
INTRODUCTION
Prostate cancer is the most common nonskin cancer in American males [1]. While organ-confined prostate tumors are usually curable, metastatic prostate cancer is highly resistant to therapeutic intervention and almost uniformly fatal. Therefore, the development of effective novel targeted therapies to inhibit prostate cancer metastasis could have a considerable impact on prostate cancer mortality.
Hepsin (HPN) is one of the most upregulated genes in human prostate cancer and is overexpressed in up to 90% of prostate tumors with levels often increased >10 fold [2-4]. Hepsin increases early in prostate cancer initiation and its high levels are maintained throughout progression and metastasis [3-6]. Hepsin belongs to the protein family of type-II transmembrane cell surface serine proteases [7, 8]. Hepsin can cleave and activate pro-uPA, pro-HGF, Laminin332 and pro-MSP [9-11]. Previous studies indicate that Hepsin overexpression plays an important role in prostate and ovarian cancer [12-15]. The levels of Hepsin expression correlate with the high Gleason score and are indicative of poor clinical outcome and relapse after prostatectomy [3-6]. Hepsin is not overexpressed in prostate cancer cell lines and overexpression of Hepsin in cells in culture results in cell detachment and downregulation of cell proliferation [16, 17]. Hepsin upregulation in the context of the prostate gland in vivo promotes SV40 large T antigen-driven prostate cancer progression and metastasis to the liver, lung and bone [12]. Furthermore, Hepsin overexpression in the LNCaP human prostate cancer cell line grown as an orthotopic xenograft in mice promotes invasive tumor growth and lymph node metastasis [18].
In this study we report the development of a novel, non-toxic, and orally bioavailable small molecule Hepsin inhibitor, HepIn-13. We show that long-term exposure to HepIn-13 blocks prostate cancer metastasis in a preclinical genetic model of metastatic prostate cancer.
RESULTS
Identification of novel small molecule Hepsin inhibitors
Hepsin is prominently overexpressed in the majority of human prostate cancers and functional in vivo studies support a causal role for Hepsin in cancer progression [12, 18, 19]. Interestingly, while most of the cancer literature is primarily focused on Hepsin in prostate cancer, analysis of publically available datasets indicates that Hepsin is frequently amplified in a variety of human cancer types, especially in ovarian serous adenocarcinoma (10%), sarcoma (7.2%), lung adenocarcinoma (5.4%), lung squamous cell carcinoma (4.5%), adenoid cystic carcinoma (5%), breast carcinoma (2.6%), as well as many other cancer types (Figure S1). We hypothesized that inhibition of Hepsin activity using small molecules would attenuate prostate cancer progression and may have therapeutic potential in other cancers with Hepsin amplification.
We have previously identified several small molecule compounds that inhibit the activity of purified recombinant Hepsin [20]. To develop and analyze therapeutically-relevant Hepsin inhibitor, we analyzed all available from ChemBridge derivatives of the lead compound #4 (Figure 1). In these studies we used recombinant human Hepsin produced in Drosophila S2 cells [21] (Figure S2). While the majority of these compounds either did not show inhibition or inhibited Hepsin with decreased potency, six compounds (HepIn-1, HepIn-8, HepIn-13, HepIn-17, HepIn-20 and HepIn-25) displayed similar or increased potency (Figure 1, A-B). IC50 values were determined by titration against Hepsin activity and HepIn-13 was found to be the most potent inhibitor with an IC50 of 0.33 µM. (Figure 1, B). Similarly to compound #4, the identified derivatives were specific for Hepsin, as they showed only minor activity against Matriptase, a serine protease highly similar to Hepsin (Figure S3).
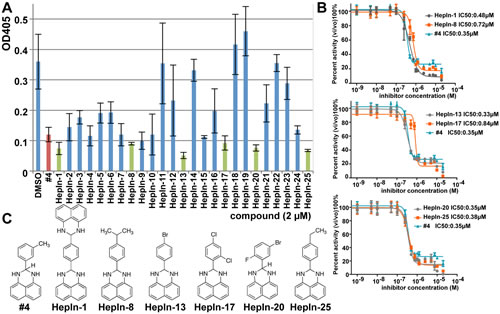
Figure 1: Identification of novel small molecule Hepsin inhibitors. (A) Attenuation of Hepsin-dependent proteolytic activity by the lead compound #4 [20] and its derivatives. Purified recombinant Hepsin was preincubated with 2 µM of the indicated compounds for 30 min. The residual percent activity of the enzyme toward the chromogenic substrate was determined using a microplate reader at 405 nm. Data are the means of three independent experiments ±SD. (B) IC50 determination for Hepsin inhibitors #4, HepIn-1, HepIn-8, HepIn-13, HepIn-17, HepIn-20, HepIn-25. Data are the means of three independent experiments ±SD. (C) Chemical structures of identified Hepsin inhibitors.
Since our Hepsin activity assay utilizes a small peptide substrate, it was necessary to analyze whether the identified compounds inhibit Hepsin-mediated cleavage of a protein substrate. It has been previously reported that Hepsin can cleave and activate pro-HGF [10, 11]. This Hepsin activity is likely to be important for prostate cancer progression, because HGF/MET signaling pathway is strongly implicated in tumor progression and metastasis in prostate cancer [22]. Thus, we analyzed whether our compounds can inhibit Hepsin-mediated cleavage of pro-HGF. We found that both the original lead compound #4 and its six derivatives inhibited Hepsin-mediated cleavage of pro-HGF (Figure S4, A-B). Therefore, we conclude that we identified several novel small molecule inhibitors, which inhibit the in vitro activity of recombinant Hepsin at sub-micromolar concentrations.
Inhibition of Cell Surface Hepsin proteolytic activity
To determine whether the identified compounds can suppress the activity of full-length Hepsin, when it is expressed on the surface of live cells, we developed a cell-based Hepsin activity assay. For this purpose, we generated HEK293 cells overexpressing full-length Hepsin (Figure 2, A). HA-tagged human pro-HGF secreted into serum-free conditioned media from stably transduced HEK293 cells was used as a protein substrate in these experiments (Figure 2, B). Hepsin overexpressing, but not the control vector-transduced cells, efficiently cleaved the HA-tagged pro-HGF (Figure 2, C). This cleavage was inhibited in the presence of Hepsin inhibitors (Figure 2, C-C’).
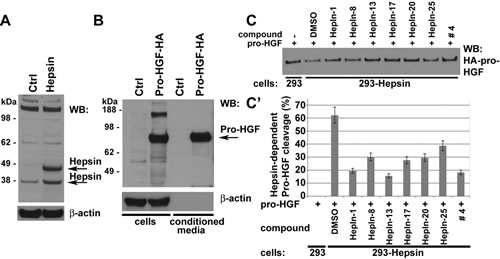
Figure 2: Hepsin inhibitors attenuate the Hepsin-mediated cleavage of pro-HGF in cell based activity assays. (A) Western blot (WB) analysis of vector control- (Ctrl) or Hepsin-expressing HEK 293 cells with anti-Hepsin and anti-β-actin antibodies. Note that Hepsin is present as both full-length (precursor) and cleaved (activated) enzymes. (B) Western blot (WB) analysis of vector control (Ctrl) or pro-HGF-HA expressing HEK 293 cells and their conditioned media with anti-HA and anti-β-actin antibodies. (C) Cell-based Hepsin activity assay. Control or Hepsin-expressing 293 cells in the presence of DMSO or 10µM of indicated compounds were cultured for 2 hours in media containing pro-HGF-HA, and the remaining uncleaved pro-HGF-HA was detected by Western blotting with anti-HA antibodies. (C’) Quantitation of data shown in C. Data are the means of three independent experiments ±SD
Pharmacokinetics and oral bioavailability of Hepsin inhibitors
To determine whether the Hepsin inhibitors were suitable for in vivo use, we analyzed them in mice. Intravenous injections of escalating amounts of the compounds did not result in any signs of acute toxicity, even when injected at the maximal practical dose of a 20 mg/kg.
To determine blood half-life of our compounds, we injected 1 mg of the compound into the tail vein and analyzed blood at different time points after injection. The compounds were extracted from blood plasma and analyzed by LCMS. To generate a standard curve, we added known concentrations of the compounds to mouse blood plasma and then extracted and analyzed them by LCMS (Figure 3, A). As expected, the blood concentration of the compounds dropped quickly immediately after the tail vein injections due to rapid tissue dissemination. The terminal blood half-life of HepIn-13 was calculated at 55 min, and similar values were obtained for other Hepsin inhibitors (Figures 3, B and S5).
To analyze the oral bioavailability, we delivered 3 mg of the test compounds by oral gavage and then analyzed blood for the appearance of the compounds 30 min after the treatment. We found that only one compound, HepIn-13, was detectable in blood after oral gavage (Figures 3, C-D and S6). Thus, we focused our further studies on HepIn-13.
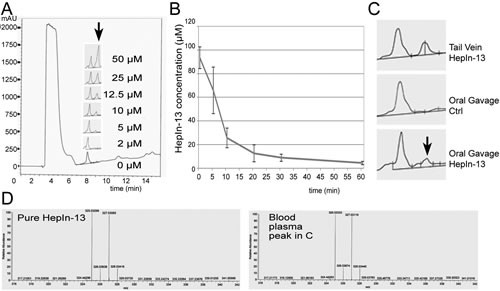
Figure 3: Pharmacokinetics and oral bioavailability of the Hepsin inhibitor HepIn-13. (A) Analytical HPLC profiles (mAU at 220 nm) of HepIn-13 which was dissolved in the mouse blood plasma at indicated concentrations and then extracted and analyzed by HPLC. (B) Blood concentrations of HepIn-13 at indicated time points after tail vein injection of 1 mg of the compound. Data are the means of three independent experiments ±SD. (C) HepIn-13 is orally bioavailable. Analytical HPLC profiles (mAU at 220 nm) of blood plasma extracts: from tail vein injected with HepIn-13, untreated and oral gavage treated mice. (D) Mass Spectrometry confirmation of the presence of HepIn-13 in blood plasma in the peak highlighted by arrow in panel C.
Molecular modeling of interactions between Hepsin and HepIn-13
The crystal structure of Hepsin has been previously determined [23]. We used in silico molecular docking approaches to model interactions between Hepsin and HepIn-13 (http://www.dockingserver.com/web). The entire extracellular region of Hepsin was used in these unsupervised docking experiments. Interestingly, the best-ranked calculated docking pose demonstrates an interaction of HepIn-13 with the catalytic pocket of Hepsin (Figure 4). Moreover, one of the amino acids of the Hepsin catalytic triad, Ser195 (Ser353 in full-length Hepsin), is predicted to interact with HepIn-13. Thus, in silico docking of Hepsin and HepIn-13 places HepIn-13 within the catalytic pocket of Hepsin and predicts an interactions with one of the amino acids of the Hepsin catalytic triad.
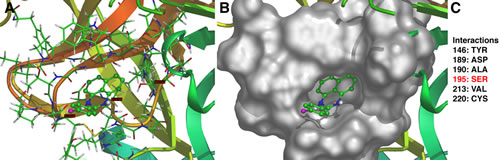
Figure 4: Predicted model of Hepsin–HepIn-13 interaction. The best-ranked calculated docking of HepIn-13 with Hepsin predicts an interaction of the compound with the Hepsin catalytic domain. (A) Ribbon model of Hepsin-HepIn-13 interaction. (B) Surface model of Hepsin-HepIn-13 interaction. (C) Amino acids of Hepsin involved in interaction. Note, Ser353(195) is a part of Hepsin catalytic triad [23]
In vivo efficacy of HepIn-13 in the mouse model of metastatic prostate cancer.
To determine whether HepIn-13 can attenuate or prevent prostate cancer progression in vivo, we performed animal studies. For this purpose, we chose to use LPB-Tag/PB-Hepsin mice, which is the only existing genetic model of metastatic prostate cancer that expresses Hepsin and consistently shows bone metastasis, which is prevalent in human prostate cancer [12]. LPB-Tag/PB-Hepsin mice upregulate expression of SV40 large T antigen and Hepsin in the prostate epithelium and develop primary prostate cancer with metastatic lesions in approximately 55% of the animals [12]. LPB-Tag/PB-Hepsin mice are derived from LPB-Tag animals (12T-7f adenocarcinoma line), which express SV40 large T antigen, but do not express Hepsin [12, 24]. LPB-Tag mice present with primary prostate tumors, but do not develop metastasis. Thus, LPB-Tag/PB-Hepsin model of prostate cancer was ideal for our analysis of Hepsin inhibitors, because the functional significance of Hepsin expression in these mice was already well established.
Considering the characteristics of the LPB-Tag/PB-Hepsin model, we anticipated that we would need to treat animals with HepIn-13 for a relatively long duration, starting at 10 weeks of age, when the animals present with low-grade prostate tumors. The most non-invasive long-term delivery of an orally bioavailable small molecule compound involves mixing the test compound with animal water or food. Thus, we first analyzed whether we can detect HepIn-13 in the animal blood, after the mice are exposed to the soft rodent chow containing varying concentrations of HepIn-13. With our limit of blood HepIn-13 detection at 1 µM, we found that exposure of the animals to rodent chow containing 0.25% of HepIn-13 resulted in the presence of HepIn-13 in blood at concentrations that are significantly higher than its IC50 (Figure S7). We also analyzed blood of mice exposed for 5 weeks to the food containing 0.25% of HepIn-13 for signs of liver toxicity; however, the levels of AST and ALT were within the normal ranges in these animals indicating absence of general liver damage. The animals did not exhibit signs of distress in this chronic dosing experiment. Thus, we decided to perform animal trials with HepIn-13 mixed with rodent chow at 0.25% and 0.1%.
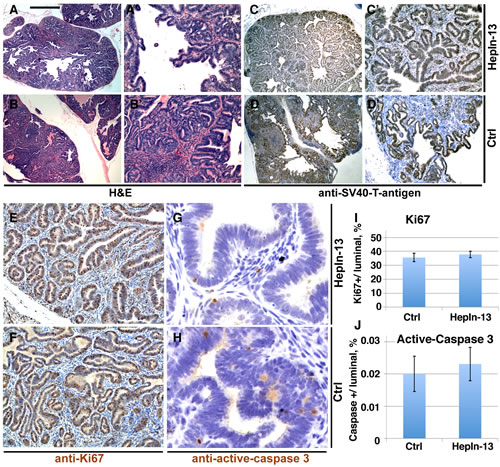
Figure 5: Primary prostate tumors in HepIn-13-treated (0.25%) and control LPB-Tag/PB-Hepsin mice. (A-B’) Hematoxylin and Eosin stainings of sections from primary prostate tumors in 23 week-old HepIn-13-treated (A, A’) and untreated control (B, B’) males. (C-D’) Immunohistochemical staining of sections from primary prostate tumors in 23 week-old HepIn-13-treated (C, C’) and control (D, D’) males with anti-SV40 large T antigen. (E-F) Immunohistochemical staining of sections from primary prostate tumors in 23 week-old HepIn-13-treated (E, G) and control (F, H) males with anti-Ki67 (E, F) and anti-active-caspase 3 (G, H) antibodies. (I) Quantitation of anti-Ki67 staining. N=3. Bar graph shows mean values ±SD. (J) Quantitation of anti-active caspase 3 staining. N=3. Bar in A represents 0.5 mm in A, B, C, D, 100 µm in A’, B’, C’, D’, 55 µm in E, F, and 15 µm in G, H.
For the animal trial, 10 week-old LPB-Tag/PB-Hepsin males were randomly assigned to one of 3 groups. One control group was exposed to soft rodent chow alone (12 mice), while the other two groups were exposed to the same rodent chow mixed with 0.1% (10 mice) or 0.25% (11 mice) of HepIn-13. Thirteen weeks later the animals were euthanized and their prostate glands, livers, lungs and bones were examined by histological staining and immunochistochemistry (Figures 5, 6). As expected, the control animal group presented with metastatic lesions in 66% of the animals, and bone metastasis in 42% of the animals (Table 1, Figure 6). While cellular proliferation, apoptotic cell death, blood vessel density (Figures 5, S8) and the size of the primary prostate tumors were not significantly changed in the group treated with 0.25% of Hepln-13 (mean tumor size 1.9±2.0 g in HepIn-13-treated versus 2.1±3.3 g in control animals), no metastasis was found in these mice (Table 1). The group exposed to 0.1% of HepIn-13 displayed intermediate metastasis phenotype (30% animals with metastasis, 20% bone metastasis). Overall, we conclude that HepIn-13 displays dose-dependent inhibition of Hepsin overexpression-relevant prostate cancer phenotypes in LPB-Tag/PB-Hepsin mice and blocks prostate cancer metastasis in this animal model.
Table 1: Incidence of metastasis and metastatic sites in control and HepIn-13-treated LPB-Tag/PB-Hepsin mice. 10 week-old LPB-Tag/PB-Hepsin mice were randomly divided into 3 groups. Mice were exposed for 13 weeks to soft rodent chow alone (Control group), or chow mixed with 0.1% (HepIn-13 0.1% group) or 0.25% (HepIn-13 0.25% group) of HepIn-13 for 13 weeks. Animals were euthanized and their prostate glands, livers, lungs and bones were analyzed by histology and IHC staining with anti-SV40 T antibodies. The number of mice displaying indicated metastatic lesions relative to the total number of mice (in parentheses) is shown. * indicates P<0.05. ** indicates P<0.01. Two-tailed Fisher’s exact test.
Metastatic lesions |
||||
Total |
Liver |
Lung |
Bone |
|
Control group |
8(12) |
8 (12) |
4 (12) |
5 (12) |
HepIn-13 (0.1%) group |
3(10) |
3 (10) |
1 (10) |
2 (10) |
HepIn-13 (0.25%) group |
0(11)** |
0 (11)** |
0 (11) |
0 (11)* |
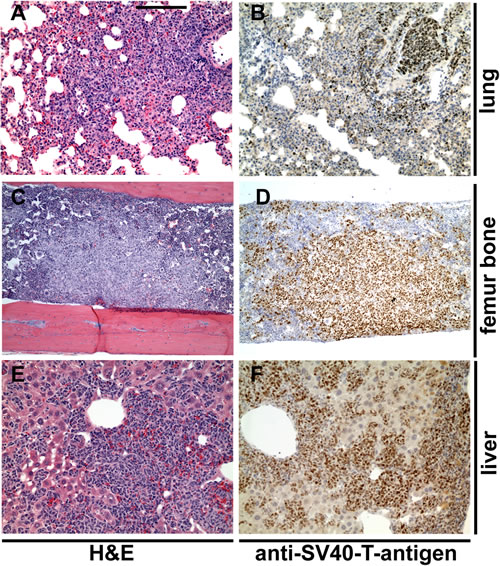
Figure 6: Metastatic lesions in control untreated LPB-Tag/PB-Hepsin mice. Hematoxylin and Eosin (A, C, E) and anti-SV40 large T antibody (brown in B, D, F) stainings of metastatic lesions in lung (A, B), bone (C, D) and liver (E, F) in 23 week-old LPB-Tag/PB-Hepsin males. Bar in A represents 200 µm in A, B, E, F, 360 µm in C, D.
DISCUSSION
We show here that HepIn-13 inhibits Hepsin and blocks prostate cancer metastasis. Importantly, HepIn-13 was able to block metastasis to the bone, which is the most common site of metastasis in human prostate cancer. Interestingly, despite the high penetrance of bone metastasis in human prostate cancer, this has been difficult to replicate in mouse models. Consistent bone metastasis was reported in mice with prostate cancer caused by deletion of PTEN/P53 and the loss and subsequent re-activation of Telomerase [25]; however, subsequent pathological analysis suggested direct bone invasion from large primary tumors rather than bona fide bone metastasis [26]. LPB-Tag/PB-Hepsin mice is a genetic model of prostate cancer which consistently develops bone metastasis [12, 26]. We have previously reported that 20% of 21 week-old LPB-Tag/PB-Hepsin mice present with femur bone lesions. In this study we used a different animal feed (soft transgenic dough) and found that by 23 weeks of age 42% of LPB-Tag/PB-Hepsin mice present with bone metastasis. All the mice with lesions in femur bones also displayed lesions in spine, indicating broad skeletal involvement in affected individuals. Since spine lesions may be the result of a direct primary tumor invasion, we believe that the presence of femur lesions in LPB-Tag/PB-Hepsin mice is more significant, because it indicates bona fide bone metastasis. Treatment of LPB-Tag/PB-Hepsin mice with Hepsin inhibitor HepIn-13 completely suppressed the development of bone metastasis, strongly suggesting that Hepsin plays an important role in the skeletal spread of prostate cancer.
The analysis of HepIn-13 in a single autochthonous model of prostate cancer is an important limitation of this study. Additional Hepsin overexpressing models of prostate cancer will need to be developed to analyze the efficacy of HepIn-13. We have crossed PB-Hepsin [12] and Pr-Cre4/PTENfl/fl [27] mice, hypothesizing that overexpression of Hepsin in PTEN-/- prostate epithelial cells could promote bone metastasis. However, we found that the synthetic Probasin (PB) promoter that was used in the generation of PB-Hepsin mice [28] is inactivated in PTEN-/- cells in vivo. Therefore, different approach to drive Hepsin expression will have to be used in the future to generate new models to analyze the efficacy of HepIn-13 in inhibition of prostate cancer bone metastasis.
While HepIn-13 is the first small molecule Hepsin inhibitor used in an animal model of prostate cancer, a protein-based Hepsin inhibitor, Kunitz domain-1, was previously used in a xenograft model of prostate cancer [18]. In that model, LnCaP-34 cells displayed a Hepsin-dependent ability to invade and develop lymph node metastasis [18]. Treatment of mice carrying LnCaP-34 tumors with Kunitz domain-1 decreased contralateral prostate invasion (46% weight reduction) and lymph node metastasis (50% inhibition) [18].
In addition to Kunitz domain-1, development of several antibodies specifically inhibiting Hepsin have been reported [13, 29, 30]. While Hepsin antibodies are likely to be useful for prostate cancer imaging, the therapeutic use of these antibodies for tumor eradication may be somewhat limited due to the prominent expression of endogenous Hepsin in hepatocytes [31]. Hepsin is not an essential gene and Hepsin knockout mice are viable and fertile [32, 33]; however, these animals exhibit hearing loss due to developmental deformities in the cochlea [34]. Moreover, Hepsin knockout mice display enlarged hepatocytes and narrowed liver sinusoids [35]. Consistent with the phenotype of Hepsin knockout mice, we did not observe any prominent deficiency in mice treated with Hepsin inhibitors; however, more careful examination will be necessary in the future.
One of possible limitation of HepIn-13 is its potential inhibitory effect on other proteases. While we found that HepIn-13 did not inhibit Matriptase, it is always possible that HepIn-13 can inhibit other enzymes. Considering the lack of toxicity associated with long-term HepIn-13 exposure in mice, it appears unlikely that HepIn-13 inhibited essential enzymes. Therefore, the potential off-target activity of HepIn-13 is unlikely to pose a significant problem.
While we found that HepIn-13 was orally bioavailable, we used relatively high concentration of HepIn-13 (0.25%) in animal food to achieve detectable levels in blood. Therefore, it appears that the oral bioavailability of HepIn-13 is relatively poor and additional modifications will be necessary to enhance the drug properties without decreasing the potency and specificity.
METHODS
Reagents
Small molecule compounds were either purchased from ChemBridge Corporation or were synthesized (HepIn-13). HepIn-13 was prepared by heating 1,8-diaminonaphthalene (1 mM) and 4-bromobenzaldehyde (1 mM) overnight in absolute ethanol. The resulting mixture was concentrated and the resulting dark brown/purple solid was recrystallized twice from 95% ethanol to yield a light purple crystalline solid (64% yield). The product was homogeneous by HPLC (>95% purity) and had 1H NMR and mass spectra consistent with the proposed structure. The chromogenic peptide pyroGlu-Pro-Arg-pNA (s-2366) was purchased from DiaPharma. Rabbit anti-human Hepsin polyclonal antibody was purchased from Cayman Chemical (#100022). Anti-Flag antibody was purchased from Sigma (F1804). Anti-HA antibody was purchased from Roche (#1867423). Goat anti-rabbit secondary antibody was purchased from Jackson ImmunoResearch. Human recombinant Matriptase was purchased from R&D Systems. Molecular biology-grade DMSO was purchased from Sigma. Purified recombinant human pro-HGF was obtained from Dr. George F. Vande Woude (Van Andel Research Institute).
Recombinant Hepsin expression purification and in vitro activity assays
Extracellular part of human Hepsin (amino acids 45-417) containing the scavenger receptor cysteine-rich (SRCR) and trypsin-like serine protease domains was expressed and purified from S2 cells using the Drosophila Inducible/Secreted Expression System (Invitrogen), as previously described [36]. The Hepsin Vmax, Km and Hepsin inhibition were determined using chromogenic substrate pyroGlu-Pro-Arg-pNA as described [20]. Briefly, the inhibition assays were performed by incubating the compounds with Hepsin in the assay buffer (30mM Tris-HCl, pH8.4; 30mM imidazole, 200mM NaCl and 1% DMSO) for 30 minutes at room temperature followed by addition of the substrate at the observed Km and incubation for 3h. The endpoint absorbance was measured using a VersaMax microplate reader (Molecular Devices). Residual Hepsin activity was observed relative to DMSO controls. IC50 was calculated by fitting the data to a four-variable nonlinear regression using GraphPad Prism5.
Recombinant DNA constructs
For generation of a Doxycycline- regulated Hepsin expression plasmid, full-length human Hepsin, retaining the last intron for transcript stabilization, was amplified by stitching PCR using both cDNA and genomic DNA templates. The resulting fragment was cloned into EcoRI/SalI sites of pTRE-Tight (Clontech). For generation of HA-tagged pro-HGF expression plasmid, mouse cDNA library was PCR amplified with oligos containing the restriction enzyme cutting sites and C-terminal HA sequence. The fragment was cloned into SacI/SalI sites of pIRES-hrGFP-2a (Stratagene). All DNA constructs were sequence verified.
Cell lines and cell culture
LNCaP, CHO, PC3 and HEK293FT cells were obtained from ATCC. LNCaP and PC3 cells were maintained in RPMI1640 supplemented with 10% FBS and penicillin/streptomycin. HEK293FT cells were maintained in DMEM supplemented with 10% FBS, nonessential amino acids and penicillin/streptomycin. CHO cells were grown in F-12 media supplemented with 10% FBS and penicillin/streptomycin. Drosophila S2 cells expressing inducible secreted extracellular part of human Hepsin were grown in Schneider’s medium (Invitrogen) containing 10% fetal bovine serum, and penicillin/streptomycin [21]. TET-OFF HEK293 cells were purchased from Clontech (C3008-1). For generation of a cell line expressing full-length human Hepsin (HEK293-Hepsin), TET-OFF HEK293 cells were stably transduced with a mixture of pTRE-Tight-hHepsin and pBabe and stable clones were selected with puromycin. For generation of a cell line expressing secreted HA-tagged full-length mouse pro-HGF (HEK293-pro-HGF-HA), HEK293 cells were stably transduced with a mixture of pIRES-hrGFP-2a (Stratagene) and pBabe and stable clones were selected with puromycin.
Cell-based Hepsin activity assay
HEK293 cells stably transduced with full-length human Hepsin were used in cell-based Hepsin activity assays. To obtain HA-tagged pro-HGF substrate, HEK293 cells stably transduced with HA-tagged pro-HGF were washed with PBS and incubated in serum-free media for 8 hours. The conditioned media containing HA-tagged pro-HGF was collected, filtered through 0.2 µm surfactant-free cellulose acetate filter, concentrated by ultrafiltration (Amicon Ultra, Millipore) and kept in aliquots at -80˚C. For the activity assay, the HEK293-Hepsin cells were plated in a 24-well plate at 200,000 cells/well. Forty-eight hrs later, cells were rinsed with serum-free media and cultured for 4 hours in serum-free media with 1% DMSO or indicated test compounds dissolved in DMSO. The conditioned serum-free media containing HA-tagged pro-HGF was added and the cells were cultured for additional 90 minutes. The resulting extracellular media was collected and the cleavage of pro-HGF was analyzed using western blotting with anti-HA antibodies.
In vivo efficacy trial in a genetic animal model of metastatic prostate cancer
LPB-Tag/PB-Hepsin mice express SV40 large T antigen and Hepsin transgenes in luminal prostate epithelium and develop prominent primary prostate cancer with metastatic lesions in liver, lung, and bone [12]. LPB-Tag/PB-Hepsin males were generated as F1 in the crossing of LPB-Tag transgenic females (12T-7f adenocarcinoma line, CD-1 background) [24] with PB-Hepsin transgenic males (C57BL/6J background), as described [12].
For the in vivo experiment, the 10-week-old LPB-Tag/PB-Hepsin males were randomly divided into 3 groups. The control group was exposed to soft Transgenic Dough Diet (Bio-Serv, S3472) with 10% of powdered rodent chow and two experimental groups were exposed to the same food mixed with either 0.1% or 0.25% of the compound HepIn-13. After 13 weeks of exposure, the animals were euthanized and their prostates, livers, lungs, and spine and femur bones were analyzed.
Determination of compound pharmacokinetics in mice
CD-1 mice (Charles River) were used in general pharmacokinetics experiments. All experiments involving mice were conducted in compliance with Fred Hutchinson Cancer Research Center Committees on Use and Care of Animals guidelines. The indicated compounds were solubilized in 1:1 Ethanol:Cremophore EL (Merck) mixture and diluted with 9 volumes of PBS. To determine the highest dose resulting in acute toxicity, escalating doses of the compounds were injected into the tail vein of adult mice and animals were monitored for 2 days. No signs of acute toxicity were observed in these experiments for all investigated compounds injected at the maximum practical dose of 20mg/kg. To determine the blood half-life, 1 mg of the indicated compound was injected into the tail vein of adult mice and the blood was collected at different time points after injection. The compounds were extracted from blood plasma with ethyl acetate, air dried, resuspended in Acetonitrile and analyzed by HPLC (Agilent Technologies, ACE 3 C8-300, 150 x 3.0mm, ACE-212-1503, A 72063). The HPLC peaks were subsequently analyzed using Mass Spectrometry (LTQ-OrbiTrap, Agilent). To test oral bioavailability, 3 mg of the indicated compound was delivered by oral gavage and blood was drawn at different time points after the treatment and analyzed for the presence of the compounds as described above. Similar approach was used to test for the presence of the indicated compounds in blood of the animals exposed to Transgenic Dough Diet (Bio-Serv, S3472) with 10% powdered rodent chow and 0.1% or 0.25% of the test compound. Levels of Aspartate transaminase and Alanine aminotransferase (AST and ALT) in blood were measured by Phoenix Central Laboratory (Mukilteo, WA).
Western blotting
Total prostate protein lysates were generated by manual grinding of frozen in liquid nitrogen prostates using prechilled on dry ice Mortar and Pestle. The resulting frozen powder was extracted with the protein lysis buffer containing: 1xPBS, 1% SDS, 1% NP40, 2% Tween 20, and 1.5 M Urea. The proteins were separated on 4-12% NuPAGE gels and transferred to PVDF membrane using the iBlot system (Invitrogen). The membrane was blocked in TBST buffer containing: 5% non-fat milk, 2% normal goat serum in 50 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.1% Tween-20 and incubated overnight at +4˚C in the same buffer containing anti-Hepsin (Cayman, 1:1000), anti-HA (Roche, 1:1000) or anti--actin (Sigma A5441, 1:20,000), antibodies. The relevant HRP-labeled secondary antibodies (1:5000) were purchased from Jackson ImmunoReserch Laboratories and the blots were developed using ECL (Pierce). Expression levels were quantified by densitometry using ImageQuantTL.
Immunohistochemistry
Tissue samples were processed, embedded in paraffin, and sectioned at 5µm. Sections were deparafinized, rehydrated, and processed for immunostaining with anti-SV40-Tag antibody (Millipore, DP02). The ABC MOM kit (Vector Laboratories) was used for immunohistorchemistry. Antibodies were detected using DAB peroxidase substrate kit and sections were counterstained with hematoxylin QS (both from Vector Laboratories).
Molecular Docking
To model the potential interaction between Hepsin and identified in this study Hepsin inhibitors, molecular docking of Hepsin and the identified small molecule-inhibitors was performed by using DockingServer [37] (www.dockingserver.com). The previously determined crystal structure of extracellular region of human Hepsin [23] and 3-dimentional structure of HepIn-13 were used in these experiments. Docking calculations were then performed by using AutoDock 4 [38] integrated in DockingServer. Docking simulations were carried out by using the Lamarckian Genetic Algorithm. The initial position, orientation, and torsions of the inhibitor molecules were set randomly, and all rotatable torsions were released during docking. Each docking experiment was derived from 100 different runs that were set to terminate after a maximum of 2500000 energy evaluations. The conformer with the lowest docking energy calculated with AutoDock 4 scoring function was selected as the final binding conformation.
Statistical Analysis
For all graphs, data presented are mean value ± SD. Statistics were calculated using GraphPad Prism 6. Statistical significance was determined by the unpaired Student’s t test, the two-tailed Fisher’s exact test (for comparison of incidence of metastasis) and Mann–Whitney test (for comparison of tumor size). P value is indicated by asterisk(s) in the figures. *, denotes P<0.05; **, P<0.01. Differences at P<0.05 and lower were considered statistically significant.
ACKNOWLEDGMENTS AND NOTES
This work has been supported, in part, by awards from the Prostate Cancer Foundation (to V. Vasioukhin), DOD Postdoctoral Fellowship Award W81XWH-11-1-0622 (to X. Tang), and grants from NIH (CA102365 and CA085859 to V. Vasioukhin). The authors thank Drs. Lawrence D. True and Maria S. Tretiakova (University of Washington) for help with anatomical pathology analysis of bone lesions in LPB-Tag/PB-Hepsin mice, Dr. Peter Nelson (FHCRC) and all members of the Vasioukhin lab for comments and suggestions, Dr. George F. Vande Woude (Van Andel Research Institute) for gift of purified recombinant human pro-HGF, Drs. Roland K. Strong, Bin Xu and Hengyu Xu (FHCRC) for help with S2 cells and chromatography. Hepsin inhibitor #4 is described in US Patent 8,450,334; held by V. Vasioukhin. The other authors declare no potential conflict of interest.
References
1. ACS. (2013). Cancer facts & figures.
2. Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA and Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001; 412(6849):822-826.
3. Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, Catalona WJ, Watson MA and Milbrandt J. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001; 61(15):5692-5696.
4. Stamey TA, Warrington JA, Caldwell MC, Chen Z, Fan Z, Mahadevappa M, McNeal JE, Nolley R and Zhang Z. Molecular genetic profiling of Gleason grade 4/5 prostate cancers compared to benign prostatic hyperplasia. The Journal of urology. 2001; 166(6):2171-2177.
5. Chen Z, Fan Z, McNeal JE, Nolley R, Caldwell MC, Mahadevappa M, Zhang Z, Warrington JA and Stamey TA. Hepsin and maspin are inversely expressed in laser capture microdissectioned prostate cancer. The Journal of urology. 2003; 169(4):1316-1319.
6. Stephan C, Yousef GM, Scorilas A, Jung K, Jung M, Kristiansen G, Hauptmann S, Kishi T, Nakamura T, Loening SA and Diamandis EP. Hepsin is highly over expressed in and a new candidate for a prognostic indicator in prostate cancer. The Journal of urology. 2004; 171(1):187-191.
7. Hooper JD, Clements JA, Quigley JP and Antalis TM. Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J Biol Chem. 2001; 276(2):857-860.
8. Szabo R and Bugge TH. Type II transmembrane serine proteases in development and disease. The international journal of biochemistry & cell biology. 2008; 40(6-7):1297-1316.
9. Moran P, Li W, Fan B, Vij R, Eigenbrot C and Kirchhofer D. Pro-urokinase-type plasminogen activator is a substrate for hepsin. J Biol Chem. 2006; 281(41):30439-30446.
10. Kirchhofer D, Peek M, Lipari MT, Billeci K, Fan B and Moran P. Hepsin activates pro-hepatocyte growth factor and is inhibited by hepatocyte growth factor activator inhibitor-1B (HAI-1B) and HAI-2. FEBS letters. 2005; 579(9):1945-1950.
11. Herter S, Piper DE, Aaron W, Gabriele T, Cutler G, Cao P, Bhatt AS, Choe Y, Craik CS, Walker N, Meininger D, Hoey T and Austin RJ. Hepatocyte growth factor is a preferred in vitro substrate for human hepsin, a membrane-anchored serine protease implicated in prostate and ovarian cancers. The Biochemical journal. 2005; 390(Pt 1):125-136.
12. Klezovitch O, Chevillet J, Mirosevich J, Roberts RL, Matusik RJ and Vasioukhin V. Hepsin promotes prostate cancer progression and metastasis. Cancer Cell. 2004; 6(2):185-195.
13. Xuan JA, Schneider D, Toy P, Lin R, Newton A, Zhu Y, Finster S, Vogel D, Mintzer B, Dinter H, Light D, Parry R, Polokoff M, Whitlow M, Wu Q and Parry G. Antibodies neutralizing hepsin protease activity do not impact cell growth but inhibit invasion of prostate and ovarian tumor cells in culture. Cancer Res. 2006; 66(7):3611-3619.
14. Pal P, Xi H, Kaushal R, Sun G, Jin CH, Jin L, Suarez BK, Catalona WJ and Deka R. Variants in the HEPSIN gene are associated with prostate cancer in men of European origin. Human genetics. 2006; 120(2):187-192.
15. Miao J, Mu D, Ergel B, Singavarapu R, Duan Z, Powers S, Oliva E and Orsulic S. Hepsin colocalizes with desmosomes and induces progression of ovarian cancer in a mouse model. International journal of cancer. 2008; 123(9):2041-2047.
16. Srikantan V, Valladares M, Rhim JS, Moul JW and Srivastava S. HEPSIN inhibits cell growth/invasion in prostate cancer cells. Cancer Res. 2002; 62(23):6812-6816.
17. Wittig-Blaich SM, Kacprzyk LA, Eismann T, Bewerunge-Hudler M, Kruse P, Winkler E, Strauss WS, Hibst R, Steiner R, Schrader M, Mertens D, Sultmann H and Wittig R. Matrix-dependent regulation of AKT in Hepsin-overexpressing PC3 prostate cancer cells. Neoplasia. 2011; 13(7):579-589.
18. Li W, Wang BE, Moran P, Lipari T, Ganesan R, Corpuz R, Ludlam MJ, Gogineni A, Koeppen H, Bunting S, Gao WQ and Kirchhofer D. Pegylated kunitz domain inhibitor suppresses hepsin-mediated invasive tumor growth and metastasis. Cancer research. 2009; 69(21):8395-8402.
19. Wu Q and Parry G. Hepsin and prostate cancer. Front Biosci. 2007; 12:5052-5059.
20. Chevillet JR, Park GJ, Bedalov A, Simon JA and Vasioukhin VI. Identification and characterization of small-molecule inhibitors of hepsin. Mol Cancer Ther. 2008; 7(10):3343-3351.
21. Kyrieleis OJ, Huber R, Ong E, Oehler R, Hunter M, Madison EL and Jacob U. Crystal structure of the catalytic domain of DESC1, a new member of the type II transmembrane serine proteinase family. The FEBS journal. 2007; 274(8):2148-2160.
22. Gherardi E, Birchmeier W, Birchmeier C and Vande Woude G. Targeting MET in cancer: rationale and progress. Nature reviews. 2012; 12(2):89-103.
23. Somoza JR, Ho JD, Luong C, Ghate M, Sprengeler PA, Mortara K, Shrader WD, Sperandio D, Chan H, McGrath ME and Katz BA. The structure of the extracellular region of human hepsin reveals a serine protease domain and a novel scavenger receptor cysteine-rich (SRCR) domain. Structure. 2003; 11(9):1123-1131.
24. Kasper S, Sheppard PC, Yan Y, Pettigrew N, Borowsky AD, Prins GS, Dodd JG, Duckworth ML and Matusik RJ. Development, progression, and androgen-dependence of prostate tumors in probasin-large T antigen transgenic mice: a model for prostate cancer. Lab Invest. 1998; 78(3):319-333.
25. Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES, Hu J, Wang W, Xiao Y, Zhang H, Zhang J, Zhang J, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012; 148(5):896-907.
26. Ittmann M, Huang J, Radaelli E, Martin P, Signoretti S, Sullivan R, Simons BW, Ward JM, Robinson BD, Chu GC, Loda M, Thomas G, Borowsky A and Cardiff RD. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 2013; 73(9):2718-2736.
27. Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X and Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003; 4(3):209-221.
28. Zhang J, Thomas TZ, Kasper S and Matusik RJ. A small composite probasin promoter confers high levels of prostate-specific gene expression through regulation by androgens and glucocorticoids in vitro and in vivo. Endocrinology. 2000; 141(12):4698-4710.
29. Koschubs T, Dengl S, Durr H, Kaluza K, Georges G, Hartl C, Jennewein S, Lanzendorfer M, Auer J, Stern A, Huang KS, Packman K, Gubler U, Kostrewa D, Ries S, Hansen S, et al. Allosteric antibody inhibition of human hepsin protease. The Biochemical journal. 2012; 442(3):483-494.
30. Ganesan R, Zhang Y, Landgraf KE, Lin SJ, Moran P and Kirchhofer D. An allosteric anti-hepsin antibody derived from a constrained phage display library. Protein engineering, design & selection : PEDS. 2012; 25(3):127-133.
31. Leytus SP, Loeb KR, Hagen FS, Kurachi K and Davie EW. A novel trypsin-like serine protease (hepsin) with a putative transmembrane domain expressed by human liver and hepatoma cells. Biochemistry. 1988; 27(3):1067-1074.
32. Wu Q, Yu D, Post J, Halks-Miller M, Sadler JE and Morser J. Generation and characterization of mice deficient in hepsin, a hepatic transmembrane serine protease. The Journal of clinical investigation. 1998; 101(2):321-326.
33. Yu IS, Chen HJ, Lee YS, Huang PH, Lin SR, Tsai TW and Lin SW. Mice deficient in hepsin, a serine protease, exhibit normal embryogenesis and unchanged hepatocyte regeneration ability. Thrombosis and haemostasis. 2000; 84(5):865-870.
34. Guipponi M, Tan J, Cannon PZ, Donley L, Crewther P, Clarke M, Wu Q, Shepherd RK and Scott HS. Mice deficient for the type II transmembrane serine protease, TMPRSS1/hepsin, exhibit profound hearing loss. The American journal of pathology. 2007; 171(2):608-616.
35. Hsu YC, Huang HP, Yu IS, Su KY, Lin SR, Lin WC, Wu HL, Shi GY, Tao MH, Kao CH, Wu YM, Martin PE, Lin SY, Yang PC and Lin SW. Serine protease hepsin regulates hepatocyte size and hemodynamic retention of tumor cells by hepatocyte growth factor signaling in mice. Hepatology. 2012; 56(5):1913-1923.
36. Beliveau F, Desilets A and Leduc R. Probing the substrate specificities of matriptase, matriptase-2, hepsin and DESC1 with internally quenched fluorescent peptides. The FEBS journal. 2009; 276(8):2213-2226.
37. Bikadi Z and Hazai E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. Journal of cheminformatics. 2009; 1:15.
38. Huey R, Morris GM, Olson AJ and Goodsell DS. A semiempirical free energy force field with charge-based desolvation. Journal of computational chemistry. 2007; 28(6):1145-1152.