
Introduction
More than a century ago, Paul Ehrlich suggested that the immune system has a fundamental role in the rejection of cancer cells. He hypothesized that clinically detectable tumors would arise much more frequently if the immune system was not permanently eliminating transformed cells [1]. This concept generated vigorous debate, but decades of intense research confirmed that the immune compartment has indeed the potential to hold tumor growth in check. Accordingly, Paul Ehrlich’s early suggestion was later refined to give rise to the immunosurveillance hypothesis of Burnet and Thomas [2], and more recently extended to the immunoediting concept by Schreiber and colleagues [3-5].
It is now evident that an effective immune response against tumors requires the activation of T lymphocytes, especially the cytotoxic CD8+ compartment. A number of tumor neoantigens have been described so far that are capable of eliciting an anti-tumor T cell response [6, 7]. Together with costimulatory signals, the recognition of such tumor antigens by the T cell receptor can lead to a profound activation of T cell effector functions that eventually are able to eradicate the tumor cells expressing this particular antigen [8].
On the other hand, it also became clear that inhibitory mechanisms counteract anti-tumor immunity. The so-called “immune checkpoints” help prevent autoimmunity, but also interfere with the attack of T cells against their tumor cell targets. Over the years, more and more regulatory pathways have been discovered that prevent successful anti-tumor T cell responses.
Two prominent members of the increasing group of immune checkpoint proteins are the T cell surface receptors CTLA-4 and PD-1.
Cytotoxic T-lymphocyte-associated protein-4 (CTLA-4) binds with high avidity to immunostimulatory CD80 and CD86 proteins on antigen-presenting cells and is thought to attenuate early T cell activation by sequestering its ligands away from the costimulatory receptor CD28 [9, 10]. CD80 and CD86 can even be removed from the surface of antigen- presenting cells by trans-endocytosis [11]. Furthermore, some reports suggest a T cell- intrinsic function of CTLA-4 by recruiting the protein phosphatases SHP2 and PP2A [12].
One study reported that CTLA-4 increased T cell motility, thereby preventing the formation of stable conjugates between T cells and antigen-presenting cells. This ultimately leads to decreased production of cytokines and T cell proliferation [13].
CTLA-4 also has a pivotal role in the activity of regulatory T cells: CTLA-4 triggering on Tregs enhances their immunosuppressive capacity [14, 15]. Recently it was shown that mature dendritic cells (DCs) secrete CTLA-4-containing microvesicles which can downregulate CD80/86 on bystander DCs, thereby negatively regulating T cell activation [16]. The fundamental role of CTLA-4 in maintaining immune homeostasis is demonstrated by the dramatic phenotype of Ctla4 knockout mice. These mice exhibit lethal lymphoproliferative disease and multiorgan tissue destruction [17, 18].
Programmed cell death protein 1 (PD-1) primarily limits the activity of T cells in peripheral tissues and plays an important inhibitory role in the tumor microenvironment as tumor cells often express high levels of its ligands, PD-L1 and PD-L2 [19-22]. PD-1 mainly exerts its negative regulatory effect by recruiting SHP2 to its cytoplasmic tail [23]. As with CTLA-4, PD-1 engagement can enhance T cell motility by blocking the T cell receptor-mediated stop signal [24]. PD-1 is also expressed on regulatory T cells and can promote their induction and maintenance [25]. In comparison to Ctla4 deficiency, the phenotype of Pdcd1 knockout mice is relatively moderate, which might have important implications in the clinical application of the respective checkpoint inhibitors [26].
During the last two decades, the E3 ubiquitin ligase Cbl-b has emerged as an intracellular immune checkpoint. Cbl-b regulates T cell activation thresholds by mediating the requirement for CD28 costimulation, and loss of Cblb leads to anergy resistance and susceptibility to autoimmunity [27, 28]. Additionally, Cbl-b contributes to the maintenance of self-tolerance by mediating the immunosuppressive effects of TGFβ, and Cblb-deficient T cells are less sensitive to TGFβ and to inhibition by Tregs [29-32]. TGFβ secreted by tumor cells or tumor-infiltrating Tregs generates an immunosuppressive milieu that contributes to immune escape of tumor cells [33]. In this context, Cblb knockout mice display enhanced responses to a TGFβ-secreting tumor compared to wild-type mice [32]. In a number of studies it was demonstrated that Cblb-/- mice can reject tumors in a T cell-dependent fashion, and the adoptive transfer of Cblb-deficient or Cblb-silenced CD8+ T cells together with a dendritic cell vaccine can significantly delay tumor growth and enhance survival rates [34-38]. Moreover, loss of Cblb or inactivation of its E3 ligase activity leads to rejection of metastatic tumors by natural killer cells [39].
The concept of using antagonists of inhibitory signals to enhance anti-tumor immune responses has found its way to the clinic with already promising results. Anti-CTLA-4 ipilimumab was the first “immune checkpoint” inhibitor that led to tumor regression and a survival benefit for patients with advanced melanoma and was therefore approved by the FDA in 2011 [40, 41].
Anti-PD-1 nivolumab was later also approved for the treatment of metastatic melanoma and a number of other cancer types. The combination of ipilimumab and nivolumab led to an improved survival benefit in metastatic melanoma patients in comparison to ipilimumab alone and was approved by the FDA in 2015 [42].
Targeting PD-1 signaling by blocking the PD-1 ligand PD-L1 is also a reasonable approach. For example, an anti-PD-L1 monoclonal antibody led to objective response rates of 6 - 17 % in melanoma, non-small-cell lung carcinoma, renal cell carcinoma, and ovarian cancer [43]. Anti-PD-L1 atezolizumab was approved by the FDA for the treatment of bladder cancer and non-small-cell lung cancer in 2016.
Nevertheless, the potency of these established checkpoint inhibitors is limited. For example, the efficacy of anti-CTLA-4 treatment depends on the immunogenicity of the tumor and can be dramatically enhanced by co-administration of a GM-CSF vaccine [44, 45]. Similarly, it has been suggested that the therapeutic benefit of PD-1 pathway blockade can be improved by combination with other approaches that induce antitumor responses [46].
Based on these data, we wanted to evaluate the efficacy of blocking PD-L1 or CTLA-4 in combination with loss of the intracellular checkpoint Cbl-b in a murine tumor model. The rationale behind this approach was that inactivating Cbl-b reduces the activation threshold for T cells and simultaneously decreases their sensitivity toward the suppressive effects of TGFβ. This should theoretically improve the efficacy of established checkpoint inhibition therapies.
In this study we confirm that loss of Cblb delays tumor growth and prolongs survival in a melanoma mouse model. Additionally, blocking CTLA-4 with a monoclonal antibody significantly boosts these effects. In contrast, however, inhibition of PD-L1-triggered signaling in Cblb-deficient mice shows no additive therapeutic benefit. Further in vitro results show that Cblb-/- T cells are less susceptible to PD-L1-mediated suppression of proliferation and IFNγ secretion. These findings suggest that Cbl-b might have a so far unappreciated role in the inhibitory PD-1 signaling pathway in T cells.
Results
CTLA-4 and PD-L1 blockade in Cblb-/- mice
Blockade of CTLA-4 and/or PD-1 has been shown to drastically diminish tumor growth and prolong survival in mice and patients in a T cell-dependent fashion [14, 40, 41]. As Cblb-/-mice are able to immunologically reject otherwise lethal tumor burdens [33-38], we wanted to investigate if this effect would be even more profound in combination with CTLA-4 or PD-L1 blockade. Therefore, we subcutaneously injected 5 x 105 B16ova cells into wild-type or Cblb-/- mice, treated them with anti-CTLA-4, anti-PD-L1 or IgG control antibodies and monitored tumor growth.
As reported in previous studies, we found that CTLA-4 blockade or Cblb ablation led to a reduction of tumor growth (Figure 1A, 1B, 1C) and extended survival compared to wild-type IgG-treated mice (Figure 2A, 2B, 2C). Blocking of CTLA-4 in Cblb-/- mice prolonged survival (Figure 2D) and attenuated tumor growth (Figure 1D) to an even more pronounced extent than in IgG-treated Cblb-deficient mice, suggesting that CTLA-4 signaling is not completely dependent on Cbl-b.
As expected, treatment of wild-type mice with anti-PD-L1 significantly reduced tumor growth (Figure 3A, 3B) and prolonged survival (Figure 4A, 4B). Surprisingly, however, blocking of PD-L1 in Cblb-/- mice had no additional effect (Figure 3D, Figure 4D). The anti-PD-L1-treated wild-type group behaved approximately the same as the Cblb-/- IgG and Cblb-/- anti-PD-L1 group (Figure 3A, Figure 4A). This suggests that PD-L1-initiated signaling is already impaired in Cblb-deficient mice.
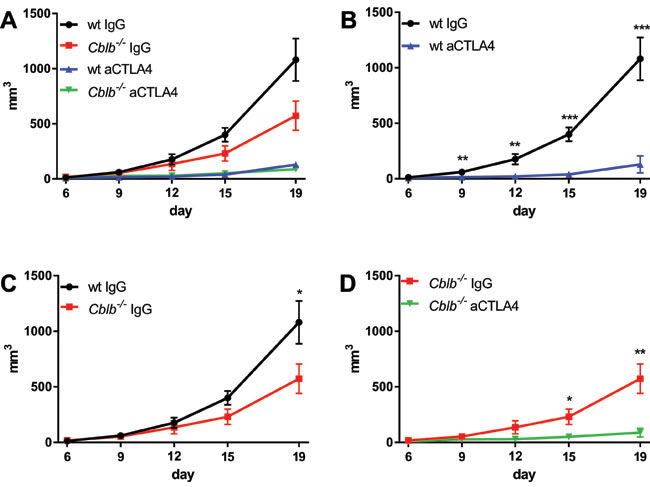
Figure 1: Kinetics of B16ova tumor growth. C57Bl/6 (wt) and Cblb-/- mice were s.c. injected with 5x105 B16ova cells and i.p. injected with 400µg anti-CTLA4 or IgG control antibody every 3rd day starting on day 0. The data are pooled from two independent experiments (n = 6-7 per group). Tumor volume is shown.
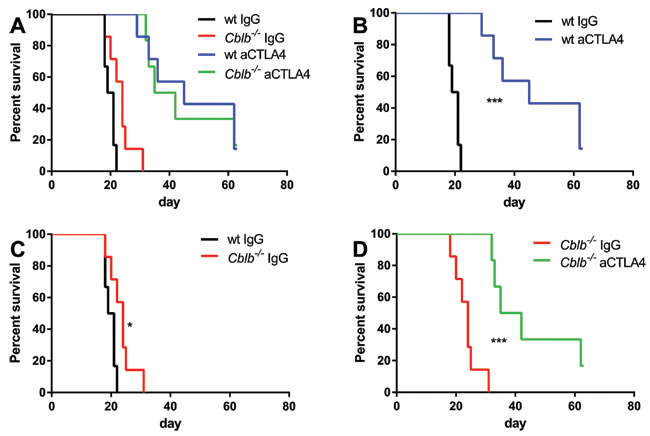
Figure 2: Survival percentages of mice from Figure 1. Mice were s.c. injected with 5x105 B16ova cells and i.p. injected with 400µg anti-CTLA4 or IgG control antibody every 3rd day from day 0 until day 18. The data shown are pooled from two independent experiments (n = 6-7 per group).
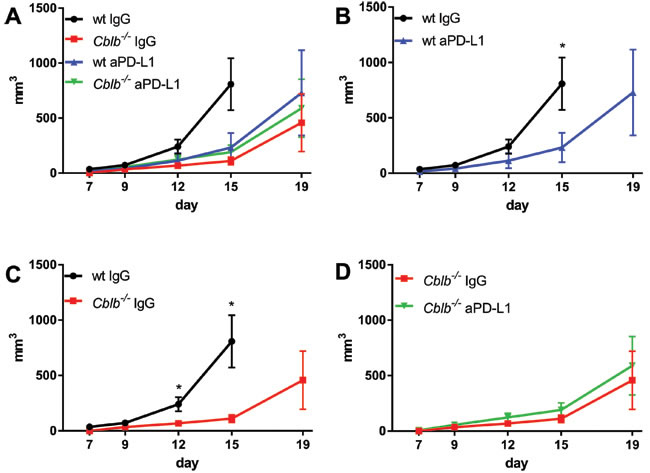
Figure 3: Kinetics of B16ova tumor growth. C57Bl/6 (wt) and Cblb-/- mice were s.c. injected with 5x105 B16ova cells and i.p. injected with 500µg anti-PD-L1 or IgG control antibody every 3rd day starting on day 0. The data are pooled from two independent experiments (n = 6-8 per group). Tumor volume is shown.
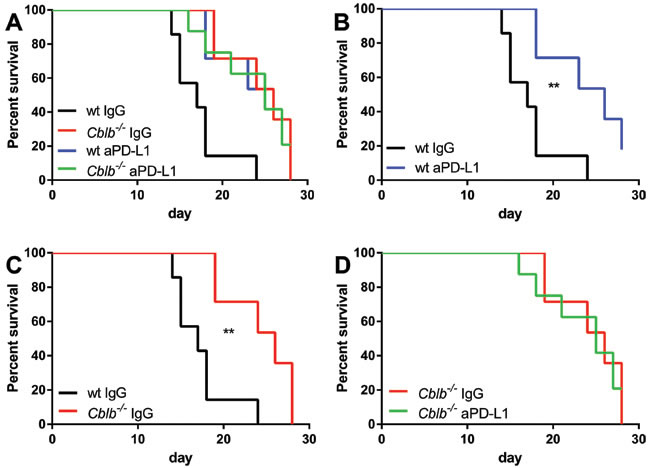
Figure 4: Survival percentages of mice from Figure 3. Mice were s.c. injected with 5x105 B16ova cells and i.p. injected with 500µg anti-PD-L1 or IgG control antibody every 3rd day from day 0 until day 18. The data shown are pooled from two independent experiments (n = 6-8 per group).
Distribution of PD-1 and CTLA-4 on T cells
In principle, the lack of any effect of blocking PD-L1 in Cblb-deficient mice could be due to diminished PD-L1 and/or PD-1 expression. To test this possibility, we performed FACS analysis of in vitro stimulated CD4+ and CD8+ T cells. PD-L1 (not shown), PD-1 and CTLA-4 expression on CD4+ and CD8+ T cells was induced upon CD3/CD28 stimulation (Figure 5). PD-1 expression on Cblb-/- CD4+ and CD8+ T cells was significantly increased compared to wild-type cells (Figure 5A, 5B), whereas CTLA-4 seemed to be elevated only in CD8+ cells (Figure 5C, 5D). We did not detect any difference in the expression of PD-L1 in either of the genotypes (data not shown).
In four independent B16ova in vivo experiments without antibody treatment, PD-L1, PD-1 and CTLA-4 were expressed on tumor-infiltrating CD4+ and CD8+ cells, and no significant differences between wild-type and Cblb knockout mice were detected: wt CD8+ PD-1+: 56.0% ± 5.7%; Cblb-/- CD8+ PD-1+: 50.3% ± 4.5%; wt CD8+ CTLA-4+: 17.1% ± 4.5%; Cblb-/- CD8+ CTLA-4+: 18.0% ± 4.2% (n = 10) (Figure 5E, 5F and data not shown).
Furthermore, PD-L1 was expressed on a large fraction of B16ova tumor cells (wt: 70.3% ± 4.6%; Cblb-/-: 84.7% ± 4.3%), whereas PD-L2 was barely detectable (wt: 0.3% ± 0.1%; Cblb-/-: 0.6% ± 0.2%) (n = 6).
Interestingly, Cblb-deficient tumor infiltrating antigen presenting cells (CD45+ MHC-II+) expressed significantly more PD-L1 on a per cell basis (not shown) and also the fraction of PD-L1 positive cells was significantly higher (wt: 85.7% ± 1.5%; Cblb-/-: 92.3% ± 1.9%) (p = 0.035; n = 6).
Cblb-deficient T cells are less susceptible to PD-L1-mediated suppression
Since we did see an effect of anti-PD-L1 treatment on tumor growth and survival in wild-type but not Cblb-/-
mice (Figure 3A), we speculated that Cbl-b could play a role in the suppressive PD-1 signaling pathway. To test this hypothesis, we performed in vitro inhibition assays using recombinant PD-L1, immobilized on the surface of culture plates. Proliferation of both CD4+ and CD8+ cells was substantially diminished by active PD-1 signaling triggered by recombinant PD-L1. However, proliferation of wild-type cells was downregulated to approximately 44% of the uninhibited control value, whereas Cblb-/- cells still showed a proliferative response of 80%, indicating that Cblb-deficient T cells were hyporesponsive to PD-L1-mediated suppression of proliferation (Figure 6A, 6B). Moreover, expression of the pro-inflammatory anti-tumor cytokine IFNγ was strongly reduced in wild-type CD8+ cells (down to 17%) but not in Cblb knockout CD8+ cells (down to 85%) (Figure 7). These results suggest that Cbl-b partly mediates the suppressive effects of PD-1 engagement. Along this line, it was reported that silencing PD-L1 in antigen presenting dendritic cells led to diminished Cbl-b protein levels in T cells, suggesting an involvement of PD-1 signaling in upregulation of Cbl-b expression [47]. However, in our experimental system we did not observe enhanced Cbl-b protein amounts after stimulation of T cells with recombinant PD-L1 (Supplementary Figure 3).
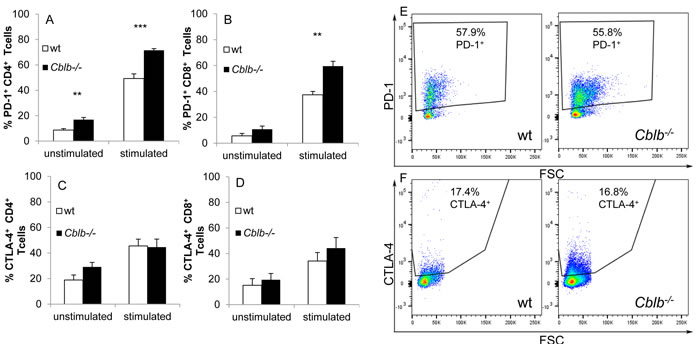
Figure 5: FACS analysis of T cells. A.-D. CD3+ T cells of C57Bl/6 (wt) or Cblb-/- mice were cultivated overnight, either unstimulated or stimulated with 3µg/ml platebound anti-CD3 and 1µg/ml soluble anti-CD28. FACS analysis of CTLA-4 (C, D) and PD-1 (A, B) expression was performed on gated CD4+ (A, C) or CD8+ (B, D) cells. Mean +/- SEM of 4-6 individual mice is shown (3 independent experiments). E, F. PD-1 and CTLA-4 expression on tumor infiltrating CD8+ T cells. Dotblots are gated on CD45+ and CD8+ cells and percentage of PD-1+ or CTLA4+ cells is shown. One out of ten mice (four independent experiments) is depicted for each genotype.
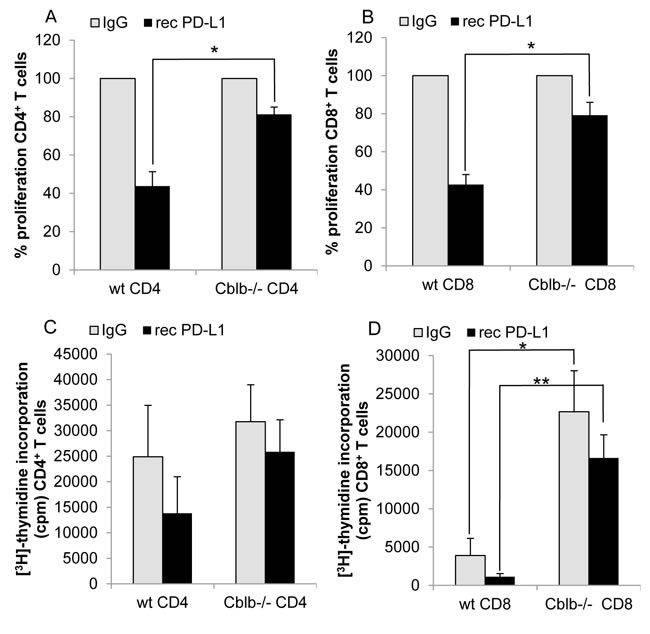
Figure 6: Proliferation of T cells. CD4+ A., C. or CD8+ B., D. T cells were stimulated with 1µg/ml platebound anti-CD3 and 0.1µg/ml soluble anti-CD28. Additionally, wells were coated with 10µg/ml recombinant PD-L1 or IgG as control. [3H]-thymidine was added on day 2 of cell culture and its incorporation measured after 16h. A, B: Control value was set to 100%; C, D: Counts per minute of [3H]-thymidine incorporation. Mean ± SEM of 4-5 individual mice is shown (3 independent experiments).
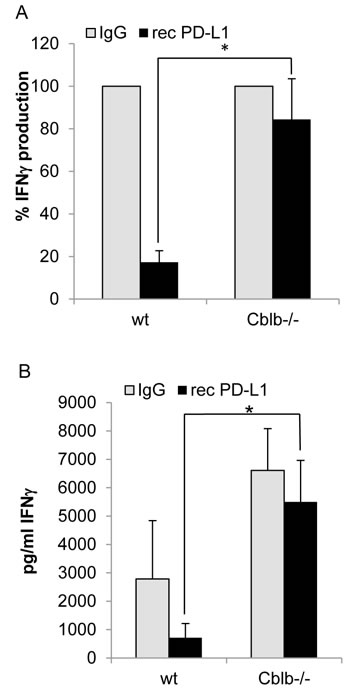
Figure 7: IFNγ production of CD8 + T cells. Cells were stimulated with 1µg/ml platebound anti-CD3 and 0.1µg/ml soluble anti-CD28. Additionally, wells were coated with 10µg/ml recombinant PD-L1 or IgG as control. Supernatants were taken on day 2 of cell culture and IFNγ was measured with Bioplex technology. A: Control value was set to 100%; B: Concentration in the supernatants. Mean ± SEM of 6-7 individual mice is shown (4 independent experiments).
Discussion
Immunotherapies against malignant neoplastic diseases emerge more and more as a promising “fifth pillar” of cancer treatment (besides surgery, radiation therapy, chemotherapy, and molecularly targeted therapies) [48]. The great expectations raised by this kind of treatment are underlined by the decision of the journal “Science” to choose cancer immunotherapy as “Breakthrough of the Year” in 2013 [49].
The rationale behind immunotherapies is to harness the immune system’s ability to eliminate structures that are recognized as “foreign”. In the case of malignant disease, these structures turn out to be cancer-associated antigens that are capable of eliciting an immune response, most notably activating cytotoxic T lymphocytes via their specific antigen receptors [50].
Unfortunately, however, T cell activity towards tumors is often compromised by inhibitory signals, the so-called “immune checkpoints”. Consequently, an important aspect of cancer immunotherapy is to block these inhibitory signals in an effort to unleash T cells to destroy malignant cells.
The E3 ubiquitin ligase Cbl-b has been shown to act as an intracellular checkpoint by interfering with a number of T cell signaling pathways [51]. Genetic loss of Cblb leads to anergy resistance and susceptibility to autoimmunity [27, 28, 52]. Cbl-b also contributes to the maintenance of self-tolerance by mediating the immunosuppressive effects of TGFβ [32]. In keeping with these observations, Cblb knockout mice reject tumors, which has been shown to crucially depend on the CD8+ T cell compartment [30, 34, 36, 38].
As proof of concept that Cbl-b may serve as a novel cancer immunotherapy target, we have already shown that adoptive transfer of Cblb-silenced polyclonal CD8+ T cells together with DC vaccination resulted in strong suppression of tumor growth with substantially prolonged overall survival, interestingly without any sign of autoimmunity [35]. Recently, it has been shown in a phase I trial that the strategy of silencing Cblb in human peripheral blood mononuclear cells followed by adoptive cell transfer is feasible and safe [53].
Considering the complexity of T cell signaling pathways, combining the inhibition of distinct immune checkpoints presumably has the benefit of targeting key regulators at different cellular levels. For example, patients with advanced melanoma show a response rate of 11% with ipilimumab (anti-CTLA-4), which is increased up to 61% when treated in combination with nivolumab (anti-PD-1) [42]. Several additional combinatorial treatments are of particular interest, such as blockade of PD-1 together with LAG-3 or TIM-3 [54, 55].
In this study, we tested the effectiveness of the strategy of inhibiting the established immune checkpoints CTLA-4 (via CTLA-4 neutralization) or PD-1 (via neutralization of the PD-1 ligand PD-L1) in combination with targeting the intracellular checkpoint Cbl-b in order to enhance T cell anti-tumor activity in a B16ova melanoma mouse model (B16ova expresses PD-L1, not shown). In wild-type mice, CTLA-4 inhibition showed a marked efficacy in terms of decreased tumor growth (Figure 1B) and prolonged survival (Figure 2B) compared to IgG-treated control mice. Similarly, anti-CTLA-4 treatment also had a significant effect in Cblb knockout mice (Figure 1D & 2D). Neutralizing PD-L1 with a monoclonal antibody also delayed tumor growth and extended survival, albeit to a lesser extent than CTLA-4 inhibition (Figure 3B & 4B). It is noticeable that in our murine melanoma model, anti-PD-L1 treatment is less effective than anti-CTLA-4 treatment even in wild-type mice. In the clinical context, blocking PD-L1/PD-1 interaction with pembrolizumab is more effective than blocking CTLA-4 with ipilimumab in the treatment of advanced melanoma [56]. This apparent discrepancy could be possibly explained by the different species, materials, and methods used. For example, the neutralizing antibodies in our study are completely different from the clinically applied immunoglobulins.
Surprisingly, anti-PD-L1 treatment was completely ineffective in Cblb knockout mice (Figure 3D & 4D), which is in striking contrast to CTLA-4 neutralization. This was not simply due to diminished PD-1 receptor expression, because PD-1 amounts were even enhanced in Cblb-deficient CD4+ and CD8+ T cells in vitro (Figure 5A & 5B) or not significantly altered on Cblb-deficient tumor infiltrating T cells, respectively (Figure 5E).
In the PD-L1 neutralization tumor experiments, the three groups “wild-type anti-PD-L1”, “Cblb-/- IgG”, and “ Cblb-/- anti-PD-L1” exhibited virtually identical benefits in terms of tumor growth and survival rates compared to the “wild-type IgG” control group (Figure 3A & 4A), possibly reflecting an involvement of Cbl-b in the PD-1 signaling pathway. However, this is not a definite proof that Cbl-b is downstream of PD-1, but this assumption is supported by in vitro inhibition assays using recombinant PD-L1. PD-L1 potently reduced the proliferation of CD4+ and CD8+ T cells (56% and 57% reduction, respectively), whereas it was much less effective in Cblb-deficient T cells (19% and 21% reduction). Furthermore, PD-1 engagement strongly impaired IFNγ secretion of wild-type CD8+ T cells (83% reduction), in striking contrast to Cblb knockout cells that were almost resistant to recPD-L1 (16% reduction). These results indicate that Cbl-b substantially mediates the suppressive effects of PD-L1/PD-1 signaling, suggesting a biochemical function of Cbl-b downstream of PD-1. Of note, during this manuscript was under revision, a paper was published confirming that Cblb-deficient T cells (and NK cells) are resistant to PD-L1/PD-1-mediated suppression [57].
Cbl-b amounts were reported to be diminished in T cells upon blockade of PD-1/PD-L1 interaction [47]. Along this line, SHP-1 and SHP-2 are recruited to PD-1 [58], and SHP-1 dephosphorylates and stabilizes Cbl-b [59]. It has also been shown that PD-1 ligation impairs PKCθ activation loop phosphorylation [60]. Impaired PKCθ activity also prolongs the half-life of Cbl-b, given that Cbl-b degradation is dependent on PKCθ [61].
Taken together, our findings suggest a novel function of Cbl-b in the PD-1 signaling pathway in effector T cells. However, whether Cbl-b exerts a specific function as an E3 ligase in the PD-1 signaling pathway remains speculative and warrants further investigation. Unraveling the detailed signaling mechanisms of the non-redundant role of Cbl-b in the PD-1 function of T cells in future studies may contribute to an improved understanding of the molecular and cellular processes that lead to autoimmunity or cancer.
Materials and Methods
Mice
Cblb knockout mice on a C57Bl/6 background were described previously [27]. Mice were maintained under SPF conditions. All animal experiments were performed in accordance with the Austrian “Tierversuchsgesetz” (BGBI. Nr.501/1989 i.d.g.F. and BMWF-66.011/0061-II/3b/2013) and were approved by the Bundesministerium für Wissenschaft und Forschung (bm:wf).
Tumor induction and in vivo antibody administration
8- to 12-week-old female mice were injected subcutaneously with 100µl PBS containing 5x105 B16ova melanoma cells into the left, ventral flank. Intraperitoneal injection of 200µl PBS containing either 0.4mg of a Syrian hamster anti-mouse CTLA4 (Clone UC10-4F10-11; BE0131), 0.5mg rat anti-mouse PD-L1 (Clone10F.9G2; BE0101), 0.4mg Syrian hamster IgG (BE0087) or 0.5mg rat IgG2b (Clone LTF-2; BE0090) (all from BioXCell, USA) was performed every 3 days starting from day 0 of B16ova challenge until day 18.
Tumor volume was calculated according to the following equation: V = π/6*length*width2. For survival analysis, mice with tumors exceeding the length limit of 15 mm were sacrificed and counted as dead.
Flow cytometry
Spleen and lymph nodes of 4-6 mice per genotype (not pooled) were mashed through a sieve, depleted of erythrocytes using the mouse erythrocyte lysing kit (R&D, WL2000) and CD3+ T-cells were isolated untouched using the Pan T Cell Isolation Kit II mouse (Miltenyi Biotech 130-095-130) according to manufacturer’s protocol.
750.000 cells per 96 flatbottom well were cultured overnight in 300µl RPMI+++ (Roswell Park Memorial Institute 1640 medium [PAA; E15-039] supplemented with 10% fetal calf serum [FCS], 2 mM l-glutamine and penicillin-streptomycin [50 U/ml]) and left unstimulated or stimulated with 3µg/ml platebound anti-CD3 (2C11, in-house made) and 1µg/ml soluble anti-CD28 (BD 553294).
Cells were harvested the next day, fixed and permeabilized (buffers from the FoxP3 staining buffer set, eBiosciences 00-5523), before staining for 20min at 4°C with specific antibodies (CD4 FITC, BD 553047; PD-1 PE, eBiosciences 12-9985-82; PD-L1 PE-Cy7, eBiosciences 25-5982-20; CTLA-4 APC, eBiosciences 17-1522-80; CD8 FITC, eBiosciences 11-0081-85), diluted 1:200 in permeabilization buffer (eBiosciences 00-8333). Data acquisition was performed on a FACSCalibur and data analysis was conducted using the FlowJo 10.0.8r1 software.
For analysis of tumor infiltrating lymphocytes, 8- to 12-week-old female mice were injected subcutaneously with 100µl PBS containing 1x105 B16ova melanoma cells into the left, ventral flank. The mice were controlled regularly and when the first tumor exceeded the length of 12mm the tumors of all mice were collected on the same day (four independent experiments: between day19 and day 22 after injection). The tumors were cut in small pieces and digested for 45 min in PBS containing 2% FCS, 2.5mM MgCl, 2.5 mg/ml collagenase D (Roche, 11088858001) and 1 mg/ml DNase I (Roche, 11284932001) at 37°C. Digested tissues were incubated 5 min at 37°C with EDTA (0.5 M) to prevent DC/T cell aggregates and mashed through a 70-µm filter and a 40-µm filter. Cells were washed, fixed, permeabilized (buffers from the FoxP3 staining buffer set, eBiosciences 00-5523) and stained for 20min at 4°C with specific antibodies (PD-1 PE, eBiosciences 12-9985-82; PD-L1 PE-Cy7, eBiosciences 25-5982-80; PD-L2 FITC, eBiosciences 11-9972-82; CD8 PerCP-Cy5.5, eBiosciences 45-0081-80; CD4 V500 BD 560782; CD45 APC-Cy7, BD 561037; CTLA-4 APC, eBiosciences 17-1522-80) diluted 1:200 in permeabilization buffer (eBiosciences 00-8333). Data acquisition was performed on a FACSVerse and data analysis was conducted using the FlowJo 10.0.8r1 software.
Analysis of proliferative responses and cytokine release
96well flatbottom wells were coated with PBS containing 10µg/ml IgG1 (HRPN; BE0088 BioXcell) or recombinant mouse PD-L1 Fc chimera protein (R&D, 1019-B7) at 37°C over night. This solution was then discarded and replaced by PBS containing 1µg/ml anti-CD3 (2C11, in-house made) and left for 4h at 37°C. After coating, 400.000 CD4+ or CD8+ cells (negative MACS sort of spleen and lymph nodes according to manufacturer’s protocol; Milteny 130-104-454 and 130-104-075) per well were cultured for 3 days in 200µl RPMI+++ and costimulated with 0.1µg/ml anti-CD28 (BD 553294). On day 2 of culture, 50µl supernatants were taken for measurement of cytokines via BioPlex technology (BioRad) and 50µl RPMI+++ containing 1 μCi [3H]-thymidine (Perkin Elmer) were added. After a 16-hour pulse, cells were harvested and [3H]-thymidine incorporation was measured with a Matrix 96 direct β counter system.
Western blot
Wells of a 24 well plate were coated with PBS containing 10µg/ml IgG1 (HRPN; BE0088 BioXcell) or recombinant mouse PD-L1 Fc chimera protein (R&D, 1019-B7) at 37°C overnight. This solution was then discarded and replaced by PBS containing 1µg/ml anti-CD3 (2C11, in-house made) and left for 3h at 37°C. After coating, 1x107 wild-type CD3+ cells (negative MACS sort of spleen and lymph nodes according to manufacturer’s protocol; Milteny 130-095-130) per well were cultured overnight in 1ml RPMI+++ and costimulated with 1µg/ml anti-CD28 (BD 553294). Cells were collected, washed once in ice-cold PBS and lysed in 30µl lysisbuffer (5mM NaP2P, 5mM NaF, 1mM Na3VO4, 5 mM EDTA, 150mM NaCl, 50mM tris [pH 7.3], 1% NP-40, aprotinin and leupeptin [50µg/ml each]) for 30min on ice. After centrifugation (15.000g, 4°C), protein lysates were subjected to Western blotting analysis with antibodies against Cbl-b (abcam 54362; dilution 1:1000) and actin (santacruz 1615; dilution 1:1000). Densitometric analysis was performed using ImageJ.
Statistics
Results are expressed as mean ± standard error of the mean (SEM). Groups were compared using the paired Student’s t-test. Overall survival was expressed using the Kaplan-Meier method and differences between groups were determined using the log-rank test. Data analysis was performed using GraphPad Prism 7.00. Significant differences are indicated as *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Acknowledgments
We thank Dr. Guenther Lametschwandtner (Apeiron Biologics, Vienna) for experimental assistance and Victoria Klepsch PhD (Medical University Innsbruck) for valuable discussions.
Conflicts of interest
The authors declare no conflict of interest.
Financial support
This work was supported by grants from the Austrian Science Fund FWF P 26892-B13 (TG) and the Christian Doppler Society (GB).
References
1. Ehrlich P. [Ueber den jetzigen Stand der Karzinomforschung]. [Article in German]. Beitraege zur experimentellen Pathologie und Chemotherapie. 1909: 117-64.
2. Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970; 13: 1-27.
3. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004; 21: 137-48. doi: 10.1016/j.immuni.2004.07.017.
4. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004; 22: 329-60. doi: 10.1146/annurev.immunol.22.012703.104803.
5. Mittal D, Gubin MM, Schreiber RD, Smyth MJ. New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr Opin Immunol. 2014; 27: 16-25. doi: 10.1016/j.coi.2014.01.004.
6. Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2015; 21: 81-5. doi: 10.1038/nm.3773.
7. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015; 348: 69-74. doi: 10.1126/science.aaa4971.
8. Nagorsen D, Scheibenbogen C, Marincola FM, Letsch A, Keilholz U. Natural T cell immunity against cancer. Clin Cancer Res. 2003; 9: 4296-303.
9. Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994; 1: 793-801.
10. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994; 1: 405-13.
11. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011; 332: 600-3. doi: 10.1126/science.1202947.
12. Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009; 229: 12-26. doi: 10.1111/j.1600-065X.2009.00770.x.
13. Schneider H, Downey J, Smith A, Zinselmeyer BH, Rush C, Brewer JM, Wei B, Hogg N, Garside P, Rudd CE. Reversal of the TCR stop signal by CTLA-4. Science. 2006; 313: 1972-5. doi: 10.1126/science.1131078.
14. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009; 206: 1717-25. doi: 10.1084/jem.20082492.
15. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008; 322: 271-5. doi: 10.1126/science.1160062.
16. Halpert MM, Konduri V, Liang D, Chen Y, Wing JB, Paust S, Levitt JM, Decker WK. Dendritic Cell-Secreted Cytotoxic T-Lymphocyte-Associated Protein-4 Regulates the T-cell Response by Downmodulating Bystander Surface B7. Stem Cells Dev. 2016; 25: 774-87. doi: 10.1089/scd.2016.0009.
17. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995; 3: 541-7.
18. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, Thompson CB, Griesser H, Mak TW. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995; 270: 985-8.
19. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002; 8: 793-800. doi: 10.1038/nm730.
20. Konishi J, Yamazaki K, Azuma M, Kinoshita I, Dosaka-Akita H, Nishimura M. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res. 2004; 10: 5094-100. doi: 10.1158/1078-0432.CCR-04-0428.
21. Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008; 8: 467-77. doi: 10.1038/nri2326.
22. Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003; 198: 851-62. doi: 10.1084/jem.20031074.
23. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000; 192: 1027-34.
24. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009; 10: 1185-92. doi: 10.1038/ni.1790.
25. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009; 206: 3015-29. doi: 10.1084/jem.20090847.
26. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999; 11: 141-51.
27. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, Le J, Ohashi PS, Sarosi I, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000; 403: 211-6. doi: 10.1038/35003228.
28. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000; 403: 216-20. doi: 10.1038/35003235.
29. Adams CO, Housley WJ, Bhowmick S, Cone RE, Rajan TV, Forouhar F, Clark RB. Cbl-b(-/-) T cells demonstrate in vivo resistance to regulatory T cells but a context-dependent resistance to TGF-beta. J Immunol. 2010; 185: 2051-8. doi: 10.4049/jimmunol.1001171.
30. Gruber T, Hinterleitner R, Hermann-Kleiter N, Meisel M, Kleiter I, Wang CM, Viola A, Pfeifhofer-Obermair C, Baier G. Cbl-b mediates TGFbeta sensitivity by downregulating inhibitory SMAD7 in primary T cells. J Mol Cell Biol. 2013; 5: 358-68. doi: 10.1093/jmcb/mjt017.
31. Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF-beta in Cbl-b-/- mice. J Immunol. 2004; 173: 1059-65.
32. Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB. Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-beta sensitivity in vitro and in vivo. J Immunol. 2006; 176: 1316-20.
33. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005; 8: 369-80. doi: 10.1016/j.ccr.2005.10.012.
34. Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007; 117: 1029-36. doi: 10.1172/JCI29472.
35. Hinterleitner R, Gruber T, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Tzankov A, Schuster M, Penninger JM, Loibner H, Lametschwandtner G, Wolf D, Baier G. Adoptive transfer of siRNA Cblb-silenced CD8+ T lymphocytes augments tumor vaccine efficacy in a B16 melanoma model. PLoS One. 2012; 7: e44295. doi: 10.1371/journal.pone.0044295.
36. Loeser S, Loser K, Bijker MS, Rangachari M, van der Burg SH, Wada T, Beissert S, Melief CJ, Penninger JM. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007; 204: 879-91. doi: 10.1084/jem.20061699.
37. Lutz-Nicoladoni C, Wallner S, Stoitzner P, Pircher M, Gruber T, Wolf AM, Gastl G, Penninger JM, Baier G, Wolf D. Reinforcement of cancer immunotherapy by adoptive transfer of cblb-deficient CD8+ T cells combined with a DC vaccine. Immunol Cell Biol. 2012; 90: 130-4. doi: 10.1038/icb.2011.11.
38. Paolino M, Thien CB, Gruber T, Hinterleitner R, Baier G, Langdon WY, Penninger JM. Essential role of E3 ubiquitin ligase activity in Cbl-b-regulated T cell functions. J Immunol. 2011; 186: 2138-47. doi: 10.4049/jimmunol.1003390.
39. Paolino M, Choidas A, Wallner S, Pranjic B, Uribesalgo I, Loeser S, Jamieson AM, Langdon WY, Ikeda F, Fededa JP, Cronin SJ, Nitsch R, Schultz-Fademrecht C, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014; 507: 508-12. doi: 10.1038/nature12998.
40. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363: 711-23. doi: 10.1056/NEJMoa1003466.
41. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003; 100: 8372-7. doi: 10.1073/pnas.1533209100.
42. Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015; 372: 2006-17. doi: 10.1056/NEJMoa1414428.
43. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366: 2455-65. doi: 10.1056/NEJMoa1200694.
44. Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998; 95: 10067-71.
45. van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999; 190: 355-66.
46. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12: 252-64. doi: 10.1038/nrc3239.
47. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, Escors D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8+ T cells. EMBO Mol Med. 2011; 3: 581-92. doi: 10.1002/emmm.201100165.
48. AACR Cancer Progress Report Writing Committee, Sawyers CL, Abate-Shen C, Anderson KC, Barker A, Baselga J, Berger NA, Foti M, Jemal A, Lawrence TS, Li CI, Mardis ER, Neumann PJ, et al. AACR Cancer Progress Report 2013. Clin Cancer Res. 2013; 19: S4-98. doi: 10.1158/1078-0432.CCR-13-2107.
49. Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013; 342: 1432-3. doi: 10.1126/science.342.6165.1432.
50. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer. 2014; 14: 135-46. doi: 10.1038/nrc3670.
51. Lutz-Nicoladoni C, Wolf D, Sopper S. Modulation of Immune Cell Functions by the E3 Ligase Cbl-b. Front Oncol. 2015; 5: 58. doi: 10.3389/fonc.2015.00058.
52. Jeon MS, Atfield A, Venuprasad K, Krawczyk C, Sarao R, Elly C, Yang C, Arya S, Bachmaier K, Su L, Bouchard D, Jones R, Gronski M, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity. 2004; 21: 167-77. doi: 10.1016/j.immuni.2004.07.013.
53. Tiozzi P KM, Alistar A, Bitting R, Neal A, Lametschwandtner G, Loibner H. Phase I clinical trial of adoptive cellular immunotherapy with APN401 in patients with solid tumors. Journal for ImmunoTherapy of Cancer. 2015; 3: 175.
54. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, Eppolito C, Qian F, Lele S, Shrikant P, Old LJ, Odunsi K. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A. 2010; 107: 7875-80. doi: 10.1073/pnas.1003345107.
55. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK, Blazar BR. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011; 117: 4501-10. doi: 10.1182/blood-2010-10-310425.
56. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015; 372: 2521-32. doi: 10.1056/NEJMoa1503093.
57. Fujiwara M, Anstadt EJ, Clark RB. Cbl-b Deficiency Mediates Resistance to Programmed Death-Ligand 1/Programmed Death-1 Regulation. Front Immunol. 2017; 8: 42. doi: 10.3389/fimmu.2017.00042.
58. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004; 173: 945-54.
59. Xiao Y, Qiao G, Tang J, Tang R, Guo H, Warwar S, Langdon WY, Tao L, Zhang J. Protein Tyrosine Phosphatase SHP-1 Modulates T Cell Responses by Controlling Cbl-b Degradation. J Immunol. 2015; 195: 4218-27. doi: 10.4049/jimmunol.1501200.
60. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004; 574: 37-41. doi: 10.1016/j.febslet.2004.07.083.
61. Gruber T, Hermann-Kleiter N, Hinterleitner R, Fresser F, Schneider R, Gastl G, Penninger JM, Baier G. PKC-theta modulates the strength of T cell responses by targeting Cbl-b for ubiquitination and degradation. Sci Signal. 2009; 2: ra30. doi: 10.1126/scisignal.2000046.