
INTRODUCTION
Selumetinib (AZD6244, ARRY-142886) is a potent, orally active inhibitor of mitogen-activated protein/extracellular signal-regulated kinase (ERK) kinase (MEK)-1/2 that suppresses the pleiotropic output of the RAF/MEK/ERK pathway [1, 2]. The kinase pathway of RAF/MEK/ERK is activated in most human tumors, often through gain-of-function mutations of RAS and RAF family members [3, 4]. In vitro studies demonstrated a tendency toward sensitivity to MEK inhibitors in tumor cell lines harboring KRAS or BRAF mutations [1–3, 5, 6]. Based on this preclinical evidence, several clinical trials have tested or are testing the efficacy of MEK inhibitors in KRAS-mutant lung cancer patients and colorectal cancer [5, 7, 8].
On the other hand, there was no absolute correlation with mutational or phospho-protein markers of BRAF/MEK, RAS, or phosphoinositide 3-kinase (PI3K) activity to MEK inhibitor response in large tumor cell panels of diverse cancer types [2]. An 18-gene signature enabling measurement of MEK functional output independent of tumor genotype has been reported [2]. There have been several studies to show that RAS pathway is activated in the absence of KRAS mutation and the RAS pathway signature is superior to KRAS mutation status for the prediction of response to RAS pathway inhibitor [9].
The aim of this study was to investigate the clinicopathologic and genomic status, especially KRAS status, of gastric cancer (GC) patients according to MEK signature in two Asian cohorts using clinical samples. In this study, we first surveyed the sensitivity to MEK inhibitor in a panel of GC cell lines and correlated with, RAS alteration, MEK signature to MEK inhibitor sensitivity. Next, we analyzed MEK signature via nanostring assay in FFPET (Formalin fixed paraffin embedded tissue) samples from advanced GC patients and then performed a correlative analysis with MEK signature status and KRAS genotype in GC.
RESULTS
MEK signature in GC cell lines
High MEK signature score is reported [2] to enrich for sensitivity to MEK inhibition in cancer cell lines, low MEK signature score is predictive of resistance, and high “compensatory resistance (Cres)” signature score predictive of resistance in the presence of high MEK signature. In an independent set of 22 cell lines of gastric tumour origin with both RNAseq expression and selumetinib pharmacology, the MEK signature was found similarly predictive of response to selumetinib (ANOVA p < 0.00054) (Figure 1). Furthermore, the Cres signature was seen to be predictive of resistance (ANOVA p < 0.0068), and the combination (MEK score – Cres score) further separated sensitivity form resistance (ANOVA p < 0.00064) (Figure 1). Interestingly, OCUM-1 and SNU-620 cells which are KRAS wild-type but high MEK signature were sensitive to selumetinib.
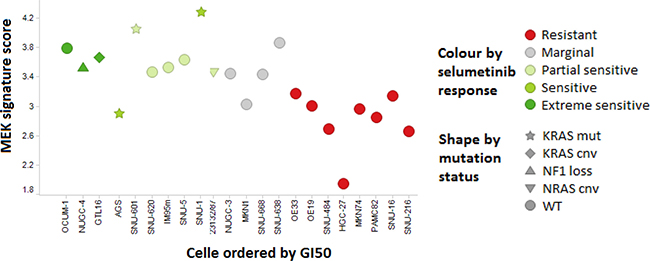
Figure 1: MEK signature and sensitivity to selumetinib in a panel of GC cell lines. Cell lines sensitive to MEK inhibition (GI50 under 3 μM; partial if TGI not reached, extreme if TGI < 5 μM) consistently show higher MEK signature score that resistant (GI50 > 20 μM) cell lines, and include all cells with known MEK pathway activating genetic alterations. Few KRAS wild type cell lines (OCUM-1, SNU-620, IM95m, open circles) with high MEK signature were sensitive to selumetinib. SNU-668 cell line was categorized as KRAS wild-type in this figure (KRAS codon 61 mutation).
MEK signature results according to KRAS status in GC specimens
First, we surveyed the incidence of KRAS amplification and mutation status in two large cohorts from previous study [10, 11]. The incidence of KRAS amplification was 1.5% (3/191) in the ACRG cohort (all Korean) and 7.5% (36/477) in the TCGA cohort (Figure 2A). The incidence of KRAS mutation was 7.2% (18/250) in the ACRG cohort and 8.8% (28/317) in the TCGA cohort.
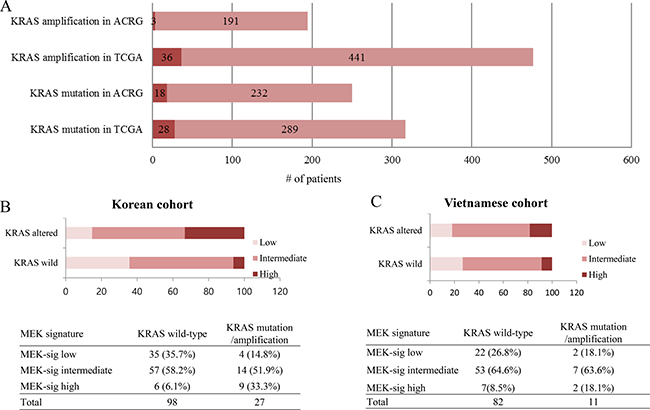
Figure 2: RAS mutation/amplification and distribution of MEK signature in GC. (A) The incidence of KRAS mutation/amplification in ACRG and TCGA. (B) Distribution of MEK signature in GC (N = 125, Korean). (C) Distribution of MEK signature in GC (N = 93, Vietnamese).
In total in the Korean cohort, 27 out of 125 patients (21.6%) showed KRAS alteration (17 (13.6%) with KRAS mutation and 10 (8.0%) with KRAS amplification) in a Korean cohort. Detected KRAS mutations were as follows; G12C (n = 2), G12D (n = 9), G12V (n = 1), A146P (n = 1), A146T (n = 1), F156L (n = 1), Q61H (n = 1), and Q61R (n = 1). Of the 125 patients, 15 (12%), 71 (56.9%), and 39 (31.2%) patients were classified as high, intermediate, and low respectively for the MEK gene expression signature score by nanostring. Of 125 patients, 87 patients were prospectively enrolled onto the VIKTORY screening program. In this patient cohort, 6 (6.9%), 50 (57.5%), and 31 (35.6%) patients had high, intermediate, and low MEK signature, respectively.
There was a statistically significant difference of distribution in MEK signature group according to KRAS status (P = 0.001) in the Korean cohort (Figure 2B). For KRAS wild-type group (n = 98), 35 (35.7%), 57 (58.2%), and 6 (6.1%) cases were classified into low, intermediate, and high MEK signature, respectively. For KRAS altered group (n = 27), 4 (14.8%), 14 (51.9%), and 9 (33.3%) cases were classified into low, intermediate, and high MEK signature, respectively. The proportion of high MEK signature was significantly higher in the KRAS altered group (33.3% in KRAS altered group vs 6.1% in KRAS wild-type group) (Figure 2B). The mean MEK signature score of KRAS wild, mutant, and amplified group was 7.27 (95% confidence interval 7.14–7.41), 7.62 (7.28–7.97), and 8.05 (7.77–8.33) (P = 0.01) (Figure 3A).
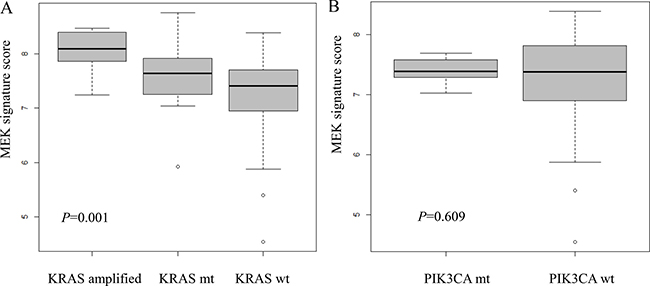
Figure 3: Distribution of MEK signature in Korean cohort according to (A) KRAS mutation or amplification; (B) PIK3CA mutation status.
In the Vietnamese cohort, the proportion of high MEK signature was higher in the KRAS altered group (18.1%) compared to KRAS wild group (8.5%) although there was no statistical significance (P = 0.48) (Figure 2C).
The association of MEK signature with BRAF and PIK3CA status
The status of BRAF and PIK3CA in each MEK signature group is summarized in Table 1. In the VIKTORY screening cohort, there were 7 (8.0%) PIK3CA mutant and 1 (1.15%) PIK3CA amplified cases. None of 8 PIK3CA altered cases belonged to high MEK signature group. The mean MEK signature score of PIK3CA wild and altered group was 7.27 (95% confidence interval 7.11–7.43), 7.40 (7.22–7.58) (Figure 3B). There was no statistical significance (P = 0.609). BRAF mutation was not identified in any of the cases.
Table 1: BRAF, and PIK3CA status 87 Korean GC patients
MEK signature |
||||
|---|---|---|---|---|
Low |
Intermediate |
High |
Total |
|
BRAF mutation |
||||
Wild |
31 (100%) |
50 (100%) |
6 (100%) |
87 |
Mutant |
0 (0%) |
0 (0%) |
0 (0%) |
0 |
PIK3CA mutation/amplification |
||||
Wild |
30 (96.8%) |
43 (86.0%) |
6 (100%) |
79 |
Mutant |
1 (3.2%) |
6 (12%) |
0 (0%) |
7 |
Amplified |
0 (0%) |
1 (2%) |
0 (0%) |
1 |
On the other hand, PIK3CA mutation was detected in 13 (14.0%) out of 93 patients in a Vietnamese cohort. Likewise, none of these patients belonged to the MEK high group. In terms of BRAF mutation, 7 (7.5%) BRAF mutated patients were identified and 1 out of 7 BRAF mutated patients belonged to the MEK high group. There was no association between MEK signature score and BRAF status (data not shown).
Clinicopathologic features of MEK low, intermediate, and high signature group
Table 2 summarizes clinicopathologic characteristics for the Korean cohort according to the MEK signature status. Of note, MEK high signature was more prevalent in well-differentiated and moderately-differentiated tumor type (Table 2). There was a higher percentage of differentiated tumors in the high MEK group (46.7%) compared to the low (12.9%) and intermediate (29.6%) MEK groups (P = 0.034). In addition, there was a higher frequency of intestinal type of Lauren classification (66.7%) in high MEK group compared to low MEK (20.5%) and intermediate MEK group (36.6%) (P = 0.012). There was no significant difference of age, sex and tumor stage. Long-term follow up data (more than 5 years) was available for the patients of the discovery set (n = 38). Disease-related death rate was 37% in low, 61.1% in intermediate, and 66.7% in high group.
Table 2: Clinicopathologic characteristics of Korean GC patients according to the MEK signature
Variable |
MEK signature |
|||
|---|---|---|---|---|
Low |
Intermediate |
High |
P Value |
|
KRAS mutation/ amplification |
0.001 |
|||
Wild |
35 (89.7%) |
57 (80.3%) |
6 (40.0%) |
|
Mutant |
4 (10.3%) |
9 (12.7%) |
4 (26.7%) |
|
Amp |
0 (0%) |
5 (7.0%) |
5 (33.3%) |
|
Age |
0.174 |
|||
< 60 |
24 (61.5%) |
41 (57.7%) |
5 (33.3%) |
|
≥ 60 |
15 (38.5%) |
30 (42.3%) |
10 (66.7%) |
|
Sex |
0.153 |
|||
Male |
23 (59.0%) |
44 (62.0%) |
13 (86.7%) |
|
Female |
16 (41.0%) |
27 (38.0%) |
2 (13.3%) |
|
WHO type |
0.034 |
|||
W/D |
1 (2.6%) |
3 (4.2%) |
1 (6.7%) |
|
M/D |
4 (10.3%) |
18 (25.4%) |
6 (40.0%) |
|
P/D or Signet ring cell |
33 (84.6%) |
48 (67.6%) |
7 (46.7%) |
|
Others |
1 (2.6%) |
2 (2.8%) |
1 (6.7%) |
|
Lauren classification |
||||
Intestinal |
8 (20.5%) |
26 (36.6%) |
10 (66.7%) |
0.012 |
Diffuse |
31 (79.5%) |
43 (60.6%) |
5 (33.3%) |
|
Mixed |
0 (0%) |
2 (2.8%) |
0 (0%) |
|
Stage |
||||
I |
0 (0%) |
1 (1.4%) |
1 (6.7%) |
0.059 |
II |
3 (7.7%) |
9 (12.7%) |
3 (20.0%) |
|
III |
8 (20.5%) |
20 (28.2%) |
7 (46.7%) |
|
IV |
28 (71.8%) |
41 (57.7%) |
4 (26.7%) |
|
DISCUSSION
MEK inhibition in combination with chemotherapy has shown beneficial effects in KRAS mutant lung cancer and biliary tract cancer [8, 12]. This strategy could be potentially adapted to GC patients [13]. Therefore, development of a robust diagnostic tool indicative of RAS pathway activity is needed for selection of patients who can benefit from MEK inhibition.
RAF-MEK-ERK pathway has profound effects on proliferative, apoptotic, and differentiation pathways [14]. Mutations occurring in RAF-MEK-ERK pathway can lead to uncontrolled regulation and aberrant signaling [14]. However, the regulation of this pathway is a rather complex process and there are also many tumor suppressor proteins interacting with this pathway such as PTEN, RKIP, PP2A, DUSP5, DUSP6, TSC1, and TSC2 [14]. Expression of activated Ras in gastric chief cells of mice resulted in metaplastic lineage transitions in stomach, which implicated that RAS signaling pathway inhibition may reverse preneoplastic metaplasia in the stomach [15].
Selumetinib (AZD 6244, ARRY-142886) is a potent, orally active inhibitor of mitogen-activated protein/extracellular signal-regulated kinase (ERK) kinase [1]. Previous in vivo and in vitro studies demonstrated a tendency toward sensitivity to selumetinib in tumors harboring KRAS or BRAF mutations compared with those without those mutations [1, 2]. However, a recent study has shown that selumetinib response does not have an absolute correlation with mutational status of BRAF, RAS, or PIK3CA [2].
Although the development and clinical trial of selumetinib has focused on KRAS-mutant non-small-cell lung cancer or BRAF-mutant melanoma, it is not clear that MEK inhibitors are specific for KRAS-mutant cancers [5]. The response rates to MEK inhibitor in clinical trials has varied. A recent study has shown that patients harbouring KRAS G12C or G12V mutations showed better response for selumetinib plus docetaxel compared with other KRAS mutations [16]. On the other hand, of twenty patients who showed response in phase II study of selumetinib in metastatic biliary tract cancers, only two patients had KRAS mutations [12]. Another study also failed to show improvement in objective response rate or progression-free survival with combination therapy of selumetinib and erlotinib over monotherapy in KRAS mutant and KRAS wild-type advanced lung cancer [17].
Clinically, it is important to develop an accurate and robust method indicative of RAS pathway activity to stratify potential patients who can benefit from selumetinib treatment. Previous in vivo and in vitro studies have suggested that gene expression signature would provide a better measure of RAS activity in cancer cells than mutation analysis [9]. In a previous data set, tumors with KRAS mutations had high signature scores [9]. However, a significant number of KRAS wild-type cell lines and tumors exhibited high RAS pathway signature scores, suggesting that these samples have upregulated RAS signaling through another mechanism [9]. In our study, we performed nanostring assays to evaluate pathway-associated gene expression and calculate MEK signature using six genes, DUSP4, DUSP6, ETV4, ETV5, PHLDA1, and SPRY2, in advanced GC patients. Our results showed that KRAS altered tumors have a tendency to have high MEK signature. However, the relationship between MEK signature and KRAS status was not absolute in our study. There is a subset of KRAS wild type GCs which revealed high MEK signature which is in agreement with previous findings that the RAS pathway signature is a high sensitivity but low specificity predictor of KRAS mutation status [9]. PIK3CA is known to mediate resistance to MEK inhibitor [2, 18]. In our cohort, no tumor harboring PIK3CA mutation had high MEK signature. A phase II selumetinib/docetaxel trial is currently ongoing for second-line metastatic GC patients as part of an umbrella trial with patient selection based on high vs low MEK signature or RAS gene alterations (clinicaltrials.gov NCT#02448290).
GC is the second most common cause of cancer-related deaths worldwide, and the prognosis of advanced GC is still poor [19]. To date, only trastuzumab along with chemotherapy showed positive survival outcomes among many targeted therapy trials [13, 20]. One study has reported a potential relationship between KRAS amplification, the activation of KRAS signaling pathway, and cell growth in GC [21]. KRAS mutation was more frequently found in intestinal type, according to the Lauren classification [22, 23]. Our study showed that MEK high signature was more prevalent in well-differentiated and moderately-differentiated tumor type and intestinal type by Lauren classification. In addition to KRAS mutation, MEK1 S72G mutation [24], which activates ERK1/2 and enhances tumorigenicity, has been shown to be hypersensitive to MEK inhibitors in GC. The correlation between MEK signature and MEK1 mutation should be investigated in future studies.
In conclusion, 6.9% of metastatic GC score high for expression of the MEK signature assessed by nanostring assay and have tendency to be enriched for KRAS genomic alterations and intestinal type tumors by Lauren classification. The correlation between MEK signature, KRAS alteration and treatment response to selumetinib should be validated in prospective clinical trials.
MATERIALS AND METHODS
Selumetinib sensitivity and MEK signature scoring in GC cell lines
MTS was used to determine cell proliferation. Cells were seeded in 96-well plates at a density to allow for logarithmic growth during the 72-hour assay and then incubated overnight at 37°C, 5% CO2. Cells were treated with concentrations of selumetinib ranging from 30 to 0.003 μmol/L for 72 hours. For the MTS endpoint, cell proliferation was measured by the CellTiter AQueous Non-Radioactive Cell Proliferation Assay (Promega) reagent as per manufacturer’s instructions. Absorbance was measured with a Tecan Ultra instrument. Expression of MEK and Cres signature genes was determined by RNA sequencing. Total RNA was extracted from cell lines and mRNA was enriched using the oligo dT magnetic beads. cDNA was synthesized from fragmented and size selected mRNA (~200 bp). Sequencing adaptors were ligated to the fragments and PCR amplified before sequenced using Illumina HiSeq 2000’s pair-end sequencing technology (2 × 90 bp). Roughly ~120 million reads were obtained for each cell line.
Sensitivity to MEK inhibition with selumetinib was defined as a GI50 under 3 μM, and further divided into partial (TGI not reached within 30 mM concentration range tested) and extreme (TGI< 5 μM) sensitivity. Resistant cell lines showed GI50> 20 μM and did not reach TGI within 30 μM concentration range tested. MEK signature was scored from RNAseq as the mean of gene centric RPKM for the 18 MEK signature genes [2]. Association between MEK signature score and categorical sensitivity to selumetinib was determined using ANOVA.
Patient population
This study was conducted in two cohorts of Asian GC patients, which consisted of a Korean cohort (n = 125) and a Vietnamese cohort (n = 93). For the training set in the Korean cohort, positive control (KRAS altered) and negative control (KRAS wild) cases were selected from 312 surgically resected GC samples that had been sequenced as previously reported [11]. Positive control group (n = 17) consisted of 8 KRAS amplified tumors and 9 KRAS mutated tumors by target sequencing. Negative control group (n = 21) was confirmed to have no KRAS mutation and amplification. The remaining 87 of 125 specimens in the Korean cohort were from the GC molecular screening program, the VIKTORY molecular screening study (NCT# 02299648). Briefly, 87 patients had MEK signature scoring results from nanostring assay, in addition to the cancer panel sequencing results (either from Ion torrent or Illumina HiSeq). Clinicopathological information of Korean GC cohort, including age, sex, WHO histologic type, Lauren classification, clinical stage, and GC-associated survival data was collected. The study was approved by the institutional review board of Samsung Medical Center (IRB no. 2014-1-136). Written informed consent form was obtained from all patients before the study. All experimental procedures were carried out in accordance with guidelines approved by Samsung Medical Center.
RNA preparation
Hematoxylin and Eosin stain was performed on one tumor section per patient and tumors were reviewed by a pathologist for tumor purity. Samples with < 50% tumor was discarded from the study. The tumor component was macro-dissected from 2 × 5 μm FFPET sections either from FFPET or fresh frozen samples and RNA was extracted using RNeasy FFPE Extraction kit (Qiagen) or QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Sample RNA was quantified using Qubit 2.0 Flourometer with the Broad Range RNA kit using the standard protocol. Samples that had < 20 ng/μl total RNA were not tested in the nanostring assay. Where available, more tissue for these samples were ordered, re-extracted, and those 20 ng/ul or greater were tested in the nanostring assay.
Nanostring assay and MEK signature calculation
Nanostring assays were performed by following the standard protocol ‘Setting up 12 nCounter Assays (MAN-C0003-03, 2008–2013). Hybridization incubations were performed between 17–18hrs. Cartridges were either read immediately or stored dark (in aluminum foil) at 4ºC until ready to be read. All cartridges were read within 2 days of preparation, on the AZ GEN2 Digital Analyzer station with high resolution selected.
To enable assessment of sample MEK signature scores against previously defined cut-points, three reference standards, representing high, medium and low MEK signature scores were run in every assay. These reference samples were selected from a previous assessment of MEK signature scores using Nanostring in 197 commercially sourced Vietnamese Gastric cancer samples (unpublished data). Up to eight test samples and a background control sample (RNase free water instead of RNA in the hybridisation reaction) using the same reagents were ran alongside the reference samples. MEK signature score adjustment was undertaken by applying a linear transformation to the calculated MEK signatures for each sample. The transformation coefficients were calculated as the linear regression coefficients calculated from the fit of the reference sample MEK signature score values generated by Samsung Medical Center against those previously generated by AstraZeneca.
Each sample was normalized individually with the three reference samples using nSolver Analysis Software version 2.5. The normalized data was exported as Log2 data and then adjusted using the method described previously. Previously we published an 18-gene mRNA expression signature reproducibly predictive of MEK pathway output and response to MEK inhibition with selumetinib and subsequently demonstrated that a. 6-gene sub-signature shows enhanced reproducibility in cell lines, primary tumor explant in vivo models and fresh/fixed tissue samples, crossing tumor type and gene expression platforms (Dry et al. 2013). For each patient sample, the 6-gene MEK signature score was calculated by taking the arithmetic average of the normalized gene expression value (nSolver values) of the 6 MEK genes; DUSP4, DUSP6, ETV4, ETV5, PHLDA1, and SPRY2 . For each patient sample, the 6-gene MEK signature score was calculated by taking the arithmetic average of the normalized gene expression value (nSolver values) of the 6 MEK genes; DUSP4, DUSP6, ETV4, ETV5, PHLDA1, and SPRY2. Then, the adjusted MEK signature score was compared against the following two cut-off values; ‘MEK-low’ (25th percentile), 7.17 and ‘MEK-high’ (90th percentile), 8.08. Where adjusted MEK score for a patient was less than or equal to the MEK-low cut-off, the patient was considered as part of the MEK-low. Where adjusted MEK score for a patient was greater than or equal to the MEK-high cut-off, the patient was considered as part of the MEK-high.
Statistical analysis
Statistical analysis was performed using SPSS version 18.0 (SPSS, Chicago, IL, USA). Contingency tables and Fisher’s exact tests were used to correlate KRAS status and MEK signature results. One way Anova test was done to compare the distribution of MEK score according to KRAS and PIK3CA status. Contingency tables and Fisher’s exact tests were also used to correlate MEK signature with KRAS status, age, sex, WHO tumor type, Lauren classification, and tumor stage. A P-value <0.05 was considered significant.
Authors’ contributions
J.L. conceived the idea and designed the study. R.B., A.S, J.R.D., D.R.H., E.K., K.K., and K.M.K conducted the experiment and analyzed the data. S.T.K., S.H.P., W.K.K., and K.M.K contributed by providing study material. S.A. and R.B wrote the main manuscript text. All authors reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
FUNDING
This work was supported by funding from the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (HI16C1990, HI14C2188). Support was also provided by a grant from the 20 by 20 Project of Samsung Medical Center (GF01140111).
REFERENCES
1. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007; 6:2209–2219.
2. Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, Chresta C, McCormack R, Byrne N, Cockerill M, Graham A, Beran G, Cassidy A, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010; 70:2264–2273.
3. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006; 439:358–362.
4. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–954.
5. Okumura S, Janne PA. Molecular pathways: the basis for rational combination using MEK inhibitors in KRAS-mutant cancers. Clin Cancer Res. 2014; 20:4193–4199.
6. Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009; 8:834–843.
7. Shimizu T, Tolcher AW, Papadopoulos KP, Beeram M, Rasco DW, Smith LS, Gunn S, Smetzer L, Mays TA, Kaiser B, Wick MJ, Alvarez C, Cavazos A, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012; 18:2316–2325.
8. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crino L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013; 14:38–47.
9. Loboda A, Nebozhyn M, Klinghoffer R, Frazier J, Chastain M, Arthur W, Roberts B, Zhang T, Chenard M, Haines B, Andersen J, Nagashima K, Paweletz C, et al. A gene expression signature of RAS pathway dependence predicts response to PI3K and RAS pathway inhibitors and expands the population of RAS pathway activated tumors. BMC Med Genomics. 2010; 3:26.
10. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–209.
11. Cristescu R, Lee J, Nebozhyn M, Kim KM, Ting JC, Wong SS, Liu J, Yue YG, Wang J, Yu K, Ye XS, Do IG, Liu S, et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat Med. 2015; 21:449–456.
12. Bekaii-Saab T, Phelps MA, Li X, Saji M, Goff L, Kauh JS, O’Neil BH, Balsom S, Balint C, Liersemann R, Vasko VV, Bloomston M, Marsh W, et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J Clin Oncol. 2011; 29:2357–2363.
13. Lee J, Kim KM, Kang WK, Ou SH. Innovative personalized medicine in gastric cancer: time to move forward. Clin Genet. 2014; 86:37–43.
14. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Basecke J, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3:954–987. doi: 10.18632/oncotarget.652.
15. Choi E, Hendley AM, Bailey JM, Leach SD, Goldenring JR. Expression of Activated Ras in Gastric Chief Cells of Mice Leads to the Full Spectrum of Metaplastic Lineage Transitions. Gastroenterology. 2016; 150:918–930.e913.
16. Janne PA, Smith I, McWalter G, Mann H, Dougherty B, Walker J, Orr MC, Hodgson DR, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios CH, et al. Impact of KRAS codon subtypes from a randomised phase II trial of selumetinib plus docetaxel in KRAS mutant advanced non-small-cell lung cancer. Br J Cancer. 2015; 113:199–203.
17. Carter CA, Rajan A, Keen C, Szabo E, Khozin S, Thomas A, Brzezniak C, Guha U, Doyle LA, Steinberg SM, Xi L, Raffeld M, Tomita Y, et al. Selumetinib with and without erlotinib in KRAS mutant and KRAS wild-type advanced nonsmall-cell lung cancerdagger. Ann Oncol. 2016.
18. Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, Sellers WR, Lengauer C, Stegmeier F. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009; 69:4286–4293.
19. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
20. Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010; 376:687–697.
21. Mita H, Toyota M, Aoki F, Akashi H, Maruyama R, Sasaki Y, Suzuki H, Idogawa M, Kashima L, Yanagihara K, Fujita M, Hosokawa M, Kusano M, et al. A novel method, digital genome scanning detects KRAS gene amplification in gastric cancers: involvement of overexpressed wild-type KRAS in downstream signaling and cancer cell growth. BMC Cancer. 2009; 9: 198.
22. Miki H, Ohmori M, Perantoni AO, Enomoto T. K-ras activation in gastric epithelial tumors in Japanese. Cancer Lett. 1991; 58:107–113.
23. Yoo J, Park SY, Robinson RA, Kang SJ, Ahn WS, Kang CS. ras Gene mutations and expression of Ras signal transduction mediators in gastric adenocarcinomas. Arch Pathol Lab Med. 2002; 126:1096–1100.
24. Sogabe S, Togashi Y, Kato H, Kogita A, Mizukami T, Sakamoto Y, Banno E, Terashima M, Hayashi H, de Velasco MA, Sakai K, Fujita Y, Tomida S, et al. MEK inhibitor for gastric cancer with MEK1 gene mutations. Mol Cancer Ther. 2014; 13:3098–3106.