
INTRODUCTION
Kidney cancer represents one of the most frequent malignant neoplasms in the United States [1]. About 80% of kidney tumors belong to the clear cell renal cell carcinoma (ccRCC) histology. Thirty percent of ccRCC patients develop metastatic disease (mRCC), which is associated with poor prognosis and short overall survival (OS) [2]. However, since the approval of the first targeted drug (sorafenib) in 2005, the therapeutic landscape has changed considerably, and OS of mRCC increased from nine months in 2005 to 30 months in 2011 [3]. Today, approved targeted drugs for ccRCC include tyrosine-kinase inhibitors (TKI), mTOR pathway inhibitors and antibodies against the vascular endothelial growth factor (VEGF). In particular, TKIs such as sunitinib, which target multiple receptors at the same time, have been highly successful in the treatment of mRCC patients [4]. Currently, ten different drugs are approved for first- and second-line therapy [5–10]. The efficacy of these agents is highest in the first-line setting with the choice of agent class being important for OS [11]. However, the optimal sequence of targeted drugs is unknown due to a lack of biomarkers for patient stratification [12, 13]. On the molecular level, ccRCC is a heterogeneous tumor displaying a broad spectrum of genetic alterations [14, 15]. Multiregion sequencing revealed intra-tumor heterogeneity of mRCC with wide subclonal diversity [16–18]. Here, we report the results of a pilot study (MORE, Molecular Renal Cancer Evolution) using whole-exome sequencing (WES) of primary and biopsy tissue samples from treatment-naïve patients at baseline and after TKI treatment respectively. Our aim was to assess whether genetic alterations that can be exploited therapeutically develop in metastatic sites during disease progression under TKI regimens.
RESULTS
Patients
Four of the 17 patients enrolled in the MORE study (Table 1) showed progression under TKI treatment. At these time points, metastasis biopsies were collected, and DNA from tumor tissues at baseline and progression was subjected to WES. Patient 1 (female, 81.3 years) progressed under sunitinib therapy after 5.4 months, and a biopsy (vaginal wall metastasis) was taken (Figure 1). Patient 2 (female, 70.5 years) progressed after 6.2 months under sunitinib therapy. After needle biopsy from the right chest wall, the therapy was continued with axitinib until she suffered a second progression after 11.8 months (Figure 1). Patient 3 (male, 46.3 years) showed no residual malignant disease after cytoreductive nephrectomy and adjuvant sunitinib treatment. At relapse after 6.1 years (73 months), a baseline biopsy of the left chest wall metastasis was taken, and treatment was continued with pazopanib. He progressed at 3.9 months and the therapy was switched to everolimus, which was discontinued after 1.8 months due to clinical progression (soft tissue metastasis). After re-biopsy of the chest wall metastasis, nivolumab treatment was initiated (Figure 1). Patient 4 (male, 58.9 years), progressed after 5.3 months sunitinib treatment, and a biopsy was collected from the os ilium. In addition, a cutaneous nodule in the left chest wall could be resected when he progressed after 6 months on axitinib (Figure 1). The therapy was continued with everolimus for 1.5 months, followed by pazopanib for 2 months. The patient died of the disease soon thereafter.
Table 1: Clinical characteristics of the four patients who experienced tumor progression
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
|
|---|---|---|---|---|
Age (y) |
81.3 |
70.5 |
46.3 |
46.3 |
Gender |
F |
F |
M |
M |
TNM |
T4N2M1 |
T1aN0M1 |
T3bN2M1 |
T3aN2M1 |
Fuhrman Grade |
G3 |
G2 |
G2 |
G3 |
Site of metastasis |
paraaortal lymph node, lung, bilataral adrenal, cava thrombus |
lung, bone, liver |
retroperineal lymph node, bone, chest wall, pleura |
lung, bone, adrenal |
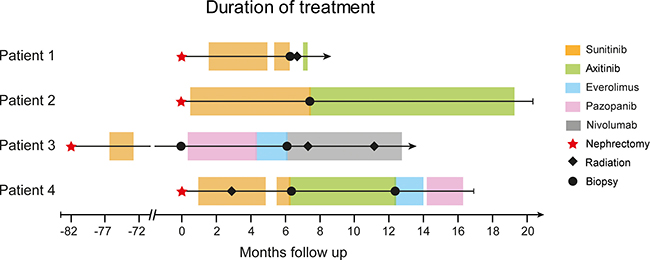
Figure 1: Swimmer plot with therapy sequences and durations of different treatment lines for each patient. The different therapies are indicated by colors and include nephrectomy, radiation therapy, and biopsies from progressive sites.
Molecular analysis at baseline and tumor progression
To evaluate clonal diversity and molecular alterations upon progression on TKI treatment of the four patients (Nr. 1-4), we performed WES of the primary tumor and biopsy samples from metastatic sites upon disease progression (Figure 1). Matched buffy coat samples were sequenced to exclude germline alterations from sequencing data. Sequencing of tumor and and matched buffy coat resulted in a mean target coverage of 124× and revealed an average of 486 non synonymous somatic mutations (Table 2, Supplementary Table 1) with a mean coverage of 104× at mutation sites. Mutations in RCC-associated genes, including VHL, BAP1, PBRM1, LRP1B, and KMT2C were further validated by Sanger sequencing. Consistent with previous reports [16, 17], the genetic compositions among the four patients were diverse (Figure 2), with alterations in genes known to be recurrently mutated in ccRCC, i.e. VHL, SETD2, PBRM1, and BAP1 [14, 15] and others which are known oncogenes or might be potential therapy targets. These selected genes are shown in Figure 3. All somatic nonsynonymous mutations and their resulting effect on transcripts and proteins are given in Supplementary Table 1.
Table 2: Sample characteristics and quality metrics of the sequencing data
Case |
Type |
Timepoint |
Site |
Target coverage [x] |
Mapped Reads [%] |
Insert Size [bp] |
Functional somatic SNVs |
|---|---|---|---|---|---|---|---|
Patient 1 |
Tumor |
Baseline |
right kidney |
106.03 |
86.11 |
166 |
720 |
Metastasis |
1. Progress |
left vaginal wall |
121.8 |
85.63 |
169 |
760 |
|
Buffy Coat |
Baseline |
blood |
109.93 |
88.83 |
171 |
- |
|
Patient 2 |
Tumor |
Baseline |
left kidney |
111.81 |
85.38 |
172 |
500 |
Metastasis |
1. Progress |
right chest wall |
110.09 |
88.96 |
174 |
580 |
|
Buffy Coat |
Baseline |
blood |
117.94 |
88.56 |
173 |
- |
|
Patient 3 |
Metastasis 1 |
Baseline |
left chest wall |
156.9 |
97.73 |
184 |
81 |
Metastasis 2 |
1. Progress |
left chest wall |
151.96 |
98.34 |
164 |
251 |
|
Buffy Coat |
Baseline |
blood |
158.27 |
97.95 |
164 |
- |
|
Patient 4 |
Tumor |
Baseline |
left kidney |
109.85 |
88.33 |
172 |
371 |
Metastasis 1 |
1. Progress |
os ilium |
117.01 |
90.06 |
176 |
342 |
|
Metastasis 2 |
2. Progress |
left chest wall |
126.32 |
87.42 |
167 |
770 |
|
BuffyCoat |
Baseline |
blood |
120.94 |
90.53 |
174 |
- |
|
Mean |
124.53 |
90.29 |
171.23 |
486 |
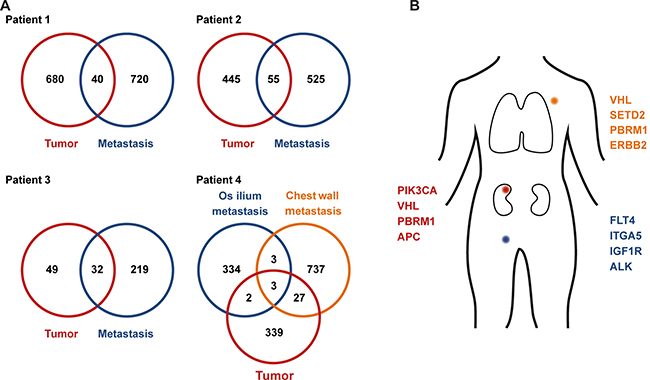
Figure 2: Clonal mutations in primary and metastatic sites. Numbers of exclusive and shared mutations identified in primary tumor tissue and metastasis biopsies (A). Coding mutations in the primary tumor and metastasis biopsies from patient 4 (B).
We found remarkable heterogeneity between primary and metastatic lesions with only a small subset of alterations present in both sites (Figure 2): WES of the primary tumor from patient 1 identified private mutations in the tumor suppressor genes PTEN and APC as well as in HRAS, BCAR1 (COSM1479104), and SETD1A (COSM126103). Forty mutations, including in VHL (COSM14311), BAP1, and STK25 were shared between primary and metastatic site (Figure 3, Patient 1). The metastasis upon progression under sunitinib carried mutations in FLT4, KMT2D, and BMP5, which were not detected in the primary tumor. We identified 55 mutations shared between primary tumor and metastasis of patient 2 (Figure 2A), including BAP1, LRP1B, and BCAR1. She had a private mutation in PIK3CA (COSM1041490) in the primary tumor. In line with previous findings suggesting that mTOR mutations occur predominantly at advanced stages of tumor evolution in subclonal metastatic populations [16, 17], the metastasis revealed a private mTOR mutation. Additional mutations were found in FGFR1 and ERBB2 (Figure 2). After cytoreductive nephrectomy and adrenalectomy of the left kidney, patient 3 was treated with sunitib and showed no evidence of residual disease or indications of progression during follow-up. When the tumor relapsed after more than six years with synchronous bone, pleural and chest wall metastases, a biopsy from the left chest wall metastasis was used as a baseline sample. After progression on pazopanib and everolimus, this metastasis was re-biopsied, and the therapy was continued with nivolumab. WES analysis showed an increase of mutational load between baseline (81 mutations) and progression (251 mutations), and 32 (39.5%) of the 81 baseline mutations were also found in the re-biopsy (Figure 3), including changes in VHL, BAP1, KMT2C, CSMD3, and FAT3. The re-biopsy revealed additional private mutations in the epigenetic regulators KMT2D (COSM1299437) and KMT2E (COSM1083684) as well as in PBRM1, one of the most frequently mutated genes in ccRCC, which codes for a subunit of the BAF chromatin remodeling complex. Furthermore, we detected a mutation in BRCA1, which was not been present at baseline. In patient 4, WES of the primary tumor and two different locations of metastases was performed (Figure 2A, 2B). Only three (RYR3, ABI3BP, and TMEM255A) of the 1445 identified nonsynonymous somatic mutations were found to be present at every site. Notably, the mutational spectrum of the primary tumor revealed a larger overlap with the chest wall metastasis than with the initial pelvic bone metastasis (30 vs. 5 mutations, respectively), including changes in VHL and PBRM1, possibly indicating an early separation of the cells giving rise to the latter metastasis. The analysis of the chest wall metastasis identified further mutations in cancer-associated genes, including SETD2 and ERBB2. In addition, with 700 somatic variants, the mutational load of the chest wall metastasis was considerably higher than in the primary tumor and the metastasis located in the os ilium (371 and 342 mutations, respectively). This suggests an early separation of cells giving rise to the chest wall lesion.
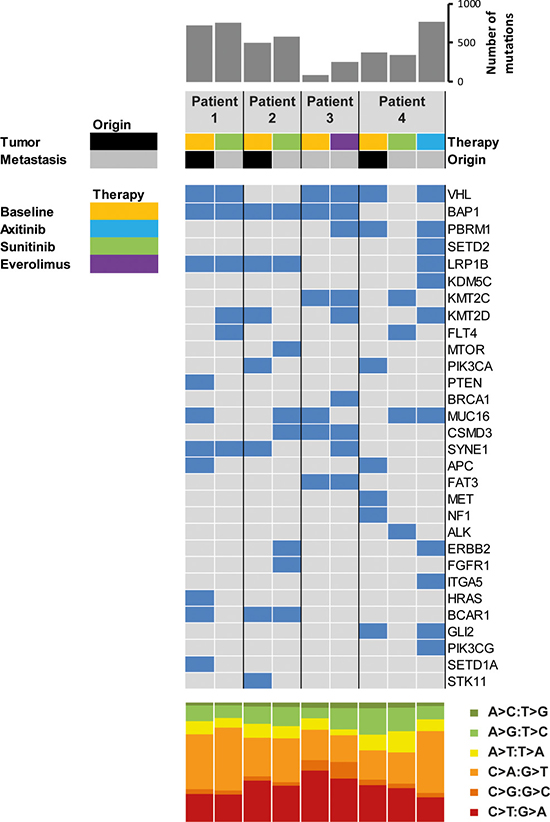
Figure 3: Coding somatic mutations identified in primary tumors and metastatic sites at baseline and upon progression. The upper histogram shows the number of nonsynonymous mutations per sample. The heatmap indicates the presence (blue) and absence (grey) of mutations in selected cancer-associated genes in primary and progressive samples of the four patients. The lower histogram shows the proportions of base substitutions for each sample.
DISCUSSION
The current pharmacological treatment strategies in ccRCC are based on targeted drugs and, more recently, immune checkpoint inhibitors. Although numerous TKIs are available, tumor heterogeneity and secondary resistance are major challenges for cancer therapy. Especially a heterogeneous phenotypic response to TKI therapy and progression has recently been found [19]. Hence, a deeper understanding of the complexity of molecular and mutational signatures of ccRCC is needed. This can be achieved with the ultra-high DNA sequencing. However, apart from large catalogues of somatic mutations in cancer including ccRCC [14, 15] provided by international sequencing projects like TCGA and ICGC, multiregional sequencing also revealed remarkable intra-tumor heterogeneity [16–18, 20] Here, we provide evidence for considerable temporal molecular heterogeneity between therapy-naïve primary tumors and metastases developing under TKI. The mutational profiles provide insights into clonal evolution occuring during tumor progression under therapy: Molecular profiling revealed recurrent genomic alterations in genes frequently altered in ccRCC [14], including VHL, SETD2, and BAP1. Deletions of the VHL tumor suppressor gene are occuring early during tumorigenesis of ccRCC [16]. We detected additional VHL mutations in baseline samples and metastatic sites in three out of four patients. Well-known alterations were found to be common to primary and metastatic sites. These included mutations in the ccRCC tumor-driving gene BAP1 in patients 1, 2, and 3. The clonality of the BAP1 mutation, which has been associated with poor prognosis and a high metastasizing potential [21] supports BAP1 as a molecular marker for ccRCC sub-classification [21].
In agreement with previous reports [16, 17], our WES results revealed that mTOR mutations occur predominately in subclonal branches in advanced disease stages (patient 2). We also identified two different ITGA5 mutations in independent metastases, but not in the primary tumor, of patient 4, suggesting parallel evolution of the two metastatic clones. In contrast to patients 1–3, most of the mutations in patient 4 were found to be private for each sample. This might be due to several reasons: Either the initial molecular features present in the primary tumor were lost in the metastases, or multiple novel mutations accumulated in the metastases, or the sequenced tissue region in primary tumor did not contain the clones that gave rise to the metastases. Although we cannot distinguish between these possibilities, all of them are consistent with a high molecular heterogeneity present early during tumor development, followed by clonal selection and/or independent evolution after dissemination of tumor cells. Supported by previous findings in breast cancer [22], the larger overlap of the primary tumor with the chest wall metastasis than with the initial pelvic bone metastasis possibly indicates an early separation of the cells giving rise to the latter metastasis.
The identification of clinically relevant mutations upon tumor progression under TKI treatment suggests that it might be possible to derive alternative targeted therapies based on molecular changes in the metastases. The analysis of the molecular patterns upon TKI progression revealed different potentially targetable mutations in all cases tested. While we detected subclonal mutations in FLT4, ITGA5, SETD2, and BRCA1 in the metastases, one patient developed mutations in mTOR and several receptor tyrosine kinase genes, including ERBB2, ERBB4, and FGFR1. Moreover, the chest wall re-biopsy of patient 3 exhibited mutations in PBRM1 and BAP1, which are mostly mutually exclusive. Their co-occurrence has been associated with aggressive tumors and poor prognosis [23, 24]. The acquired BRCA1 mutation, which was not detected in the baseline of patient 3, represents another impairment of DNA repair, which might indicate that PARP inhibitors could be beneficial in this case. Activity of PARP in BRCA-mutated ovarian and prostate cancer is well established and olaparib is clinically approved [25, 26]. Activity of PARP inhibitors in BRCA-mutated ccRCC has not yet been reported. However, the molecular principle remains similar through different entities, and an individual treatment approach after failure of approved substances may be warranted. Alternatively, BRCA1 mutations giving rise to impaired DNA repair may result in increased expression of neoantigens, a potential marker of sensitivity to immune checkpoint inhibitors. Furthermore, VEGFR3/FLT4 inhibitors that are currently in early clinical development may be active in patients carrying VEGFR3/FLT4 mutations, respectively [27]. Mutations in mTOR may be particularly sensitive to mTOR inhibitory drugs. Finally, several drugs targeting the FGFR are currently under clinical development.
To the best of our knowledge, this study is the first to examine molecular alterations associated with ccRCC therapy resistance in tissue DNA in a longitudinal fashion. The molecular data provide evidence for various routes leading to drug resistance of ccRCC subclones with acquired mutations in known therapy target genes on metastatic progression.
The major limitation of our study is its small sample size due to the low progression rate to date, and our findings have to be corroborated in larger studies. To this end, MORE is open and actively recruiting, and we will provide an extension of the current data set in due time. However, our results agree with comparable precision medicine approaches in other tumor entities in that the progression of patients under therapy is very specific and restricted to the individual case. In other words, highly recurrent targetable mutations cannot be expected from the analysis of many more cases. In contrast, the serial molecular analysis of tumors from individual ccRCC patients progressing under therapy might indicate ways leading to therapy resistance and support tailored treatment decisions.
MATERIALS AND METHODS
The MORE protocol
The MORE (Molecular Renal Cancer Evolution) study is a prospective clinical study designed to investigate molecular alterations in metastatic ccRCC progressing under first-line TKI. The primary objective of MORE is to characterize molecular alterations in metastatic lesions compared to baseline tissue in order to understand the individual factors leading to therapy resistance and tumor progression. Secondly, we hope to identify potential molecular targets for personalized therapies for the progressive patients in order to improve individual patient care. The study was approved by the ethics committee at the Heidelberg University Medical Faculty (S-539/2013) and listed on clinicaltrials.gov (NCT02208128) and the national study register (DRKS0006193). Patients with metastatic disease and no prior systemic therapy were eligible for the study. At baseline, tissue samples from debulking primary tumor surgery or biopsy samples were taken. After standard of care first and second line therapies, biopsies of progressing metastases were sampled.
Patients and samples
Seventeen patients were classified as low risk according to the MSKCC [8] prognostic score and had pure clear cell histology at baseline and progression. These included 11 male and six female patients with a median age of 65 years (range: 46 to 82 years) at the date of initial diagnosis. Four patients had initial tumor stage T1, one patient T2, nine patients T3, and two patients T4 disease. Sixteen patients had already developed metastatic disease at diagnosis. Of the 17 recruited patients, four progressed under TKI treatment and were subjected to the molecular analyses (Table 1). Selection criteria for these four patients were the disease progression and the availability of high quality DNA from progressive metastases. Six primary tumor and three normal adjacent tissue specimens from each patient were taken upon resection and immediately frozen in liquid nitrogen. Biopsies were taken during tumor progression. The histology of all tissue samples were confirmed by a board-certified pathologist. Unstained and unembedded samples with a median tumor cell fraction of 70% (range: 40–90%) were subjected to DNA isolation, followed by WES.
Isolation, quantification, and quality control of genomic DNA
Fresh frozen surgical tumor tissue and biopsy samples (10–30 mg) were mechanically disrupted and homogenized using the TissueRuptor (Qiagen, Hilden, Germany). Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s protocol. As germline control, genomic DNA from matched blood cells was isolated using the QIAamp DNA Blood Mini Kit (Qiagen). DNA concentrations were determined using the Qubit (Thermo Fisher Scientific, Waltham, MA, USA), and DNA integrity was assessed using the TapeStation (Agilent Technologies, Santa Clara, CA, USA).
Library construction and exome sequencing
Sequencing libraries were prepared from 200 ng genomic DNA. Prior to library preparation, all DNA samples were sheared to an average fragment length of 150 bp using the Covaris S220 ultrasonicator. Exome-enriched sequencing libraries were prepared using the Agilent SureSelectXT Human All Exon V5+UTR kit (low input protocol). Library sizes and qualities were evaluated before and after capture by Bioanalyzer 2100 analysis using the High Sensitivity DNA Kit (Agilent) and quantified using the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific). All libraries were subjected to 100 bp paired-end sequencing on the Illumina HiSeq 2000 v3 at the DKFZ Genomics and Proteomics Core Facility. Selected variants were further validated by bidirectional Sanger sequencing on an ABI 3130 Genetic Analyzer (Thermo Fisher Scientific, Waltham, USA) using the BigDye Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific, Waltham, USA) as described previously [28].
NGS data analysis and variant calling
Sequence data analysis and mutation calling was performed using the One Touch Pipeline (OTP), a fully automated computational platform for sequence analysis. Briefly, raw FASTQ data files and metadata were loaded into OTP. After quality score estimation using FastQC, the files were aligned to the human genome (hg19) using BWA [29]. All aligned files were subsequently merged with Picard. Files belonging to one same sample were combined into one file, which was quality controlled using a variety of in-house tools and standard SAMtools [30]. PCR duplicates were removed, and only unique reads were used for variant calling and target coverage estimation. After processing of bam files, an in-house SNV calling pipeline, consisting of three different steps (calling, annotation, and filtering) was used based on each tumor-control comparison. For variant calling, samtools, mpileup, and bcftools were used. Called variants were annotated using the dbSNP and COSMIC databases and filtered for somatic variants in protein coding regions with a mutant allele frequency of > 2.5%. If a mutation was found in only one of the matched primary/metastatic samples from the same patient, unfiltered somatic and non synonymous exonic variants were called independent of their allele frequencies.
Data availability
FASTQ files are available at the European Genome-Phenome Archive (https://ega-archive.org/) under the accession number EGAS00001001861.
Abbreviations
ccRCC, clear cell renal cell carcinoma; MORE, Molecular Renal Cancer Evolution; mRCC, metastatic renal cell carcinoma; OS, overall survival; OTP, One Touch Pipeline; SD, stable disease; TKI, tyrosine kinase inhibitor; VEGF, vascular endothelial growth factor; WES, whole-exome sequencing.
Author contributions
Study design: SP, CG; patient treatment: SP, MH, DJ, CG; study coordination: HS, SP, CG; sample processing: SDi, YJD, SDu, ALR, A-LV, AS; data analysis: ER, SDi, MS; manuscript writing: SDi, HS, SP, CG.
ACKNOWLEDGMENTS
We thank Christina Geörg for sample management and the Genomics and Proteomics Core Facility at DKFZ for sequencing analyses.
CONFLICTS OF INTEREST
The authors have declared no conflicts of interest.
FUNDING
This work was supported by the DKFZ-Heidelberg Center for Personalized Oncology (DKFZ-HIPO) and funded through HIPO project number 045.
REFERENCES
1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9–29.
2. Heng DY, Xie W, Regan MM, Warren MA, Golshayan AR, Sahi C, Eigl BJ, Ruether JD, Cheng T, North S, Venner P, Knox JJ, Chi KN, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol. 2009; 27:5794–5799.
3. Manola J, Royston P, Elson P, McCormack JB, Mazumdar M, Négrier S, Escudier B, Eisen T, Dutcher J, Atkins M, Heng DY, Choueiri TK, Motzer R, Bukowski R, and International Kidney Cancer Working Group. Prognostic model for survival in patients with metastatic renal cell carcinoma: results from the international kidney cancer working group. Clin Cancer Res. 2011; 17:5443–5450.
4. Schmid TA, Gore ME. Sunitinib in the treatment of metastatic renal cell carcinoma. Ther Adv Urol. 2016; 8:348–371.
5. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt-Wolf IG, et al, and Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007; 356:2271–2281.
6. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, Chevreau C, Filipek M, Melichar B, Bajetta E, Gorbunova V, Bay JO, Bodrogi I, et al, and AVOREN Trial investigators. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007; 370:2103–2111.
7. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, et al, and RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008; 372:449–456.
8. Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009; 27:3584–3590.
9. Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarba JJ, Chen M, McCann L, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010; 28:1061–1068.
10. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG, Negrier S, Ou YC, Castellano D, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011; 378:1931–1939.
11. Motzer R, Barrios C, Kim T, Falcon S, Cosgriff T, Graydon Harker W, Pittman K, Sabbatini R, Rha S, Flaig T, Page R, Bavbek S, Beck J, et al. Record-3: Phase II randomized trial comparing sequential first-line everolimus (EVE) and second-line sunitinib (SUN) versus first-line SUN and second-line EVE in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2013; 31:abstr 4504.
12. Escudier B, Gore M. Sequencing therapy in metastatic renal cell cancer. Semin Oncol. 2013; 40:465–471.
13. Calvo E, Grunwald V, Bellmunt J. Controversies in renal cell carcinoma: treatment choice after progression on vascular endothelial growth factor-targeted therapy. Eur J Cancer. 2014; 50:1321–1329.
14. Creighton CJ, Morgan M, Gunaratne PH, Wheeler DA, Gibbs RA, Gordon Robertson A, Chu A, Beroukhim R, Cibulskis K, Signoretti S, Vandin Hsin-Ta Wu F, Raphael BJ, Verhaak RG, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–49.
15. Scelo G, Riazalhosseini Y, Greger L, Letourneau L, Gonzalez-Porta M, Wozniak MB, Bourgey M, Harnden P, Egevad L, Jackson SM, Karimzadeh M, Arseneault M, Lepage P, et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat Commun. 2014; 5:5135.
16. Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, Martinez P, Phillimore B, Begum S, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014; 46:225–233.
17. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–892.
18. Serie DJ, Joseph RW, Cheville JC, Ho TH, Parasramka M, Hilton T, Thompson RH, Leibovich BC, Parker AS, Eckel-Passow JE. Clear Cell Type A and B Molecular Subtypes in Metastatic Clear Cell Renal Cell Carcinoma: Tumor Heterogeneity and Aggressiveness. Eur Urol. 2016.
19. Crusz SM, Tang YZ, Sarker SJ, Prevoo W, Kiyani I, Beltran L, Peters J, Sahdev A, Bex A, Powles T, Gerlinger M. Heterogeneous response and progression patterns reveal phenotypic heterogeneity of tyrosine kinase inhibitor response in metastatic renal cell carcinoma. BMC Med. 2016; 14:185.
20. Hoefflin R, Lahrmann B, Warsow G, Hubschmann D, Spath C, Walter B, Chen X, Hofer L, Macher-Goeppinger S, Tolstov Y, Korzeniewski N, Duensing A, Grullich C, et al. Spatial niche formation but not malignant progression is a driving force for intratumoural heterogeneity. Nat Commun. 2016; 7:ncomms11845.
21. Brugarolas J. Molecular genetics of clear-cell renal cell carcinoma. J Clin Oncol. 2014; 32:1968–1976.
22. Hosseini H, Obradovic MM, Hoffmann M, Harper KL, Sosa MS, Werner-Klein M, Nanduri LK, Werno C, Ehrl C, Maneck M, Patwary N, Haunschild G, Guzvic M, et al. Early dissemination seeds metastasis in breast cancer. Nature. 2016.
23. Kapur P, Pena-Llopis S, Christie A, Zhrebker L, Pavia-Jimenez A, Rathmell WK, Xie XJ, Brugarolas J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol. 2013; 14:159–167.
24. Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, Yamasaki T, Zhrebker L, Sivanand S, Spence P, Kinch L, Hambuch T, Jain S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012; 44:751–759.
25. Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, Matei D, Macpherson E, Watkins C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012; 366:1382–1392.
26. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, Boysen G, Porta N, Flohr P, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med. 2015; 373:1697–1708.
27. Alam A, Blanc I, Gueguen-Dorbes G, Duclos O, Bonnin J, Barron P, Laplace MC, Morin G, Gaujarengues F, Dol F, Herault JP, Schaeffer P, Savi P, et al. SAR131675, a potent and selective VEGFR-3-TK inhibitor with antilymphangiogenic, antitumoral, and antimetastatic activities. Mol Cancer Ther. 2012; 11:1637–1649.
28. Pfarr N, Kriegsmann M, Sinn P, Klauschen F, Endris V, Herpel E, Muckenhuber A, Jesinghaus M, Klosterhalfen B, Penzel R, Lennerz JK, Weichert W, Stenzinger A. Distribution of MED12 mutations in fibroadenomas and phyllodes tumors of the breast—implications for tumor biology and pathological diagnosis. Genes Chromosomes Cancer. 2015; 54:444–452.
29. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760.
30. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079.