympus). The data showed the means and the s.e.m of three independent experiments. The comparisons between two classes were performed by two sample unpaired t-test.
Cell proliferation assay.
Cell proliferation was assessed by cell counting using an automated cell counter (Bigital Bio) and by Sulforhodamine B (SRB) assay as described [58]. The comparisons between two classes were performed by two-sample unpaired t-test. Cisplatin (Teva) was added 24 hours post- plating at the indicated concentrations. The data showed the means and the s.e.m of three independent experiments. The comparisons between two classes were performed by two-sample paired t-test. The IC50 (concentration of drug inhibiting 50% of cell growth) was calculated applying non-linear fitting dose response.
Annexin V evaluation.
Cells (1x105) were plated in 6-well plate and treated with cisplatin (Teva) 100 uM for 16 hours. The data were acquired and analyzed using Guava instrument (Millipore). The data showed the means and the s.e.m of three independent experiments. The comparisons between two classes were performed by two-sample paired t-test.
Bromodeoxyuridine (BrdU) assay.
BrdU Flow Kit (BD Pharmingen) was used to quantify cells that were actively synthesizing DNA. Cells were pulsed with 1 (Ca-Ski and RO82) or 2 (EKVX) hours of BrdU incorporation, washed and released for additional 1 hour in complete medium. Stained cells were measured and analyzed with flow cytometer (Beckman Coulter). The data showed the means and the s.e.m of three independent experiments. The comparisons between two classes were performed by two-sample paired t-test.
Fluorescent in situ hybridization (FISH).
FISH analyses were performed on cells smeared over a positively charged slide and on human non small cell lung cancer samples that were arrayed on a tissue microarray. BAC DNA probes used for FISH were extracted using QIAprep Midiprep (Qiagen), verified by sequence tagged site content mapping and labeled with Spectrum Orange deoxyuridine triphosphate or Spectrum Green deoxyuridine triphosphate (Vysis, Downers Grove, IL) by use of a Nick Translation Kit (Vysis) according to the manufacturer’s instructions. The FISH hybridization signals were analyzed in an Olympus BX51 microscope coupled to a charge-coupled device camera COHU 4912 (Olympus). The images captured were analyzed using the Mac Probe software (PowerGene Olympus). A minimum of 100 nuclei were counted.
Real-time PCR.
Total RNA was extracted using EZ1 RNA cell mini kit (Qiagen) on a Bio-Robot EZ1 (Qiagen) according to the manufacturer’s instructions and 500 ng of RNA was retro-transcribed using Super Script III RT (Invitrogen) with random primers. Relative quantification of gene expression was performed in triplicate using TaqMan assays on Demand on a ABI Prism 7900HT Sequence Detection System (Applera) by comparative Ct method, using the HPRT gene (HPRT PDAR, 4326321E) (Applied Biosystem) as endogenous reference control and shOT and siOT cells as calibrators. The list of genes investigated and primer sets used is in Supplemental Table 4. Quantitative PCR was also used to determine YAP1 DNA copy number status using relative quantification method with standard curves. The RNaseP gene (RNaseP, 4401631 Applied Biosystem) was used as a reference gene. Real time PCR data were analyzed by the SDS software 2.3, in order to obtain the Ct and the standard deviation. In each experiment, standard curves were analyzed for both reference gene (RNaseP) and target gene using serial diluitions of a normal control DNA. Using the standard curves of reference gene and target gene, the mean Ct of individual samples were converted to nanogram equivalent of template DNA. The obtained values were converted to target gene copy number by normalizing over a normal donor DNA as calibrator.
Wound healing assay.
Exponentially growing cells (8x105) were plated in 60 mm dish. Cell monolayer was scratched using a pipette tip. Complete medium was added after washing in PBS, wound closure was monitored every 24 hours and pictures were taken by bright-field microscopy (Olympus).
Time-lapse video microscopy.
Cells (3×103) were plated in 24-well plate. After overnight cell adhesion, cells were incubated at 37°C in 5% CO2 atmosphere in the Leica Time Lapse AF6000LX workstation equipped with the Leica DMI 6000 motorized microscope and an environmental chamber for the proper setting of temperature humidity and CO2 concentration. The allows the acquisition of the Images were collected every 5 minutes for 12 hours using AF6000 Software (Leica) and were analyzed with IM2000 software (Leica), to obtain the total distance covered (micron) by the cells. The data showed the means and the s.e.m of three independent experiments. The comparisons between two classes were performed by two-sample paired t-test.
Chemotaxis experiments.
Transwell permeable supports, 6.5 mm diameter inserts, 8.0 um pore size, polycarbonate membranes (Corning Inc.) were used to perform migration assay. Cells (105) were seeded in the upper chamber of the transwell insert in serum-free medium (Sigma). The lower chamber of the transwell was filled with 600 uL of culture medium containing 10% of fetal bovine serum. Cells were incubated at 37°C and 5% CO2 for 16 h, then transwells were removed and stained with 0.1% Crystal Violet (Sigma) in 25% methanol. Non-migrated cells were scraped off the top of the transwell with a cotton swab. Migrated cells were quantified by eluting crystal violet with 1% SDS and reading the absorbance at 550 nm using the microplate reader Infinite 200 (TECAN). The data showed the means and the s.e.m from one representative experiment out of three. The comparisons between two classes were performed by two-sample paired t-test.
Bioinformatic analyses.
YAP1 target genes in 11q22-amplified cancer cell lines were analyzed by gene expression profiling experiments. Class comparison analysis performed using the BRB Array Tool software from the US National Cancer Institute (linus.nci.nih.gov/BRBArrayTools.html) and comparing YAP1 proficient cells versus YAP1-silenced cells. Differential expression levels of individual probe sets were considered statistically significant if p <0.001 by random variance t tests. A multivariate permutation test was applied to provide 90% confidence that the false discovery rate was less than 10%. Such stringent significance threshold was used to limit the number of false-positive findings. The statistically significantly modulated genes identified were functionally annotated using Ingenuity bioinformatic tool.
The analysis of YAP1 copy number (deletion or amplification) status in independent datasets was performed using Oncomine (www.oncomine.org) and Sanger Centre (www.sanger.ac.uk/genetics/CGP/CellLines) database repositories, two cancer microarray databases which contain gene copy number data collected from the literature and proprietary datasets. The comparison between YAP1 deletion and amplification was performed in order to test the null hypothesis that the two anomalies are expected at the same frequency by Chi-square test.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We thank Dr. Gustavo Baldassarre, Aviano National Cancer Institute, Italy for assistance in video time lapse microscopy. We acknowledge the technical support by Luca Meldoli in flow cytometry experiments.
Funding
We acknowledge the unrestricted grant support of Associazione Italiana per la Ricerca sul cancro (AIRC) to PM.
REFERENCES
1. Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development. 2011; 138:9-22.
2. Edgar BA. From cell structure to transcription: Hippo forges a new path. Cell 2006; 124:267-273.
3. Yu FX, Guan KL. The Hippo pathway: regulators and regulations. Genes Dev. 2013; 27:355-371.
4. Pan D. The hippo signaling pathway in development and cancer. Dev. Cell. 2010; 19:491-505.
5. Sudol M, Bork P, Einbond A, Kastury K, Druck T, Negrini M, Huebner K, Lehman D. Characterization of the mammalian YAP (Yes-associated protein) gene and its role in defining a novel protein module, the WW domain. J. Biol. Chem. 1995; 270:14733-14741.
6. Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A, Oren M, Sudol M, Cesareni G, Blandino G. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J. Biol. Chem. 2001; 276:15164-15173.
7. Strano S, Monti O, Pediconi N, Baccarini A, Fontemaggi G, Lapi E, Mantovani F, Damalas A, Citro G, Sacchi A, Del Sal G, Levrero M, Blandino G. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA Damage. Mol. Cell 2005; 18:447-459.
8. Levy D, Reuven N, Shaul Y. A regulatory circuit controlling Itch-mediated p73 degradation by Runx. J. Biol. Chem. 2008; 283:27462-27468.
9. Basu S, Totty NF, Irwin MS, Sudol M, Downward J. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 2003; 11:11-23.
10. Matallanas D, Romano D, Yee K, Meissl K, Kucerova L, Piazzolla D, Baccarini M, Vass JK, Kolch W, O’neill E. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 2007; 27:962-975.
11. Yuan M, Tomlinson V, Lara R, Holliday D, Chelala C, Harada T, Gangeswaran R, Manson-Bishop C, Smith P, Danovi SA, Pardo O, Crook T, Mein CA, Lemoine NR, Jones LJ, Basu S. Yes-associated protein (YAP) functions as a tumor suppressor in breast. Cell Death Differ. 2008; 15:1752-1759.
12. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, Fan ST, Luk JM, Wigler M, Hannon GJ, Mu D, Lucito R, Powers S, Lowe SW. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006; 125:1253-1267.
13. Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, Deng CX, Brugge JS, Haber DA.. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc. Natl. Acad. Sci. U. S. A. 2006; 103:12405-12410.
14. Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 2007; 17:2054-2060.
15. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007; 130:1120-1133.
16. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, Daidone MG, Dupont S, Basso G, Bicciato S, Piccolo S. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011; 147:759-772.
17. Reardon DA, Michalkiewicz E, Boyett JM, Sublett JE, Entrekin RE, Ragsdale ST, Valentine MB, Behm FG, Li H, Heideman RL, Kun LE, Shapiro DN, Look AT. Extensive genomic abnormalities in childhood medulloblastoma by comparative genomic hybridization. Cancer Res. 1997; 57:4042-4047.
18. Imoto I, Yang ZQ, Pimkhaokham A, Tsuda H, Shimada Y, Imamura M, Ohki M, Inazawa J. Identification of cIAP1 as a candidate target gene within an amplicon at 11q22 in esophageal squamous cell carcinomas. Cancer Res. 2001; 61:6629-6634.
19. Imoto I, Tsuda H, Hirasawa A, Miura M, Sakamoto M, Hirohashi S, Inazawa J. Expression of cIAP1, a target for 11q22 amplification, correlates with resistance of cervical cancers to radiotherapy. Cancer Res. 2002; 62:4860-4866.
20. Dai Z, Zhu WG, Morrison CD, Brena RM, Smiraglia DJ, Raval A, Wu YZ, Rush LJ, Ross P, Molina JR, Otterson GA, Plass C. A comprehensive search for DNA amplification in lung cancer identifies inhibitors of apoptosis cIAP1 and cIAP2 as candidate oncogenes. Hum. Mol. Genet. 2003; 12:791-801.
21. Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RC, Albertson DG. Rare amplicons implicate frequent deregulation of cell fate specification pathways in oral squamous cell carcinoma. Oncogene. 2005; 24:4232-4242.
22. Baldwin C, Garnis C, Zhang L, Rosin MP, Lam WL. Multiple microalterations detected at high frequency in oral cancer. Cancer Res. 2005; 65:7561-7567.
23. Hermsen M, Alonso Guervós M, Meijer G, van Diest P, Suárez Nieto C, Marcos CA, Sampedro A. Chromosomal changes in relation to clinical outcome in larynx and pharynx squamous cell carcinoma. Cell. Oncol. 2005; 27:191-198.
24. Modena P, Lualdi E, Facchinetti F, Veltman J, Reid JF, Minardi S, Janssen I, Giangaspero F, Forni M, Finocchiaro G, Genitori L, Giordano F, Riccardi R, Schoenmakers EF, Massimino M, Sozzi G. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J. Clin. Oncol. 2006; 24:5223-5233.
25. Scotto L, Narayan G, Nandula SV, Arias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M, Murty VV. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008; 47:755-765.
26. Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, Yatabe Y, Hasegawa Y, Shimokata K, Horio Y, Hida T, Sekido Y. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis. 2008; 29:2139-2146.
27. Fernandez-L A, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, Taylor MD, Kenney AM. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev. 2009; 23:2729-2741.
28. Hélias-Rodzewicz Z, Pérot G, Chibon F, Ferreira C, Lagarde P, Terrier P, Coindre JM, Aurias A. YAP1 and VGLL3, encoding two cofactors of TEAD transcription factors, are amplified and overexpressed in a subset of soft tissue sarcomas. Genes Chromosomes Cancer. 2010; 49:1161-1171.
29. Choschzick M, Tabibzada AM, Gieseking F, Woelber L, Jaenicke F, Sauter G, Simon R. BIRC2 amplification in squamous cell carcinomas of the uterine cervix. Virchows Arch. 2012; 461:123-128.
30. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007; 8:221-233.
31. LaCasse EC, Baird S, Korneluk RG, MacKenzie AE. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene. 1998; 17:3247-3259.
32. Gadkar-Sable S, Shah C, Rosario G, Sachdeva G, Puri C. Progesterone receptors: various forms and functions in reproductive tissues. Front. Biosci. 2005; 10:2118-2130.
33. Betticher DC, Heighway J, Hasleton PS, Altermatt HJ, Ryder WD, Cerny T, Thatcher N. Prognostic significance of CCND1 (cyclin D1) overexpression in primary resected non-small-cell lung cancer. Br. J. Cancer 1996; 73:294-300.
34. Zhang YJ, Jiang W, Chen CJ, Lee CS, Kahn SM, Santella RM, Weinstein IB. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 1993; 196:1010-1016.
35. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999; 18:2551-2562.
36. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, Halder G, Lai ZC, Guan KL. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007; 21:2747-2761.
37. Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J. Cell Biol. 1991; 114:1285-1294.
38. Aleem E, Berthet C, Kaldis P. Cdk2 as a master of S phase entry: fact or fake? Cell. Cycle. 2004; 3:35-37.
39. Bates S, Rowan S, Vousden KH. Characterisation of human cyclin G1 and G2: DNA damage inducible genes. Oncogene. 1996; 13:1103-1109.
40. Demetrick DJ, Zhang H, Beach DH. Chromosomal mapping of the genes for the human CDK2/cyclin A-associated proteins p19 (SKP1A and SKP1B) and p45 (SKP2). Cytogenet. Cell Genet. 1996; 73:104-107.
41. Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992; 11:961-971.
42. Tsai LH, Harlow E, Meyerson M. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus E1A-associated p33 kinase. Nature. 1991; 353:174-177.
43. Virtanen I, Korhonen M, Kariniemi AL, Gould VE, Laitinen L, Ylänne J. Integrins in human cells and tumors. Cell Differ. Dev. 1990; 32:215-227.
44. Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR. Protein kinases of the Hippo pathway: regulation and substrates. Semin. Cell Dev. Biol. 2012; 23:770-784.
45. Hollander MC, Alamo I, Jackman J, Wang MG, McBride OW, Fornace AJ Jr. Analysis of the mammalian gadd45 gene and its response to DNA damage. J. Biol. Chem. 1993; 268:24385-24393.
46. Mattei MG, Borg JP, Rosnet O, Marme D, Birnbaum D. Assignment of vascular endothelial growth factor (VEGF) and placenta growth factor (PLGF) genes to human chromosome 6p12-p21 and 14q24-q31 regions, respectively. Genomics. 1996; 32:168-169.
47. Dittmer J. The biology of the Ets1 proto-oncogene. Mol. Cancer. 2003; 2:29.
48. Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001; 411:342-348.
49. Colozza M, Azambuja E, Cardoso F, Sotiriou C, Larsimont D, Piccart MJ. Proliferative markers as prognostic and predictive tools in early breast cancer: where are we now? Ann. Oncol. 2005; 16:1723-1739.
50. Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003; 13:65-70.
51. Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J. Cell. Sci. 2011; 124:3189-3197.
52. Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005; 17:559-564.
53. Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol. Cell. Biochem. 2003; 253:269-285.
54. Lockwood WW, Coe BP, Williams AC, MacAulay C, Lam WL. Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int. J. Cancer. 2007; 120:436-443.
55. Roman E, Meza-Zepeda LA, Kresse SH, Myklebost O, Vasstrand EN, Ibrahim SO. Chromosomal aberrations in head and neck squamous cell carcinomas in Norwegian and Sudanese populations by array comparative genomic hybridization. Oncol. Rep. 2008; 20:825-843.
56. Tian W, Yu J, Tomchick DR, Pan D, Luo X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. U. S. A. 2010; 107:7293-7298.
57. Brenca M, Rossi S, Lorenzetto E, Piccinin E, Piccinin S, Rossi FM, Giuliano A, Dei Tos AP, Maestro R, Modena P. SMARCB1/INI1 Genetic Inactivation Is Responsible for Tumorigenic Properties of Epithelioid Sarcoma Cell Line VAESBJ. Mol. Cancer. Ther. 2013; 12:1060-1072.
58. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006; 1:1112-1116.