
INTRODUCTION
Lung cancer remains a leading cause of cancer-related deaths worldwide. Oncogenic EGFR activation, commonly observed in non-small cell lung cancer (NSCLC), drives cell proliferation and survival. Indeed, EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib have become an important therapeutic modality for the treatment of NSCLC patients with EGFR mutations [1–3]. However, innate or acquired resistance limits the efficacy of kinase inhibitors. The presence of KRas, BRaf and ERBB2 mutations predicts primary resistance to EGFR inhibitors [4, 5]. Acquired resistance to EGFR inhibitors has been associated most frequently with selection for secondary EGFR mutations, such as T790M in exon 20 [6, 7] and with MET amplification [8]. Second and third generation EGFR inhibitors have been developed and novel combination of these inhibitors with other therapies have been tested to overcome therapeutic resistance [9]. Additionally, drugs that target MET are in clinical trials to overcome MET-amplification-mediated resistance to EGFR inhibitors [10].
MET amplifications have been found in ~20% of NSCLC patients with acquired resistance to gefitinib or erlotinib [8, 11]. MET-amplified lung cancer cells do not respond to EGFR inhibitors [12], but are highly sensitive to MET inhibition [13, 14]. Several MET kinase inhibitors have been evaluated in clinical trials [15], however acquired resistance remains a serious limitation of these drugs. Although the mechanisms of resistance to MET targeting drugs are not well understood, it has been shown that a switch to EGFR dependency can underlie the resistance to MET kinase inhibitors. Thus, one approach to optimize the anticancer activity and to prevent the growth of resistant clones in MET-amplified lung cancer cells is the combined therapy with MET and EGFR inhibitors [16, 17]. However, lung cancer cells with amplified MET and overexpressed EGFR frequently show limited response to combined treatment [16, 18], underscoring the necessity to identify additional mechanism of resistance.
In addition to cell-autonomous mechanisms of resistance, factors in the tumor microenvironment have been shown to block the response to therapy [19, 20]. Stroma-induced resistance to targeted therapy is frequently mediated by hepatocyte growth factor (HGF)-induced activation of MET [19–21]. HGF activates pro-survival signaling and promotes epithelial-mesenchymal transition of cancer cells, hindering the response to therapy. HGF has been shown to block the response of lung cancer cells to EGFR targeting drugs [22, 23] and increased levels of serum HGF predict poor progression-free survival and overall survival in NSCLC patients treated with EGFR inhibitors [24, 25]. Surprisingly, it has recently been shown that MET-amplified lung cancer cells become dependent on HGF for survival upon pharmacologic MET inhibition [26]. This indicates that both HGF and MET need to be targeted in MET-amplified NSCLC to prevent or to delay the onset of resistance.
Neutralizing anti-HGF antibodies that are being tested, alone or in combination with other therapies, appear to be well tolerated in clinical trials [27, 28]. They have shown encouraging results, yielding partial responses in cancer patients. However, there are currently no small molecule HGF inhibitors available for clinical trials. We developed and characterized two distinct series of low molecular weight inhibitors of HGF, which act as inhibitors of pro-HGF activation [29, 30].
HGF is secreted as a single chain inactive precursor called pro-HGF. Proteolytic conversion of pro-HGF to the heterodimeric active HGF is required to trigger ligand-dependent MET signaling. This is achieved by one of the trypsin-like serine proteases, matriptase, hepsin or HGF activator (HGFA), which are expressed by tumor cells [31]. A proteolytically inert mutant form of pro-HGF (uncleavable-HGF) or a truncated form of HGF that lacks the β-chain (NK4) displayed a competitive antagonism with HGF for MET binding. Both mutants suppressed proliferation, migration, and invasion of cancer cells and inhibited tumor growth and metastasis in vivo [32–35]. We demonstrated that triplex inhibitors of matriptase, hepsin and HGFA prevent activation of pro-HGF and inhibit HGF/MET signaling. SRI31215, a novel inhibitor of pro-HGF activation, blocked signaling between tumor-promoting fibroblasts and colon cancer cells and overcame fibroblast-mediated resistance to EGFR inhibitors in colon cancer cells [30, 36].
In this study, we established that recombinant HGF or HGF-producing fibroblasts protect MET-amplified NSCLC cells from MET-targeting therapy by maintaining AKT and ERK signaling in MET-inhibited cells. We demonstrated that novel inhibitors of HGF activation overcome fibroblast-mediated resistance to MET inhibition in MET-amplified lung cancer cells. This highlights the need to include agents blocking the biological activity of HGF, such as inhibitors of pro-HGF activation, into therapeutic regimens for MET-amplified NSCLC patients.
RESULTS
HGF rescues MET-amplified lung cancer cells from MET tyrosine kinase inhibition
Whereas MET-amplified lung cancer cells do not respond to EGFR inhibition, their growth is strongly inhibited by MET tyrosine kinase inhibitors, confirming that these cells are addicted to MET signaling [13, 14]. Accordingly, the selective MET tyrosine kinase inhibitor, JNJ38877605, inhibits the viability of two MET-amplified NSCLC cell lines, EBC-1 and H1993 (Figure 1A and 1B). Inhibition of HGF using an HGF-specific neutralizing antibody had no impact on the viability of EBC-1 or H1993 cells (Figure 1A and 1B), confirming that MET amplification triggers ligand-independent MET signaling. We showed that JNJ38877605 inhibits the viability of EBC-1 cells in a dose- and time-dependent manner (Supplementary Figure 1A and 1B). In contrast, A549 cells, which do not harbor amplified MET, did not respond to the growth-inhibitory effect of JNJ38877605 (Figure 1C). This demonstrates that MET-amplified lung cancer cells display selective sensitivity to MET kinase inhibition.
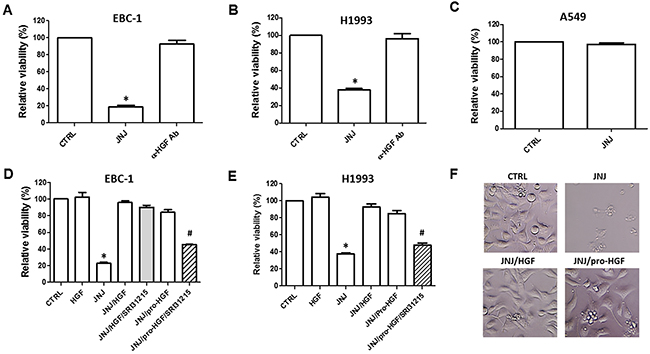
Figure 1: HGF rescues MET-amplified lung cancer cells from MET inhibition. EBC-1 (A) and H1993 (B) MET-amplified lung cancer cells, and A549 cells (C) which do not have amplified MET were treated with JNJ38877605 (JNJ) (25 nM) or HGF neutralizing antibody (α-HGF Ab). Cell viability was determined after 72 h by CellTiter Glo®. (D and E) EBC-1 and H1993 cells were treated with JNJ38877605 (25 nM), HGF (100 nM) or pro-HGF (100 nM) as indicated. Cell viability was determined by CellTiter Glo®. (F) Representative images of EBC-1 cells treated with JNJ38877605 alone or with HGF or pro-HGF after 72 h of treatment.
HGF has been shown to mediate resistance to several targeted therapeutic agents such as inhibitors of EGFR, BRaf and HER2 [20, 37]. Here we show that HGF also blocks the response to JNJ38877605, a specific MET kinase inhibitor, in MET-amplified EBC-1 and H1993 cells (Figure 1D-1F). HGF increased the survival of MET-inhibited cells in a dose-dependent manner (Supplementary Figure 1C). Pro-HGF, the inactive precursor of HGF, was as potent as HGF in protecting EBC-1 and H1993 cells from JNJ38877605-induced cell death (Figure 1D-1F), confirming that these cell lines express enzyme(s) that can activate pro-HGF into its mature form.
These data indicated that MET-amplified NSCLC cells rely on HGF for survival upon pharmacological inhibition of MET. We have shown earlier that SRI31215, a triplex inhibitor of matriptase, hepsin and HGFA, (Figure 2), inhibits pro-HGF activation [36]. Here we show that SRI31215 blocks the pro-survival activity of pro-HGF in EBC-1 and H1993 cells (Figure 1D and 1E). Consistent with its mode of action, SRI31215 does not impact the pro-survival activity of mature HGF (Figure 1D) [36].
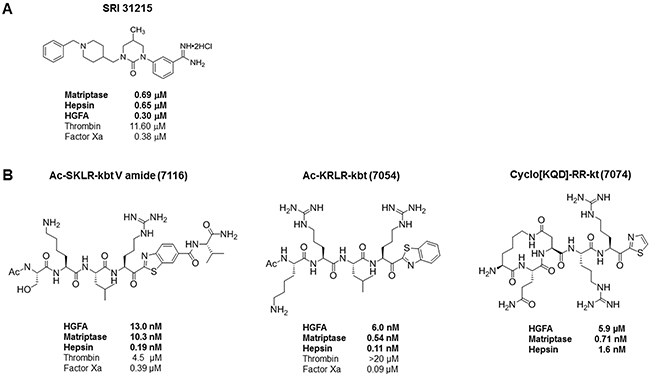
Figure 2: Molecular structure and activity of SRI31215 (A) and peptide inhibitors of pro-HGF activation, ZFH7054, ZFH7074 and ZFH7116 (B). The IC50 values for matriptase, hepsin and HGFA are indicated in bold.
The levels of matriptase and hepsin, two of the pro-HGF converting enzymes, are significantly elevated in lung cancer compared to normal lung tissue (Supplementary Figure 2). Moreover, the levels of HGF are increased in lung cancer tissue compared to normal lung mucosa (Supplementary Figure 2), suggesting that inhibition of pro-HGF activation is a valid approach to inhibit HGF/MET signaling.
Inhibition of pro-HGF activation overcomes fibroblast-mediated resistance to MET kinase inhibition
HGF is a key growth factor in the tumor micro-environment that inhibits the response of cancer cells to targeted therapy [20]. Cancer-associated fibroblasts are the main source of pro-HGF in the tumor microenvironment, therefore we tested whether inhibitors of pro-HGF activation can overcome fibroblast–mediated resistance to MET-targeted therapy. These experiments were performed in the absence of serum, which contains enzymes that can cleave and activate pro-HGF.
Consistent with our finding that HGF and pro-HGF block the activity of JNJ38877605 (Figure 1D and 1E), conditioned medium from WI38 lung fibroblasts which produce pro-HGF (Supplementary Figure 3A) rescued EBC-1 and H1993 cells from JNJ38877605-induced cell death (Figure 3A and 3B). We showed that fibroblasts with silenced HGF (Supplementary Figure 3A) failed to inhibit the response of EBC-1 cells to JNJ38877605 (Figure 3A). Accordingly, HGF neutralizing antibodies blocked the ability of fibroblasts to protect EBC-1 cells from JNJ38877605-induced cell death (Figure 3A). These experiments established that fibroblasts inhibit JNJ38877605-induced cell death through HGF. Consistent with published data [17], HGF or fibroblasts did not further stimulate MET signaling (Supplementary Figure 3B) or promote the growth of MET-amplified EBC-1 (Figure 3A).
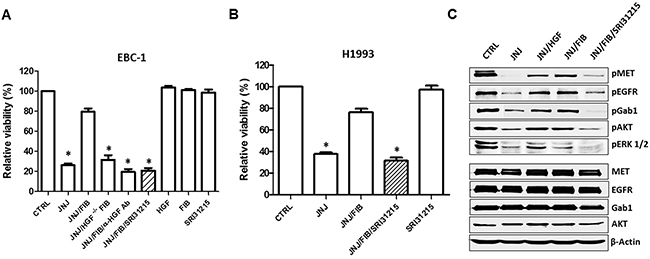
Figure 3: Inhibition of pro-HGF activation overcomes fibroblast-mediated resistance to MET tyrosine kinase inhibition. (A) EBC-1 cells were treated with JNJ38877605 (25 nM) alone or in the presence of conditioned medium (CM) from WI38 fibroblasts (FIB). CM was also prepared from fibroblasts with silenced HGF (HGF-/- FIB), or from fibroblasts cultured with HGF neutralizing antibody (α-HGF Ab) or SRI31215 (10 μM) as indicated. Cell viability was determined by CellTiter Glo®72 h after treatment. (B) H1993 cells were treated with JNJ38877605 (25 nM), fibroblast CM (FIB) and SRI31215 (10 μM) as indicated and cell viability was determined after 72 h. (C) Serum-starved EBC-1 cells were treated with JNJ38877605, recombinant HGF (100 nM), FIB and SRI31215 (10 μM) as indicated for 6 hours. Cell lysates were analyzed by immunoblotting for phospho- and total MET, EGFR, Gab1, AKT and ERK 1/2. *, p < 0.05 compared to JNJ/FIB.
Like HGF neutralizing antibodies, SRI31215 blocked the ability of fibroblasts to rescue EBC-1 and H1993 cells from JNJ38877605-induced cell death (Figure 3A and 3B). Accordingly, the substrate-based peptide inhibitors of matriptase, hepsin and HGFA, ZFH7054, ZFH7074 and ZFH7116 (Figure 2) restored the sensitivity of EBC-1 cells to JNJ38877605 in the presence of fibroblasts and overcame the ability of pro-HGF to rescue EBC-1 cells from MET inhibition (Supplementary Figure 3C). Treatment of EBC-1 cells with SRI31215, ZFH7054, ZFH7074 or ZFH7116 alone did not have any impact on the viability of EBC-1 cells (Supplementary Figure 3D), confirming that these compounds do not target cancer cells directly, but inhibit the crosstalk between cancer cells and fibroblasts.
To understand the mechanism whereby HGF elicits resistance to JNJ38877605, EBC-1 cells were treated with JNJ38877605 alone or in the presence of recombinant HGF or fibroblasts and SRI31215. As expected, JNJ38877605 inhibited the expression of p-MET, p-EGFR, p-Gab1, p-ERK 1/2 and p-AKT (Figure 3C). We demonstrated that HGF or pro-HGF-producing fibroblasts restored phosphorylation of MET, EGFR and Gab1 in JNJ38877605-treated EBC-1 cells. Accordingly, HGF or fibroblasts reactivated ERK1/2 and AKT signaling upon pharmacological MET inhibition by JNJ38877605. Importantly, SRI31215 blocked fibroblast-induced MET and EGFR signaling and AKT and ERK activation in MET-inhibited cells (Figure 3C), consistent with its ability to sensitize EBC-1 and H1993 cells to JNJ38877605 (Figure 3A and 3B). The levels of total MET, EGFR, Gab1 and AKT were not altered by the treatments (Figure 3C). Treatment of cells with HGF, fibroblasts or SRI31215 alone did not alter the expression or the activity of MET, EGFR, Gab1 or AKT (Supplementary Figure 3B).
Collectively, our data show that fibroblasts elicit resistance to MET-targeted therapy through HGF in MET-amplified lung cancer cells. We showed that HGF is sufficient to sustain MET and EGFR signaling and downstream activation of ERK and AKT upon MET inhibition. Inhibitors of pro-HGF that we developed blocked the pro-survival activity of fibroblasts, indicating that dual inhibition of MET and HGF is required to overcome resistance to MET-targeted therapy in MET-amplified NSCLC.
HGF supports the migration of MET-amplified EBC-1 lung cancer cells upon MET tyrosine kinase inhibition
MET activation promotes growth, survival and stimulates epithelial-mesenchymal transition which leads to enhanced migration of cancer cells [38]. Indeed, the migration of MET-amplified EBC-1 cells was completely inhibited by the MET tyrosine kinase inhibitor, JNJ38877605 (Figure 4). The inhibitory effect of JNJ38877605 on the migration of EBC-1 cells was prevented by either recombinant HGF or by WI38 lung fibroblasts (Figure 4). This result established that MET-amplified NSCLC cells become dependent on HGF not only for survival, but also for migration when MET is inhibited.
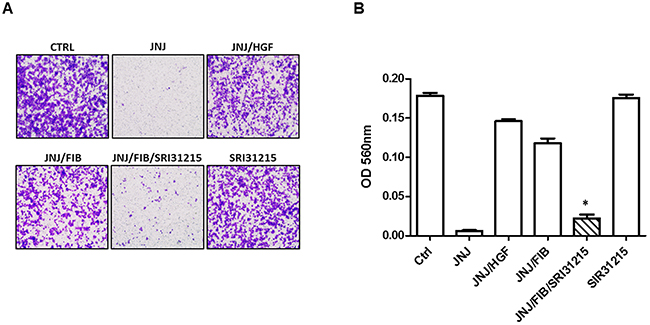
Figure 4: HGF upholds the migration of MET-addicted EBC-1 lung cancer cells upon MET tyrosine kinase inhibition. Migration of EBC-1 cells was assessed by a transwell migration assay. EBC-1 cells were treated with JNJ38877605 (25 nM) in the absence or presence of recombinant HGF or conditioned medium from fibroblasts (FIB) and SRI31215 (10 μM) for 24 h. (A) Representative images of stained cells that migrated across the polycarbonate membrane. (B) Relative migration of cells was determined by the optical density (OD) of stained cells at 560 nm.*, p < 0.05 compared to JNJ/FIB.
We have shown previously that SRI31215 impedes crosstalk between colon cancer cells and fibroblasts and thus inhibits the tumor-promoting activities of fibroblasts in colon cancer [36]. Here we demonstrate that SRI31215 blocks the ability of fibroblasts to restore the migration of MET-inhibited EBC-1 cells (Figure 4). The peptide inhibitors of pro-HGF activation, ZFH7054, ZFH7074 and ZFH7116 (Figure 2) which target the three pro-HGF activating enzymes [29], also blocked fibroblast-mediated migration of MET-inhibited EBC-1 cells (Supplementary Figure 4). Treatment of cancer cells with inhibitors of pro-HGF activation alone had no impact on the migration of EBC-1 cells (Figure 4 and Supplementary Figure 4).
Thus, while HGF (or fibroblasts) do not further stimulate MET signaling (Supplementary Figures 3B and 5A) or promote the viability (Figure 3) in MET-amplified cancer cells, our results established that MET-amplified NSCLC cells become addicted to HGF upon pharmacological MET inhibition. HGF-producing fibroblasts promote the survival and migration of MET-amplified lung cancer cells when MET is inhibited. We demonstrated that novel inhibitors of pro-HGF processing block crosstalk between tumor cells and fibroblasts and thus restrain the tumor-promoting activity of fibroblasts.
HGF-induced signaling in MET-inhibited cells
To understand how HGF signals in MET-amplified lung cancer cells when MET kinase activity is inhibited, we used the Proteome Profiler Human Phospho-RTK array (R&D Systems) to investigate the phosphorylation profiles of 49 different receptor tyrosine kinases. Additionally, we used Phospho-Kinase Array (R&D Systems) containing 46 kinases and their substrates.
Inhibition of MET kinase by JNJ38877605 blocked MET phosphorylation, however we showed that JNJ38877605 also significantly reduced phosphorylation of EGFR, RON (Figure 5A), AXL and RET (Supplementary Figure 5A). This suggests that MET cross-phosphorylates these RTKs in MET-amplified EBC-1 cells. Indeed, MET interacts with several other receptor tyrosine kinases (RTKs) and MET heterodimers with EGFR, RON, HER2 and HER3 have been found abundant in MET-amplified cancer cells [39].
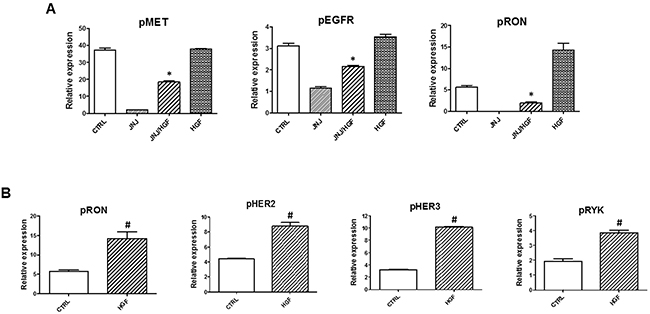
Figure 5: HGF-induced signaling in MET inhibited NSCLC cells. EBC-1 cells were treated with JNJ38877605 (25 nM) or HGF (100 nM) alone, or with a combination of JNJ38877605 and HGF for 6 hours. Cell lysates were prepared and subjected to antibody-based human phospho-receptor tyrosine kinase (RTK) array as described in methods to analyze the phosphorylation profiles of receptor tyrosine kinases. *, p < 0.05 compared to JNJ38877605 (JNJ); #, p < 0.05 compared to CTRL.
HGF restored phosphorylation of MET, EGFR and RON in MET-inhibited EBC-1 cells (Figure 5A, Supplementary Figure 5D), but did not rescue phosphorylation of AXL or RET (Supplementary Figure 5A) in JNJ38877605-treated cells. The array analysis confirmed that HGF restored activation of ERK and AKT in MET-inhibited cells (Supplementary Figure 5B). In addition, we found that WNK1, a prosurvival kinase that signals through ERK5 and Wnt [40], is highly activated in MET-amplified EBC-1 cells (Supplementary Figure 5C). Inhibition of MET kinase by JNJ38877605 significantly reduced phosphorylation of WNK1 in EBC-1 cells and HGF restored its activity in MET inhibited cells. In contrast, the levels of HSP60 were not restored by HGF in MET- inhibited cells (Supplementary Figure 5C). This indicates that novel mechanisms, such as WNK1 activation, may contribute to prosurvival activity of HGF in MET-amplified NSCLC cells and maintain the tumor-promoting crosstalk between tumor cells and cancer- associated fibroblasts. The potential role of WNK1 in HGF signaling and in HGF/fibroblast-dependent resistance to targeted therapy needs to be confirmed in other NSCLC cell lines.
How does HGF signal in MET-inhibited cells? It is possible that due to MET overexpression in MET-amplified cells there is always some residual MET in MET- inhibited cells, which responds to HGF stimulation. Indeed, we showed that HGF reactivates MET in JNJ38877605-treated cells (Figure 3 and Figure 5). However, we cannot exclude the possibility that HGF, at least in part, promotes survival of MET- amplified cells in a MET-independent manner. In fact, HGF stimulation increased phosphorylation of MET-related receptor RON, HER2, HER3 and RYK, which is likely to contribute to the pro-survival activity of HGF (Figure 5B).
DISCUSSION
The tumor-microenvironment plays a major role in resistance to targeted therapy. In this study we demonstrated that HGF, commonly produced by cancer-associated fibroblasts, confers resistance to MET kinase inhibitors in MET-amplified NSCLC cells. We showed that a novel inhibitor of pro-HGF activation, SRI31215, blocks the crosstalk between cancer cells and fibroblasts and overcomes resistance to anti-MET therapy in MET-amplified NSCLC cells.
The availability of targeted therapeutic options, such as EGFR inhibitors, has improved the management of patients with EGFR mutant NSCLC. However, only a fraction of patients shows a good clinical response to EGFR inhibitors and virtually all patients develop resistance to therapy [41–43]. Acquired resistance to EGFR inhibitors has been associated most frequently with selection for secondary EGFR mutations, such as T790M in exon 20 [6, 7] and MET amplifications [8]. MET-amplified lung cancers, which display ligand-independent MET activation, are addicted to MET signaling and are extremely sensitive to MET inhibition [13]. Nevertheless, acquired resistance to MET kinase inhibitors remains a major limitation of these drugs. In some cases, a switch to EGFR dependency has been shown to underlie the resistance to MET kinase inhibitors [16, 17]. However, some lung cancer cells with amplified MET and overexpressed EGFR fail to respond to combined treatment with EGFR and MET inhibitors or develop resistance to dual EGFR/MET inhibition [44]. Indeed, resistance to dual EGFR/MET inhibition has recently been reported in a MET-amplified NSCLC patient [45].
The tumor microenvironment has been shown to elicit resistance to therapy. HGF, which is commonly overexpressed in the tumor microenvironment, is a frequent source of resistance to targeted therapy [19–21]. HGF activates MET, expressed on tumor cells, and drives resistance to EGFR-targeted therapy by triggering ERK and PI3K/AKT pro-survival signaling and by promoting epithelial-mesenchymal transition [20]. Elevated levels of circulating HGF are associated with a mesenchymal tumor type, poor patient prognosis and resistance to EGFR inhibitors in lung cancer patients [11, 25]. Notably, EGFR inhibitors have been shown to elevate plasma levels of HGF in NSCLC patients [46], which may be due to therapy-induced recruitment of fibroblasts. HGF is also induced in airway epithelium by smoking [47], by far the biggest cause of lung cancer.
Here we demonstrated that HGF also drives resistance to anti-MET therapy in MET-amplified lung cancer cells (Figure 1 and Figure 3). Thus, MET-amplified NSCLC cells become addicted to HGF upon MET inhibition. HGF reactivates MET, EGFR and RON phosphorylation and restores AKT, ERK and WNK1 signaling upon MET inhibition (Figure 3, Figure 5, Supplementary Figure 5), promoting the survival of MET-inhibited cancer cells. Thus, simultaneous inhibition of HGF and MET may be required to prevent resistance to targeted therapy in MET-amplified NSCLC cells.
Neutralizing anti-HGF antibodies that are being tested, alone or in combination with other therapies, appear to be well tolerated. They have shown encouraging results, yielding partial response in cancer patients [28, 48–51]. There are currently no known small molecule inhibitors of HGF available, which have several advantages compared to therapeutic antibodies [52]. We developed SRI31215, a novel small molecule inhibitor of pro-HGF activation, which is the rate-limiting step in HGF/MET signaling [30]. HGF is synthesized and secreted by tumors or, more commonly, by stromal cells as an inactive precursor, pro-HGF. SRI31215 is a triplex inhibitor of the three serine proteases, matriptase, hepsin and HGFA, each of them being sufficient for the processing of pro-HGF into the biologically active ligand [31]. Structurally distinct triplex inhibitors of matriptase, hepsin and HGFA that we developed in parallel (Figure 2) exert similar biological activity as SRI31215 (Supplementary Figures 3 and 4). SRI31215 and the peptide inhibitors mimic the activity of Hepatocyte Growth Factor Activator Inhibitors (HAI-1/2), which act as endogenous inhibitors of pro-HGF activation, and inhibit matriptase, hepsin and HGFA [31, 53]. While HGF-activating enzymes are upregulated in lung cancer (Supplementary Figure 2), the levels of HAI-1/2 are commonly reduced in cancer tissue, resulting in increased activation of HGF. Reduced expression of the HAIs is associated with advanced stage disease and poor prognosis in many cancers [54–60].
We demonstrated previously that SRI31215 effectively inhibits HGF-dependent MET activation, proliferation, epithelial-mesenchymal transition and migration of cancer cells and overcomes fibroblast-mediated resistance to EGFR inhibitors in colon cancer cells [36]. Here we established that SRI31215, and peptide-based inhibitors of pro-HGF activation, block the tumor-promoting activity of fibroblasts and overcome the resistance of MET-amplified lung cancer cells to anti-MET therapy (Figures 3 and 4). Our preliminary data indicate that SRI31215 also overcomes resistance to double MET/EGFR inhibition (data not shown).
Several MET kinase inhibitors have been developed and have entered clinical trials. In preclinical studies cancer cells rapidly develop resistance to MET kinase inhibitors, which will probably translate to resistance in patients. It has been demonstrated that discontinuation of MET tyrosine kinase inhibitors results in increased and prolonged MET activation, coupled to enhanced tumor growth in vitro and in vivo [61], suggesting that MET inhibitors will need to be combined with other therapies [62].
The mechanisms whereby cancer cells develop resistance to MET kinase inhibitors are not well understood. Recently, a lung adenocarcinoma patient with MET amplification developed resistance to dual anti-EGFR/MET therapy due to an acquired METD1228V mutation [45]. We demonstrated that HGF, commonly overexpressed in the tumor microenvironment, confers resistance to MET- targeted therapy in MET-amplified NSCLC cells. Thus, small molecule inhibitors of pro-HGF activation such as those described herein, offer a new approach to restore sensitivity to MET or MET/EGFR inhibition in MET-amplified NSCLC patients. Plasma levels of HGF can serve as a biomarker to select lung cancer patients that would benefit from drugs that counteract the biological activity of HGF.
In summary, our data underscore a key role of the tumor microenvironment in acquired resistance to MET kinase inhibitors in MET-amplified NSCLC cells. We demonstrated that MET-amplified NSCLC cells become addicted to HGF under pharmacological MET inhibition. Because HGF is frequently overexpressed in the tumor microenvironment, our work highlights the need to include inhibitors of biological activity of HGF into therapeutic regimen in MET-amplified NSCLC patients.
MATERIALS AND METHODS
Materials
Primary antibodies used were anti-MET, anti-pMET, anti-EGFR, anti-pEGFR, anti-GAB1, anti-pGAB1, anti-AKT, anti-pAKT, anti-pERK1/2, (Cell Signaling Technology) and anti-β-actin (Sigma). The antibody arrays used were Proteome Profiler Human Phospho-Receptor Tyrosine Kinase (RTK) (ARY001B) and Phospho-Kinase Array (ARY003B) kits from R&D Systems. Recombinant HGF and pro-HGF were purchased from R&D Systems. The MET kinase inhibitor, JNJ38877605 was purchased from Selleck Chemicals (Selleckchem). HGF-specific and nonspecific (NSP) siRNAs were purchased from Dharmacon.
Cell culture
A549, WI38 and H1993 cells were obtained from ATCC and EBC-1 cells were purchased from XenoTech, KS. EBC-1, A549 and WI38 cells were maintained in minimum essential medium (MEM) and H1993 cells were cultured in RPMI medium. Growth media were supplemented with 10% fetal bovine serum, L-glutamine and antibiotics and cells were maintained under standard cell culture conditions at 37°C and 5% CO2. Conditioned medium from fibroblasts was prepared from 18Co fibroblasts as reported before [36]. Briefly, fibroblasts were maintained in complete medium until they reached confluence. The presence of HGF in conditioned media was confirmed by immunoblotting and by ELISA as described before [36]. Confluent cultures were rinsed and maintained in serum-free MEM medium for another 36 hours. Cell supernatants were collected, centrifuged and used immediately or were aliquoted and stored at -80°C.
Transwell migration assay
Cytoselect cell migration assay kit was purchased from Cell Biolabs and the assay was performed according to the manufacturer’s instructions. EBC-1 cells were seeded in transwells (8 μM pore size inserts) at a density of 7.5X104 cells/well in serum-free media. Transwells were placed in wells (24-well format) containing 10% serum-containing media and incubated for 24h. Cells that migrated were stained for 10 minutes at room temperature, transferred to wells containing 200 μl of extraction buffer and incubated for 10 minutes on a shaker. The optical density (OD) of stained cells was measured at 560 nm.
Viability assays
CellTiter Glo® luminescent assay kit from Promega was used to assess cell viability. Cells were seeded in 96-well plates at a density of 1X104 cells per well and treated as indicated. Cell viability was assessed 24, 48 and 72 hours after treatment, following the manufacturer’s protocol.
Immunoblotting
Immunoblotting was performed using standard procedures. Proteins were separated by SDS-PAGE. Membranes were blocked with 5% non-fat milk in Tris-buffered saline with Tween 20 (TBST) for 1 hour at room temperature (RT), and immunoblotted with primary antibodies in 5% BSA-TBST overnight at 4°C. Following washing with TBST buffer, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour and then washed with TBST. Membranes were developed using the ECL chemiluminescent detection system (Amersham).
Phosphokinase arrays
The phosphorylation profiles of kinases and their protein substrates were determined using Proteome Profiler Human Phospho-Receptor Tyrosine Kinase (RTK) and Human Phospho-Kinase Array kits (R&D Systems), following the manufacturers protocol. Serum-starved EBC-1 cells were treated with JNJ38877605 for 6h in the absence or presence of recombinant HGF. Cells were rinsed with PBS and then solubilized with lysis buffer at a concentration of 1X107 cells/mL. Array membranes that had been spotted with capture antibodies were blocked with blocking buffer and then incubated with 300 μg of cell lysates overnight. Arrays were washed to remove unbound proteins followed by incubation with detection antibodies conjugated to horseradish peroxidase. Phosphorylated proteins were detected using chemiluminescent reagents and quantified using the Image Quant LAS 4000 array analysis software (GE Healthcare Life Sciences).
HGF silencing
EBC-1 cells were seeded in a 6-well plate DMEM growth medium. Upon reaching 80-90% confluence, growth medium was changed to Opti-MEM and cells were transfected with HGF-specific siRNA (Dharmacon) using Lipofectamine 2000 (Invitrogen). Non-specific (NSP), nontargeting siRNA (Dharmacon) was used as a negative control. Both HGF and NSP siRNAs were delivered at a final concentration of 25 nM. Growth medium was changed to DMEM without antibiotics after overnight transfection. HGF silencing was confirmed by immunoblotting.
Statistical analyses
All experiments were repeated at least three times. Results are expressed as the mean ± SEM as indicated. Statistical analyses using two-tailed student’s t-test were performed with the GraphPad Prism 5.0 software. p < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
The work in the author’s lab was supported by the Alabama Innovation Fund. We thank Georg Wisniewski for reading the manuscript and for helpful suggestions.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497-1500.
2. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004; 305:1163-1167.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129-2139.
4. Rosell R, Bivona TG, Karachaliou N. Genetics and biomarkers in personalisation of lung cancer treatment. Lancet. 2013; 382:720-731.
5. Ayoola A, Barochia A, Belani K, Belani CP. Primary and acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer: an update. Cancer Invest. 2012; 30:433-446.
6. Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005; 352:786-792.
7. Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005; 2:e73.
8. Bean J, Brennan C, Shih JY, Riely G, Viale A, Wang L, Chitale D, Motoi N, Szoke J, Broderick S, Balak M, Chang WC, Yu CJ, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci U S A. 2007; 104:20932-20937.
9. Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014; 11:473-481.
10. Scagliotti G, von Pawel J, Novello S, Ramlau R, Favaretto A, Barlesi F, Akerley W, Orlov S, Santoro A, Spigel D, Hirsh V, Shepherd FA, Sequist LV, et al. Phase III multinational, randomized, double-blind, placebo-controlled study of tivantinib (ARQ 197) plus erlotinib versus erlotinib alone in previously treated patients with locally advanced or metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2015; 33:2667-2674.
11. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, Lindeman NI, Murphy C, Akhavanfard S, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010; 17:77-88.
12. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039-1043.
13. Smolen GA, Sordella R, Muir B, Mohapatra G, Barmettler A, Archibald H, Kim WJ, Okimoto RA, Bell DW, Sgroi DC, Christensen JG, Settleman J, Haber DA. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006; 103:2316-2321.
14. Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on MET for growth and survival. Cancer Res. 2007; 67:2081-2088.
15. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012; 12:89-103.
16. McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010; 70:1625-1634.
17. Zhang YW, Staal B, Essenburg C, Lewis S, Kaufman D, Vande Woude GF. Strengthening context-dependent anticancer effects on non-small cell lung carcinoma by inhibition of both MET and EGFR. Mol Cancer Ther. 2013; 12:1429-1441.
18. Presutti D, Santini S, Cardinali B, Papoff G, Lalli C, Samperna S, Fustaino V, Giannini G, Ruberti G. MET gene amplification and MET receptor activation are not sufficient to predict efficacy of combined MET and EGFR inhibitors in EGFR TKI-resistant NSCLC cells. PLoS One. 2015; 10:e0143333.
19. Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015; 25:198-213.
20. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, Cooper ZA, Chapman PB, Solit DB, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012; 487:500-504.
21. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013; 501:346-354.
22. Donev IS, Wang W, Yamada T, Li Q, Takeuchi S, Matsumoto K, Yamori T, Nishioka Y, Sone S, Yano S. Transient PI3K inhibition induces apoptosis and overcomes HGF-mediated resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer Res. 2011; 17:2260-2269.
23. Wang W, Li Q, Yamada T, Matsumoto K, Matsumoto I, Oda M, Watanabe G, Kayano Y, Nishioka Y, Sone S, Yano S. Crosstalk to stromal fibroblasts induces resistance of lung cancer to epidermal growth factor receptor tyrosine kinase inhibitors. Clin Cancer Res. 2009; 15:6630-6638.
24. Arrieta O, Cruz-Rico G, Soto-Perez-de-Celis E, Ramirez-Tirado LA, Caballe-Perez E, Martinez-Hernandez JN, Martinez-Alvarez I, Soca-Chafre G, Macedo-Perez EO, Astudillo-de la Vega H. Reduction in hepatocyte growth factor serum levels is associated with improved prognosis in advanced lung adenocarcinoma patients treated with afatinib: a phase II trial. Targeted Oncol. 2016; 11:619-629.
25. Kasahara K, Arao T, Sakai K, Matsumoto K, Sakai A, Kimura H, Sone T, Horiike A, Nishio M, Ohira T, Ikeda N, Yamanaka T, Saijo N, Nishio K. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin Cancer Res. 2010; 16:4616-4624.
26. Pennacchietti S, Cazzanti M, Bertotti A, Rideout WM 3rd, Han M, Gyuris J, Perera T, Comoglio PM, Trusolino L, Michieli P. Microenvironment-derived HGF overcomes genetically determined sensitivity to anti-MET drugs. Cancer Res. 2014; 74:6598-6609.
27. Patnaik A, Weiss GJ, Papadopoulos KP, Hofmeister CC, Tibes R, Tolcher A, Isaacs R, Jac J, Han M, Payumo FC, Cotreau MM, Ramanathan RK. Phase I ficlatuzumab monotherapy or with erlotinib for refractory advanced solid tumours and multiple myeloma. Br J Cancer. 2014; 111:272-280.
28. Tabernero J, Elez ME, Herranz M, Rico I, Prudkin L, Andreu J, Mateos J, Carreras MJ, Han M, Gifford J, Credi M, Yin W, Agarwal S, et al. A pharmacodynamic/pharmacokinetic study of ficlatuzumab in patients with advanced solid tumors and liver metastases. Clin Cancer Res. 2014; 20:2793-2804.
29. Han Z, Harris PK, Karmakar P, Kim T, Owusu BY, Wildman SA, Klampfer L, Janetka JW. α-Ketobenzothiazole serine protease inhibitors of aberrant HGF/c-MET and MSP/RON kinase pathway signaling in cancer. ChemMedChem. 2016; 11:585-599.
30. Venukadasula PK, Owusu BY, Bansal N, Ross LJ, Hobrath JV, Bao D, Truss JW, Stackhouse M, Messick TE, Klampfer L, Galemmo RA Jr. Design and synthesis of nonpeptide inhibitors of hepatocyte growth factor activation. ACS Med Chem Lett. 2016; 7:177-181.
31. Kawaguchi M, Kataoka H. Mechanisms of hepatocyte growth factor activation in cancer tissues. Cancers (Basel). 2014; 6:1890-1904.
32. Mazzone M, Basilico C, Cavassa S, Pennacchietti S, Risio M, Naldini L, Comoglio PM, Michieli P. An uncleavable form of pro-scatter factor suppresses tumor growth and dissemination in mice. J Clin Invest. 2004; 114:1418-1432.
33. Michieli P, Mazzone M, Basilico C, Cavassa S, Sottile A, Naldini L, Comoglio PM. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 2004; 6:61-73.
34. Matsumoto K, Nakamura T. Mechanisms and significance of bifunctional NK4 in cancer treatment. Biochem Biophys Res Commun. 2005; 333:316-327.
35. Murakami M, Nagai E, Mizumoto K, Saimura M, Ohuchida K, Inadome N, Matsumoto K, Nakamura T, Maemondo M, Nukiwa T, Tanaka M. Suppression of metastasis of human pancreatic cancer to the liver by transportal injection of recombinant adenoviral NK4 in nude mice. Int J Cancer. 2005; 117:160-165.
36. Owusu BY, Bansal N, Venukadasula PK, Ross LJ, Messick TE, Goel S, Galemmo RA, Klampfer L. Inhibition of pro-HGF activation by SRI31215, a novel approach to block oncogenic HGF/MET signaling. Oncotarget. 2016; 7:29492-29506. doi: 10.18632/oncotarget.8785.
37. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, Ribas A, Li J, Moffat J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012; 487:505-509.
38. Graveel CR, Tolbert D, Vande Woude GF. MET: a critical player in tumorigenesis and therapeutic target. Cold Spring Harb Perspect Biol. 2013; 5:a009209.
39. Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer. 2011; 105:807-813.
40. Moniz S, Jordan P. Emerging roles for WNK kinases in cancer. Cell Mol Life Sci. 2010; 67:1265-1276.
41. Russo A, Franchina T, Ricciardi GR, Picone A, Ferraro G, Zanghi M, Toscano G, Giordano A, Adamo V. A decade of EGFR inhibition in EGFR-mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget. 2015; 6:26814-26825. doi: 10.18632/oncotarget.4254.
42. Boolell V, Alamgeer M, Watkins DN, Ganju V. The evolution of therapies in non-small cell lung cancer. Cancers (Basel). 2015; 7:1815-1846.
43. Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. 2015; 16:e447-e459.
44. Yamaoka T, Ohmori T, Ohba M, Arata S, Kishino Y, Murata Y, Kusumoto S, Ishida H, Shirai T, Hirose T, Ohnishi T, Sasaki Y. Acquired resistance mechanisms to combination Met-TKI/EGFR-TKI exposure in Met-amplified EGFR-TKI resistant lung adenocarcinoma harboring an activating egfr mutation. Mol Cancer Ther. 2016; 15:3040-3054.
45. Bahcall M, Sim T, Paweletz CP, Patel JD, Alden RS, Kuang Y, Sacher AG, Kim ND, Lydon C, Awad MM, Jaklitsch MT, Sholl LM, Janne PA, Oxnard GR. Acquired MET D1228V mutation and resistance to MET inhibition in lung cancer. Cancer Discov. 2016; 6:1334-1341.
46. Tanaka H, Kimura T, Kudoh S, Mitsuoka S, Watanabe T, Suzumura T, Tachibana K, Noguchi M, Yano S, Hirata K. Reaction of plasma hepatocyte growth factor levels in non-small cell lung cancer patients treated with EGFR-TKIs. Int J Cancer. 2011; 129:1410-1416.
47. Chen JT, Lin TS, Chow KC, Huang HH, Chiou SH, Chiang SF, Chen HC, Chuang TL, Lin TY, Chen CY. Cigarette smoking induces overexpression of hepatocyte growth factor in type II pneumocytes and lung cancer cells. Am J Respir Cell Mol Biol. 2006; 34:264-273.
48. Van Cutsem E, Eng C, Nowara E, Swieboda-Sadlej A, Tebbutt NC, Mitchell E, Davidenko I, Stephenson J, Elez E, Prenen H, Deng H, Tang R, McCaffery I, et al. Randomized phase Ib/II trial of rilotumumab or ganitumab with panitumumab versus panitumumab alone in patients with wild-type KRAS metastatic colorectal cancer. Clinical Cancer Res. 2014; 20:4240-4250.
49. Martin LP, Sill M, Shahin MS, Powell M, DiSilvestro P, Landrum LM, Gaillard SL, Goodheart MJ, Hoffman J, Schilder RJ. A phase II evaluation of AMG 102 (rilotumumab) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol. 2014; 132:526-530.
50. Gordon MS, Sweeney CS, Mendelson DS, Eckhardt SG, Anderson A, Beaupre DM, Branstetter D, Burgess TL, Coxon A, Deng H, Kaplan-Lefko P, Leitch IM, Oliner KS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clinical Cancer Res. 2010; 16:699-710.
51. Rosen PJ, Sweeney CJ, Park DJ, Beaupre DM, Deng H, Leitch IM, Shubhakar P, Zhu M, Oliner KS, Anderson A, Yee LK. A phase Ib study of AMG 102 in combination with bevacizumab or motesanib in patients with advanced solid tumors. Clin Cancer Res. 2010; 16:2677-2687.
52. Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006; 6:714-727.
53. Kawaguchi M, Takeda N, Hoshiko S, Yorita K, Baba T, Sawaguchi A, Nezu Y, Yoshikawa T, Fukushima T, Kataoka H. Membrane-bound serine protease inhibitor HAI-1 is required for maintenance of intestinal epithelial integrity. Am J Pathol. 2011; 179:1815-1826.
54. Hoshiko S, Kawaguchi M, Fukushima T, Haruyama Y, Yorita K, Tanaka H, Seiki M, Inatsu H, Kitamura K, Kataoka H. Hepatocyte growth factor activator inhibitor type 1 is a suppressor of intestinal tumorigenesis. Cancer Res. 2013; 73:2659-2670.
55. Saleem M, Adhami VM, Zhong W, Longley BJ, Lin CY, Dickson RB, Reagan-Shaw S, Jarrard DF, Mukhtar H. A novel biomarker for staging human prostate adenocarcinoma: overexpression of matriptase with concomitant loss of its inhibitor, hepatocyte growth factor activator inhibitor-1. Cancer Epidemiol Biomarkers Prev. 2006; 15:217-227.
56. Oberst MD, Johnson MD, Dickson RB, Lin CY, Singh B, Stewart M, Williams A, al-Nafussi A, Smyth JF, Gabra H, Sellar GC. Expression of the serine protease matriptase and its inhibitor HAI-1 in epithelial ovarian cancer: correlation with clinical outcome and tumor clinicopathological parameters. Clinical Cancer Res. 2002; 8:1101-1107.
57. Zeng L, Cao J, Zhang X. Expression of serine protease SNC19/matriptase and its inhibitor hepatocyte growth factor activator inhibitor type 1 in normal and malignant tissues of gastrointestinal tract. World J Gastroenterol. 2005; 11:6202-6207.
58. Nakamura K, Abarzua F, Kodama J, Hongo A, Nasu Y, Kumon H, Hiramatsu Y. Expression of hepatocyte growth factor activator inhibitors (HAI-1 and HAI-2) in ovarian cancer. Int J Oncol. 2009; 34:345-353.
59. Hamasuna R, Kataoka H, Meng JY, Itoh H, Moriyama T, Wakisaka S, Koono M. Reduced expression of hepatocyte growth factor activator inhibitor type-2/placental bikunin (HAI-2/PB) in human glioblastomas: implication for anti-invasive role of HAI-2/PB in glioblastoma cells. Int J Cancer. 2001; 93:339-345.
60. Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, Latif F, Maher ER. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer Res. 2005; 65:4598-4606.
61. Pupo E, Ducano N, Lupo B, Vigna E, Avanzato D, Perera T, Trusolino L, Lanzetti L, Comoglio PM. Rebound effects caused by withdrawal of MET kinase inhibitor are quenched by a MET therapeutic antibody. Cancer Res. 2016; 76:5019-5029.
62. Finisguerra V, Prenen H, Mazzone M. Preclinical and clinical evaluation of MET functions in cancer cells and in the tumor stroma. Oncogene. 2016; 35:5457-5467.
63. Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, Murphy SE, Yang P, Pesatori AC, et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008; 3:e1651.
64. Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, Lizyness ML, Kuick R, Hayasaka S, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002; 8:816-824.