
Introduction
Tumor heterogeneity has different meanings in different clinical disciplines. To the anatomical pathologist, it is related to observable histologic differences in regions of a tumor; to the molecular pathologist, it relates to mutational variation between cells of the same tumor; and to the medical oncologist, it is related to therapeutic response. Few cancers demonstrate tumor heterogeneity as clearly as lung adenocarcinoma. Morphologically, lung adenocarcinomas are typically heterogeneous, with in excess of 90% of resected tumors comprised of multiple histologic subtypes [1-3].
The existence of intratumoral mutational heterogeneity in lung adenocarcinoma has been debated since activating EGFR mutations were found to predict response to EGFR tyrosine kinase domain inhibitors. It is clinically evidenced after the selective pressure of effective chemotherapy or molecular targeted therapy when tumors later progress. Whilst Jiang et al reported evidence indicating EGFR mutational heterogeneity in at least one patient [4], Yatabe et al concluded that for EGFR mutations, this was likely to be non-existent, or at least exceedingly rare. The conclusions of Yatabe et al were based on multiple random samples from five EGFR mutant lung adenocarcinomas without reference to areas of variation in intratumoral morphology. Their hypothesis was that previously reported cases of intratumoral mutational heterogeneity were false negative results associated with low tumor purity, insensitive direct sequencing techniques and pseudo-heterogeneity caused by variation of mutant allele copy number across the tumor [5]. A subsequent Korean study has examined EGFR mutations in different subtypes of ten adenocarcinomas and found no evidence of heterogeneity in the two to three selectively dissected tissues from each tumor [6].
The concept of branching evolution, where all cells in a given tumor harbor common truncal mutations, but different parts of the tumor (or its metastases) develop diverging branch mutations, is now more or less axiomatic, courtesy of evidence from next generation sequencing [7]. However, a corresponding morphological phenotype has not been identified for these divergent genotypes.
In 2011, the International Association for the Study of Lung Cancer, the American Thoracic Society and the European Respiratory Society presented a thoroughly revised lung adenocarcinoma classification [3]. It recognizes the following common histologic patterns or subtypes - lepidic (cancer cells growing along but not invading through alveolar walls), acinar (glandular structures invading adjacent stroma), papillary (branching structures of fibrovascular stroma covered by invasive cancer cells), solid (sheets or nests of cancer cells containing cytoplasmic mucin) and the newly added micropapillary subtype (papillary tufts of tumor cells without fibrovascular cores either lying apparently free in alveolar spaces or surrounded by thin fibrous septa, often at a tumor’s edge). A single subtype in a tumor is rare, and any or all the above subtypes can be present in the same tumor. Up to now, there has been no understanding of the molecular basis, if any, of these subtypes.
A study by Sica et al suggested that the predominant histologic patterns present in resected lung adenocarcinomas could be equated with tumor grade (N.B. there has never been an internationally recognized grading system for resected lung adenocarcinoma). Based on metastatic potential they concluded that the lepidic pattern correlates with grade 1 (low metastatic potential); acinar and papillary patterns with grade 2 (moderate metastatic potential); and micropapillary and solid with mucin patterns with grade 3 (high metastatic potential) [8].
With its current model of companion testing of a genomic target and therapy with the corresponding inhibitor, effective precision medicine is dependent on finding any actionable mutation. This may be a negative or a positive predictor. In view of the prognostic influence of subtype on survival [1] and the possibility of corresponding molecular heterogeneity, we investigated whether the distinct histologic subtype domains in lung adenocarcinoma harbor important genomic differences.
Results
Mutation testing
Twenty-nine adenocarcinoma specimens were considered suitable for the study, having discrete regions of different subtypes that could be targeted by core punch for extraction of DNA. Two tumors were lepidic predominant, eleven were acinar predominant, three were papillary predominant, five were solid predominant and eight were micropapillary predominant. Twenty-six of the 29 tumors had higher grade micropapillary and/or solid components that could be cored for DNA extraction in addition to one or more lesser grade subtypes.
Tumors were screened for mutations in the hotspots of the EGFR, KRAS, BRAF and TP53 genes. In 24 of the tumors we identified mutations in one or more of the above genes; one of these being a case of pleomorphic carcinoma (50% giant cell/spindle cell, 30% adenocarcinoma, 20% squamous cell carcinoma) with an EGFR mutation and another a pleomorphic carcinoma comprising solid, micropapillary and giant cell components with heterozygous allelic KRAS mutations of codon 12. Five additional FFPE tumors had none of the common mutations on screening, but had well-demarcated domains of at least two histologic subtypes (table 1). A total of 63 histologic domains of differing subtypes were sampled.
Table 1:Morphologic and molecular characteristics of the 29 resected lung adenocarcinoma samples. *Bold type denotes samples with intratumoral mutational differences mapping to histologic subtypes of higher metastatic potential. #Denotes excess copy number of EFGR mutation present only in the micropapillary subtype. ϕDenotes pleomorphic component of tumor with giant cell and/or spindle cell morphology.
Patient Tumor Sample |
Subtypes screened |
EGFR |
KRAS |
BRAF |
1 |
Acinar Micropapillary |
- |
G12D G12D |
- |
2 |
Acinar Micropapillary |
Exon 19 del Exon 19 del |
- |
- |
3 |
Acinar Micropapillary |
Exon 19 del Exon 19 del |
- |
- |
4 |
Acinar Micropapillary |
Exon 19 del Exon 19 del |
- |
- |
5* |
Micropapillary Acinar |
- |
- |
V600E WT |
6 |
Acinar Solid Papillary |
- |
- |
- |
7 |
Acinar Lepidic |
- |
G12V G12V |
- |
8 |
Acinar Solid Papillary |
L858R L858R L858R |
- |
- |
9 |
Pleomorphicϕ Acinar Micropapillary |
L858R L858R L858R# |
- |
- |
10 |
Lepidic Micropapillary |
- |
- |
- |
11 |
Lepidic Micropapillary |
Exon 19 del Exon 19 del |
- |
- |
12 |
Lepidic Micropapillary |
- |
- |
- |
13 |
Lepidic Micropapillary |
L858R L858R |
- |
- |
14 |
Lepidic Papillary |
- |
G12C G12C |
- |
15* |
Lepidic Acinar |
- |
WT G12C |
WT K601N |
16 |
Papillary Micropapillary |
Exon 19 del Exon 19 del |
- |
- |
17 |
Papillary Micropapillary |
Exon 20 ins Exon 20 ins |
- |
- |
18 |
Papillary Micropapillary |
- |
- |
- |
19* |
Papillary Micropapillary Solid |
- |
WT WT G12S |
- |
20 |
Papillary Micropapillary |
- |
- |
G469V G469V |
21* |
Papillary Micropapillary |
- |
WT G12V |
- |
22 |
Papillary Micropapillary |
- |
G13D G13D |
G464V G464V |
23 |
Solid Micropapillary |
- |
- |
- |
24 |
Solid Micropapillary |
- |
- |
- |
25 |
Solid Micropapillary |
- |
- |
- |
26 |
Solid, Micropapillary, Pleomorphicϕ |
- |
G12R/G12C G12R/G12C G12R/G12C |
- |
27 |
Solid Acinar |
- |
G12C G12C |
- |
28 |
Micropapillary Acinar |
- |
G12C G12C |
- |
29 |
Micropapillary Papillary |
- |
G12C G12C |
- |
All mutations identified by HRM, were confirmed by Sanger sequencing. Nine tumors were found to have EGFR mutations (five with exon 19 deletions, three with L858R point mutations and one with an exon 20 insertion). Two of the tumors with exon 19 deletions were co-mutated with TP53. Eleven tumors were found to have KRAS mutations (ten at codon 12 and one at codon 13) and two of these had co-mutations in BRAF (one exon 11 and one exon 15). A total of four tumors (including those co-mutated with KRAS) had BRAF mutations (G464V and G469V in exon 11, and V600E and K601N in exon 15). Five tumors were mutated in TP53 hotspots (all different), including the two co-mutated with KRAS and one co-mutated with BRAF V600E. Mutations in EGFR were mutually exclusive with mutations in KRAS or BRAF. Five tumors were wild type for the four genes tested.
Mutation status by histologic subtype domain
Out of the 24 tumors with at least one confirmed mutation, intratumoral mutational heterogeneity mapping to a specific histologic subtype was discovered in four cases. Whilst none of the nine EGFR mutated tumors were found to be heterogenous, three of eleven KRAS mutated tumors (27%) and two of four BRAF (50%) were. In all such cases the mutation was present in a higher grade subtype than the wild type component within the same tumor.
In the eleven tumors with known KRAS codon 12 or 13 mutations, three were wild type in the predominant subtype, but KRAS mutant in a different morphologic subtype of higher grade. The three tumors all had a point mutation in codon 12 (G12C, G12S and G12V). In one of the predominant papillary adenocarcinomas, the papillary and micropapillary subtypes were KRAS wild type, but a KRAS mutation was present in the solid with mucin subtype. This solid-with-mucin component was the subtype found to be invading both pleura and lympho-vascular spaces, despite being the minority pattern. The patient had pleural recurrence at 8 months and died 2 years later. A second patient with KRAS wild type papillary predominant tumor and KRAS mutant micropapillary subtype had no evidence of disease after 1 year of follow-up. The percentages of these two tumors comprised of the KRAS mutant subtype were in the range of 35-40%. The third such tumor with mutational heterogeneity was in a patient with a good prognosis lepidic predominant adenocarcinoma. This tumor was KRAS wild type in the lepidic subtype but KRAS mutant in the minority acinar subtype. There was also a co-mutation in BRAF (K601N) in the acinar component. This patient survived 10 years without evidence of relapse.
Of the four primary tumors with BRAF mutations, the tumor from one patient was wild type in the acinar pattern but mutant (V600E) in the higher grade predominant micropapillary pattern (fig. 1). The mutant micropapillary subtype was also the dominant pattern present in mediastinal lymph node metastases of this patient. Three areas from two separate lymph nodes were cored, and all were found to be BRAF mutant (micropapillary involvement was so widespread in the node that it was not possible to core any pure acinar or solid component). The patient relapsed with malignant pleural effusion two months later and died of metastatic disease 17 months later.
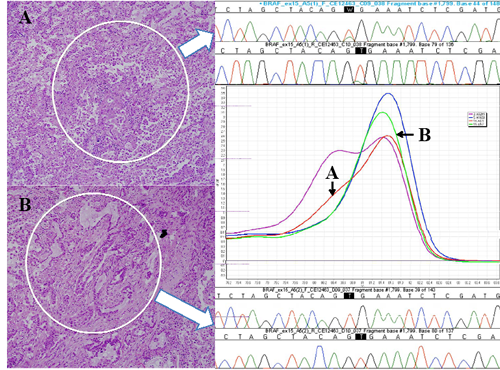
Fig 1: Photomicrographs of the cored areas marked with a dotted circle. A, Micropapillary pattern; B, Acinar pattern (H&E, x200 magnification). High resolution melting curves generated for BRAF wild type (green) and mutant (red) corresponding to subtype (arrowed). Sanger sequencing trace for each subtype is shown (arrowed). High resolution melting curves for positive control DNA from A02M1 V600E mutant melanoma cell line (magenta) and negative control DNA from H1650 BRAF wild type lung cancer cell line (blue) are also shown.
The primary tumor from a second patient was BRAF wild type in the predominant lepidic pattern, but mutant (K601N) only in the minority acinar pattern. This was the same tumor mentioned above with a KRAS G12V co-mutation in the minority acinar subtype (patient sample 15 in table 1) with the patient surviving 10 years without relapse.
The remaining two tumors had a BRAF mutation in exon 11 (G464V and G469V) present in all histologic subtypes tested.
Importantly, all nine tumors with previously identified EGFR mutations in exons 19, 20 or 21 had the relevant mutation identified in all histologic subtype domains tested, i.e. no intratumoral mutational heterogeneity was identified with respect to EGFR mutations consistent with an early origin of this mutation.
Mutant Allele-Specific Imbalance (MASI)
Whilst no differences were detected in EGFR mutations between histologic subtypes, a marked imbalance of copy number of the mutant allele was identified in one tumor (patient 9 in table 1). The mutant allele copy number, as assessed by chromogenic in situ hybridization, was very high in the micropapillary subtype, but normal in the acinar subtype and pleomorphic regions of the same tumor. No other instances of MASI were identified that mapped to histologic domains.
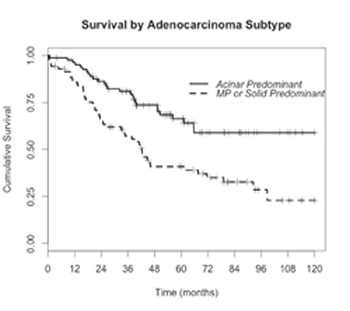
Fig 2: Kaplan-Meier curves (re-plotted from our own data in Russell et al, JTO 2011) showing the significantly poorer survival outcomes after surgical resection for micropapillary and solid predominant adenocarcinoma compared to the common acinar predominant. (Hazard ratio=2.34, p<0.0005, logrank test). MP=Micropapillary.
Discussion
Intratumoral mutational heterogeneity has been reported in renal cell carcinoma [11], colorectal carcinoma [12] and breast carcinoma [13]. However mutational heterogeneity mapping to a histologic phenotype has only been reported in uncommon biphasic lung cancers. In a study of pleomorphic carcinomas of the lung, one out of six cases had a KRAS mutation detected only in the adenocarcinoma component [14]. Pleomorphic carcinomas are uncommon biphasic tumors composed of an epithelial component (e.g. adenocarcinoma, squamous cell carcinoma or large cell carcinoma) associated with a spindle- and/or giant-cell component [15]. In another study of KRAS and EGFR mutations in adenosquamous lung carcinoma, one out of three tumors had an EGFR mutation detected only in the glandular (adeno) component and not in the squamous component [16]. This is not surprising as lung adenocarcinomas and lung squamous cell carcinomas have markedly different genomic and gene expression profiles [17,18]. In accordance with Swanton’s trunk and branch evolutionary model [7], the two histotypes could have diverged from their pluripotential stem cell origin by the acquisition of an EGFR mutation in one subclone (leading to invasive adenocarcinoma), but a different genomic event in the eventual squamous or pleomorphic component.
We have now provided evidence that intratumoral mutational heterogeneity exists in some lung adenocarcinomas, with additional mutations predictably mapping to a histologic subtype of higher metastatic potential. We propose that the acquisition of a KRAS or BRAF mutation may drive a more aggressive phenotype that is mirrored by a higher-grade histologic morphology. This could, in some cases, explain the histologic heterogeneity that typifies over 90% of lung adenocarcinoma cases, and the consistent observation that the grade of the predominant subtype is strongly associated with survival outcome (fig. 2) and tumor relapse [1-3,19,20]. KRAS mutations, found in about 30% of lung adenocarcinomas [21], are an important negative predictor for EGFR, ALK and ROS1 tyrosine kinase inhibitor therapy by virtue of their mutual exclusivity with genomic aberrations of these genes. Although tumor selection in this study was intentionally skewed to those with both mutations and multiple discrete subtype domains, we readily demonstrated intratumoral mutation heterogeneity in three out of eleven KRAS mutant tumors. Likewise BRAF mutational heterogeneity was found in two of the four tumors suitable for separate subtype sampling. In total, four of 14 tumors (29%) harboring either KRAS or BRAF mutations were found to have intratumoral mutational heterogeneity with mutations mapping to higher grade histologic subtype domains. Of particular interest is the case of BRAF V600E mutation found in micropapillary subtypes in the primary and metastatic nodes but not in the acinar subtype of the primary.
Even if there were very low levels of mutation below the sensitivity of our methodology, the result remains significant, as the tumor purity was high and, by selection and study design, roughly equivalent between the subtype samples from the same tumor. Our mutation screening identified the EGFR mutation in all morphologic patterns of all tumors tested, including a single case of pleomorphic carcinoma with MASI demonstrated on EGFR chromogenic in situ hybridization. This may be an alternative form of progression or resistance in such tumors.
We believe that these findings have far reaching biological and clinical implications not only for lung cancer, but also for tumors in general. Our results call into question the suggestion that KRAS and BRAF mutations are always early events in the evolution of lung adenocarcinoma, consistent with the conclusion of Sugio et al that KRAS can be a late event in the pathogenesis of lung cancer [22]. This is difficult to reconcile with the observation that atypical adenomatous hyperplasia (AAH), the presumed dysplastic precursor of adenocarcinoma, has a higher incidence of KRAS mutation than adenocarcinoma-in-situ and much higher than invasive adenocarcinoma [23]. This suggests that KRAS mutated AAH may not progress to adenocarcinoma in the same way proposed for EGFR mutated AAH [23-25], and that KRAS mutations occur in adenocarcinoma de novo via an alternative pathogenesis [26].
The results shown here are reminiscent of the tumor evolution model proposed by Fearon & Vogelstein [27] for colorectal cancer in which KRAS mutations were a later change in the tumor. Our results support this for some lung adenocarcinomas, particularly in the patient with lepidic predominant adenocarcinoma. A tumor of pure lepidic component is now considered adenocarcinoma-in-situ [3], so the fact that KRAS mutations were identified only in the minority invasive acinar component and not in the lepidic component suggests an evolutionary event in this patient.
Histologic subtype domains with aggressive phenotypes such as the micropapillary pattern are not limited to lung adenocarcinoma. They are also seen in breast [28,29], urothelial [30,31], gastric [32,33] and colorectal carcinoma [34,35]. Two studies reported differences in DNA ploidy in the micropapillary component of urothelial carcinoma [30,31]. That fact that the presence of even small foci of this subtype has been reported to have an association with metastatic potential and poor outcome could suggest that the association of more malignant morphological subtypes with additional genetic events found in this study may be generalizable to other solid tumors.
Another consideration raised by these findings is the sensitivity of clinical mutation screening. In the case of KRAS or BRAF mutant tumors, it may not just be admixture of stromal cells that causes a false negative result on mutation testing. Rather, it may be the presence of tumor cells of a lesser grade morphologic subtype all being wild type or diluting the mutant allele percentage below test sensitivity.
These findings underline the importance of the role of comprehensive histologic subtyping as precision medicine for lung cancer advances beyond its current focus on EGFR and ALK inhibition. There is increasing dependence on fine needle aspiration cytology and fine core biopsies to minimise patient discomfort. As we found that some of the mutant KRAS or BRAF domains comprise the minority of the primary tumor, this finding also casts some doubt as to whether small biopsies and cytological specimens from the primary tumor are sufficient to reliably detect mutations other than the ubiquitous EGFR.
Another possible implication for the future of precision medicine is that even if newer targeted therapy for BRAF or KRAS mutations [36] was successfully introduced for lung cancer, it may treat only the more aggressive subtype in some tumors. This could allow the formerly less aggressive phenotype to emerge and progress, or give the appearance of partial response despite excellent biological control of the life-threatening component. As a result of our findings, we would recommend formal incisional or excisional biopsies of primary tumors or sampling of metastases to ensure that all representative subtypes are included in mutation screening.
Our results give further support to evidence that activating EGFR mutations are an early genomic aberration in lung adenocarcinoma [5,6,37]. Isolated reports of intratumoral EGFR mutational heterogeneity often used less sensitive methods of detection or have been dismissed as MASI, i.e. heterogeneous distribution of mutant EGFR amplification within the tumor. One of the EGFR mutant tumors demonstrated marked MASI that mapped to the micropapillary subtype. The differential expansion of this clone may represent an escape mechanism from therapeutic tyrosine kinase inhibition. Sequencing of EGFR often shows an imbalance in favor of the mutant allele. Based on our results, clinicians can be confident, however, that even small biopsies submitted to HRM screening for EGFR mutation will result in appropriate initial therapy.
Lung adenocarcinoma is perhaps the archetype of the heterogeneous tumor. However our findings in lung cancer may have implications applicable to other solid tumors as molecular heterogeneity may not always be reflected by overt histological heterogeneity. As genomic biomarkers become increasingly linked to targeted therapies, the understanding of tumor heterogeneity will become ever more important.
Materials and Methods
Tumor Tissue Block Selection and Preparation
Four-micron sections were cut from formalin-fixed paraffin embedded tumor blocks of resected adenocarcinomas and stained with haematoxylin and eosin (H&E). Comprehensive histologic subtyping [3] was performed whereby the amount of each adenocarcinoma subtype present in a tumor was estimated in 5% increments followed by classification according to the predominant histologic subtype. Only tumors with discrete areas of at least 2mm diameter of separate subtypes were short-listed for the study so that it was possible to core each independent subtype with a 2mm biopsy punch. The sample set was enriched for mutations by (i) selecting cases that had previously been called as having mutations of EGFR, KRAS, BRAF and/or TP53 in the clinical or research setting and (ii) selecting tumors with a significant micropapillary subtype component.
The H&E stained sections were used to identify tumors demonstrating different, well-demarcated histologic patterns, with at least 50% tumor cell purity for each pattern. After matching and superimposing each H&E stained section on the glass slide to the surface of the corresponding FFPE tumor block, the specific subtypes of interest were separately sampled with 2mm diameter dermatology core punches. No attempt was made to further microdissect tumor cells, as the intention was to test the full histologic architecture of each particular subtype.
In all tumors, the predominant subtype and at least one other pattern were targeted. We therefore isolated multiple high purity samples of tumors with individual lepidic, papillary, acinar, solid with mucin or micropapillary morphology. Where present in a sufficient quantity, any giant cell or spindle cell patterns of pleomorphic carcinoma were also separately punched. Confirmation of consistent tumor morphology in the Z-plane was achieved by taking a section from the surface of the punched tumor block, staining it with H&E, followed by further cutting at least 1mm deeper into the block and taking another section for H&E staining.
Deparaffinization and DNA extraction
Using a blade, the punched tumor specimen was diced finely and placed in a screw-cap Eppendorf tube after removing excess paraffin from the underside of the core. Deparaffinization was achieved by treatment with 800 µL xylene, incubation for 5min, vortexing, incubation for 2 min then centrifuging for 1 min at 8000 rpm. This was repeated. Xylene was removed and samples were washed with 800 µL of 100% ethanol. This was centrifuged at 8000 rpm for 1 min and decanted. 800 µL of 70% ethanol was added. This was centrifuged at 8000 rpm for 1 min, with supernatant removed carefully by pipette. Tumor tissue was air-dried in the open on a heat block at 55ºC for 15 minutes.
DNA extraction was achieved using the Qiagen DNeasy Tissue and Blood kit following the manufacturer’s instructions with the following modifications: Samples were heated to 98ºC for 15 minutes then incubated at 56ºC for 3 days after addition of 36 µL of proteinase K (Worthington, NJ) at 20 mg/ml concentration.
The eluted DNA sample was stored at 4ºC and concentration was measured using a NanoDrop ND1000 spectrophotometer (Thermo Scientific). Working solutions of 5ng/μl were prepared for use in PCR and sequencing reactions.
Extracted DNA was amplified and scanned for mutations by high resolution melting (HRM) using the assay conditions previously described for KRAS and EGFR [9]. The region surrounding codon 600 in BRAF exon 15 was also screened by HRM using primers tagged with m13 sequences [10] and custom primers were constructed for BRAF exon 11 based on known hotspots using the following m13 tagged primers, generating a 143 bp amplicon; forward - 5’-tgtaaaacgacggccagtACTTGGTAGACGGGA CTCGAG-3’ and reverse - 5’-caggaaacagctatgaccTGTCACAATGTCACCA CATTACATAC-3’. BRAF exon 11 and exon 15 PCR products were directly used as templates for sequencing reaction. Mutations detected by HRM screening in EGFR exons 18-21, KRAS exon 2 and BRAF exons 11 and 15 were all confirmed by Sanger sequencing.
The St Vincent’s Hospital Human Research Ethics Committee approved this study (HREC-A 030/12).
Acknowledgments
We thank the Victorian Cancer Agency for funding patients’ screening for EGFR & KRAS mutations and ALK rearrangements with the aim of creating a translational research platform. We also thank Professor Bert Vogelstein and Professor David Thomas for their invaluable peer review and feedback. This study was supported by grants to GMW from the St Vincent’s Hospital Research Endowment Fund, the ANZCTS Foundation and donations of family and friends of S. O’Toole. This study was also supported by grants to AD from the National Health and Medical Research Council, Cancer Australia and the Cancer Council of Victoria. GMW was supported by NHMRC Scholarship GNT1038699 and University of Melbourne Gordon-Taylor Scholarship. HD was supported by a fellowship from the Cancer Council of Victoria. This research was supported in part by Operational Infrastructure Funding Program of the Victorian Government.
Statement of author contributions:
G.M.W conceived the project. G.M.W. and N.A. resected all lung adenocarcinomas, ordered mutation screening and, in collaboration with P.A.R., maintained clinical databases. G.M.W., H.D., J.W., T.J & M.W. performed experiments and data analysis. G.M.W., V.R. & P.A.R. selected tumors and identified morphological tumor subtypes for analysis. A.D. & H.D. advised on and supervised laboratory work. G.M.W. & A.D. wrote the manuscript with input from all authors.
References
1. Russell PA, Wainer Z, Wright GM, Daniels M, Conron M, Williams RA. Does lung adenocarcinoma subtype predict patient survival?: A clinicopathologic study based on the new International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary lung adenocarcinoma classification. J Thorac Oncol 2011; 6: 1496–504.
2. Motoi N, Szoke J, Riely GJ, Seshan VE, Kris MG, Rusch VW, Gerald WL, Travis WD. Lung adenocarcinoma: modification of the 2004 WHO mixed subtype to include the major histologic subtype suggests correlations between papillary and micropapillary adenocarcinoma subtypes, EGFR mutations and gene expression analysis. Am J Surg Pathol 2008; 32: 810–27.
3. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, Garg K, Austin JHM, Asamura H et al. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 2011; 6: 244–85.
4. Jiang S-X, Yamashita K, Yamamoto M, Piao C-J, Umezawa A, Saegusa M, Yoshida T, Katagiri M, Masuda N, Hayakawa K, Okayasu I. EGFR genetic heterogeneity of non small cell lung cancers contributing to acquired gefitinib resistance. Int J Cancer 2008; 123: 2480–6.
5. Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol 2011; 29: 2972–7.
6. Sun P-L, Seol H, Lee HJ, Yoo SB, Kim H, Xu X, Jheon S, Lee C-T, Lee J-S, Chung J-H. High incidence of EGFR mutations in Korean men smokers with no intratumoral heterogeneity of lung adenocarcinomas: correlation with histologic subtypes, EGFR/TTF-1 expressions, and clinical features. J Thorac Oncol 2012; 7: 323–30.
7. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res 2012; 72: 4875–82.
8. Sica G, Yoshizawa A, Sima CS, Azzoli CG, Downey RJ, Rusch VW, Travis WD, Moreira AL. A grading system of lung adenocarcinomas based on histologic pattern is predictive of disease recurrence in stage I tumors. Am J Surg Pathol 2010; 34: 1155–62.
9. Do H, Krypuy M, Mitchell PL, Fox SB, Dobrovic A. High resolution melting analysis for rapid and sensitive EGFR and KRAS mutation detection in formalin fixed paraffin embedded biopsies. BMC Cancer 2008; 8: 142–2.
10. Do H, Dobrovic A. Dramatic reduction of sequence artefacts from DNA isolated from formalin-fixed cancer biopsies by treatment with uracil- DNA glycosylase. Oncotarget 2012; 3: 546-58.
11. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366: 883–92.
12. Diaz LA, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012; 486: 537–40.
13. Russnes HG, Navin N, Hicks J, Borresen-Dale A-L. Insight into the heterogeneity of breast cancer through next-generation sequencing. J Clin Invest 2011; 121: 3810.
14. Pelosi G, Scarpa A, Manzotti M, Veronesi G, Spaggiari L, Fraggetta F, Nappi O, Benini E, Pasini F, Antonello D, Iannuci A, Maisonneuve P, Vale G. K-ras gene mutational analysis supports a monoclonal origin of biphasic pleomorphic carcinoma of the lung. Mod Pathol 2004; 17: 538–46.
15. Travis WD, Brambilla E, Muller-Hermelink HK, Harris CC, editors. World Health Organization Classification of Tumours. Lyon: IARC Press; 2004.
16. Tochigi N, Dacic S, Nikiforova M, Cieply KM, Yousem SA. Adenosquamous carcinoma of the lung: a microdissection study of KRAS and EGFR mutational and amplification status in a western patient population. Am J Clin Pathol 2011; 135: 783–9.
17. Newnham GM, Conron M, McLachlan S, Dobrovic A, Do H, Li J, Opeskin K, Thompson N, Wright GM, Thomas DM. Integrated mutation, copy number and expression profiling in resectable non-small cell lung cancer. BMC Cancer 2011; 11: 93.
18. Clinical Lung Cancer Genome Project (CLCGP) and Network Genomic Medicine (NGM). A genomics-based classification of human lung tumors. Sci Transl Med 2013; 5: 209ra153.
19. Warth A, Muley T, Meister M, Stenzinger A, Thomas M, Schirmacher P, Schnabel PA, Budczies J, Hoffman H, Weichert W. The Novel Histologic International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society Classification System of Lung Adenocarcinoma Is a Stage-Independent Predictor of Survival. J Clin Oncol 2012; 30: 1438-46.
20. Russell PA, Barnett SA, Walkiewicz M, Wainer Z, Conron M, Wright GM, Gooi J, Knight S, Wynne R, Liew D, John T. Correlation of Mutation Status and Survival with Predominant Histologic Subtype According to the New IASLC/ATS/ERS Lung Adenocarcinoma Classification in Stage III (N2) Patients. J Thorac Oncol 2013; 8: 461–8.
21. Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao M-S. K-ras Mutations in Non-Small-Cell Lung Carcinoma: A Review. Clin Lung Cancer 2006; 8: 9–9.
22. Sugio K, Kishimoto Y, Virmani AK, Hung JY, Gazdar AF. K-ras mutations are a relatively late event in the pathogenesis of lung carcinomas. Cancer Res 1994; 54: 5811–5.
23. Sakamoto H, Shimizu J, Horio Y, Ueda R, Takahashi T, Mitsudomi T, Yatabe Y. Disproportionate representation ofKRAS gene mutation in atypical adenomatous hyperplasia, but even distribution ofEGFR gene mutation from preinvasive to invasive adenocarcinomas. J Pathol 2007; 212: 287–94.
24. Sakuma Y, Matsukuma S, Yoshihara M, Nakamura Y, Nakayama H, Kameda Y, Tsuchiya E, Miyagi Y. Epidermal growth factor receptor gene mutations in atypical adenomatous hyperplasias of the lung. Mod Pathol 2007; 20: 967–73.
25. Yoshida Y, Shibata T, Kokubu A, Tsuta K, Matsuno Y, Kanai Y, Asamura H, Tsuchiya R, Hirohashi S. Mutations of the epidermal growth factor receptor gene in atypical adenomatous hyperplasia and bronchioloalveolar carcinoma of the lung. Lung Cancer 2005; 50: 1–8.
26. Li Z-H, Zheng J, Weiss LM, Shibata D. c-k-ras and p53 mutations occur very early in adenocarcinoma of the lung. Am Journal Path 1994; 144: 303.
27. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61: 759–67.
28. Paterakos M, Watkin WG, Edgerton SM, Moore DH, Thor AD. Invasive micropapillary carcinoma of the breast: a prognostic study. Hum Pathol 1999; 30: 1459–63.
29. Walsh MM, Bleiweiss IJ. Invasive micropapillary carcinoma of the breast: eighty cases of an underrecognized entity. Hum Pathol 2001; 32: 583–9.
30. Amin MB, Ro JY, el-Sharkawy T, Lee KM, Troncoso P, Silva EG, Ordóñez NG, Ayala AG. Micropapillary variant of transitional cell carcinoma of the urinary bladder. Histologic pattern resembling ovarian papillary serous carcinoma. Am J Surg Pathol 1994;18:1224–32.
31. Oh YL, Kim KR. Micropapillary variant of transitional cell carcinoma of the ureter. Pathol Int 2000; 50: 52–6.
32. Ushiku T, Matsusaka K, Iwasaki Y, Tateishi Y, Funata N, Seto Y, Fukayama M. Gastric carcinoma with invasive micropapillary pattern and its association with lymph node metastasis. Histopathology 2011; 59: 1081–9.
33. Ohtsuki Y, Kuroda N, Yunoki S, Murakami S, Mizukami Y, Okada Y, Iguchi M, Lee G-H, Furihata M. Immunohistochemical analysis of invasive micropapillary carcinoma pattern in four cases of gastric cancer. Med Mol Morphol 2013; 46: 114–21.
34. Haupt B, Ro JY, Schwartz MR, Shen SS. Colorectal adenocarcinoma with micropapillary pattern and its association with lymph node metastasis. Mod Pathol 2007; 20: 729–33.
35. Verdú M, Román R, Calvo M, Rodón N, García B, González M, Vidal A, Puig X. Clinicopathological and molecular characterization of colorectal micropapillary carcinoma. Mod Pathol 2011; 24: 729–38.
36. Jänne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crinò L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol 2012; 14: 38–47.
37. Yoo SB, Chung J-H, Lee HJ, Lee C-T, Jheon S, Sung SW. Epidermal growth factor receptor mutation and p53 overexpression during the multistage progression of small adenocarcinoma of the lung. J Thorac Oncol 2010; 5: 964–9.