
INTRODUCTION
The cell survival oncoprotein Akt1, also known as protein kinase B (PKB), is frequently hyperactivated in human cancers. Akt1 is recruited to the plasma membrane in the presence of phosphoinositide triphosphate (PI-3,4,5-P). Akt1 plays a central role in the ability of external signals to promote cell survival by preventing cytochrome c release from mitochondria (1-3) and maintaining mitochondrial membrane integrity by increasing hexokinase (HK) association with mitochondria (4). In mammalian cells, activating growth factors and oncogenes stimulate Akt1 kinase activity to promote anti-apoptotic signaling (4). Three separate genes with high sequence identity encode the major isoforms of Akt1/PKB (Akt1/PKBα, Akt2/PKBβ, Akt3/PKBγ). Substrate specificity of the Akt1 isoforms is similar, although Akt1 is the predominant isoform expressed in most tissues. Constitutive activation of Akt1 kinase occurs in human cancer through deletion and mutation of the tumor suppressor gene PTEN, the phosphatase that negatively regulates Akt1, through amplification of the Akt1 genes, or through amplification of the catalytic subunit of PI3 kinase (5-7). The pro-proliferative and prosurvival effects induced by Akt1 kinase are conducted through regulation of caspase 9, I B kinase , Bad, and induction of the GSK3/cyclin D1 signaling pathways (reviewed in: (8, 9)).
ErbB2/ErbB3 receptor activation, which occurs frequently in breast cancer, induces PI3K and Akt1 kinase activity (10, 11). The ErbB2 oncogene is amplified in up to 30% of human breast cancers and is associated with poor patient prognosis in response to chemotherapeutic agents. ErbB2 induces Akt1 activity, cellular growth and therapeutic resistance (12). The activation of ErbB2 is an early event in human breast cancer with ErbB2 overexpressed in up to 80% of primary ductal carcinoma in situ lesions (13). MicroRNAs (miRNAs) are 21-22 nucleotide molecules that regulate the stability or translational efficiency of targeted messenger RNAs. Derived from nuclear precursor RNAs, initial processing occurs by the inter-nuclease Drosha to release pre-miRNA of 60-70 nucleotides in length from pri-miRNA. Subsequent transport to the cytoplasm by exportin-5 results in processing by the inter-nuclease Dicer to generate the ~22 nucleotide mature miRNA (14-16).The base pairing interactions between miRNAs and their target mRNAs, often within the 3’ untranslated region (3’UTR) of target genes, results in the degradation of target mRNAs (17, 18) or inhibition of their translation (19). To date more than 2,000 miRNAs have been identified or predicted in humans (miRBase Sequence Database Version 20.0 released in Jun. 2013). It has been proposed that as each vertebrate miRNA may bind to as many as 200 gene targets, miRNAs potentially control the expression of about one-third of human mRNAs (20).
Several independent lines of evidence support a role for miRNAs in human cancer (21-25). miRNA encoding genes are frequently located at fragile sites, and in minimal regions of loss of heterozygosity, minimal regions of amplification, and in common breakpoint regions involved in cancers (26). Aberrant expression of miRNAs or mutations of miRNA genes have been described in many types of tumors. Let-7 abundance is reduced in several cancers including lung cancer (27), and let-7 was reported to regulate tumor growth by targeting the ras gene (28). miR-15a and miR16-1 were deleted and/or down-regulated in ~ 70% of patients with chronic lymphocytic leukemia (29). miR-15a/16-1 induced apoptosis by inhibiting BCL-2 (30). The miR-34 family is an important component of the p53 tumor suppressor network (25). The human miR-17/20 cluster’s genomic location, chromosome 13q31, correlates with loss of heterozygosity in a number of different cancers including breast cancer (31, 32). The expression and function of miRNA varies by cell type. The miR-17/20 cluster functions as a tumor suppressor in human breast cancer by decreasing AIB1 and cyclin D1 expression (33, 34). In contrast, in both lung cancer and lymphomas, expression of this miRNA cluster was increased, enhancing cell growth (22, 35). The onset and progression of tumorigenesis involves evasion of apoptotic signals, sustained cellular proliferation and the ability to promote tumor neoangiogenesis. As the same miRNA performs different functions through distinct pathways dependent on the tissue or cell type, it is important to understand the mechanisms by which miRNA regulates the cellular apoptosis and thereby tumorigenesis.
RESULTS
miR-17/20 sensitized stress signal-induced apoptosis in breast cancer cells.
Our previous studies demonstrated the suppression of cellular proliferation in human breast cancer cells by miR-17/20. In order to determine the potential role of miR-17/20 in regulating breast cancer cell apoptosis, MCF-7 cells were transduced with a retrovirus encoding the miR-17/20 cluster. The cells were treated either with 0.05 μM doxorubicin for 24 hours or with UV radiation and then analyzed for apoptosis by TUNEL assay. Apoptotic cells were increased in miR-17/20 overexpressing MCF-7 cells compared to control cells. Thus, miR-17/20 increased MCF-7 sensitivity to DNA damage inducing agents after doxorubicin or UV radiation (Fig. 1A and Supplemental Fig. S1).
In order to corroborate the effects of miR-17/20 on apoptosis, anti-miR-17 and anti-miR-20a were applied to block the function of endogenous miR-17 and miR-20a. In MCF-7 cells, doxorubicin-induced cellular apoptosis, assessed by TUNEL analysis, was abolished by anti-miR treatment in miR-17/20 transduced MCF-7 cells (Fig. 1B).
In order to identify the mechanisms by which miR-17/20 regulates cellular apoptosis, analysis was conducted of apoptosis-regulating pathways and target genes (p53, p57, Cyto C, Caspase 3, Caspase 9, Bax, and PARP). The expression of p53 and p27KIP1 increased in miR-17/20 transduced MCF-7 cells. p57 expression was unchanged by miR-17/20 treatment (Fig. 1C). Doxorubicin treatment of MCF-7 cells enhanced miR-17/20 induction of p53 (Fig. 1D). Consistent with the observation, transfection of MCF-7 cells with anti-miR-17 or anti-miR-20a decreased p53 expression (Fig. 1E).
The effect of miR-17/20 was next assessed in the NAFA cell line which was derived from an ErbB2-induced mouse mammary tumor. miR-17/20 transduction increased the sensitivity of NAFA cells to doxorubicin-induced apoptosis as assessed by TUNEL staining (Fig. 1F). The induction of apoptosis in miR-17/20 transduced NAFA cells was associated with induction of p27, Bax, p-γ-H2A, caspase 9 and cleaved PARP (Fig. 1G and Supplementary Figure S2). Similarly, miR-17/20 induced apoptosis in MCF-7 cells was associated with induction of Bax, Cyto C, Bcl-xs, caspase 3 and cleavage of caspase 9 and PARP (Fig. 2A).
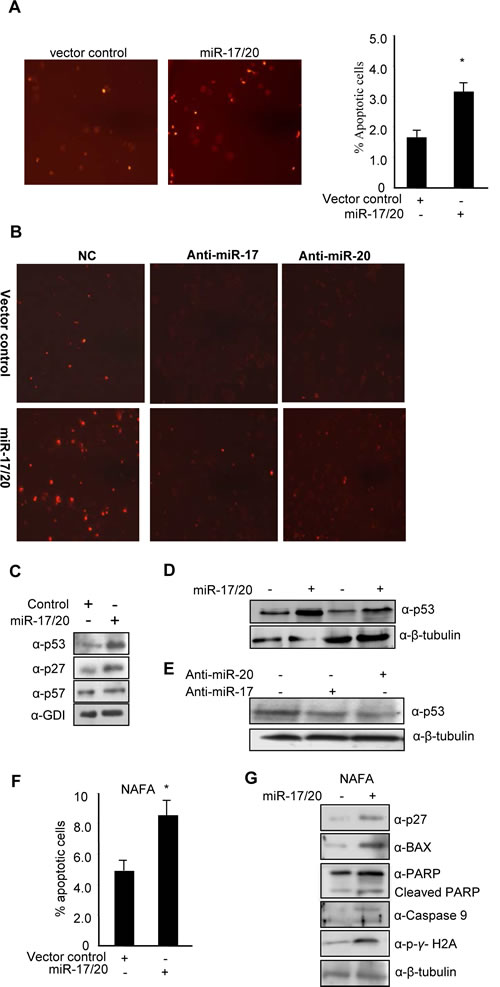
Figure 1: miR-17/20 sensitizes doxorubicin-induced apoptosis, and increase p53 expression in breast cancer cells. A, Tunnel Assay on miR-17/20 or control transduced MCF-7 cells after treatment with doxorubicin at 0.05uM for 24h. B, Tunnel Assays on anti-miR-17-, anti-miR-20a- and negative control-transfected MCF-7 cells after treatment with doxorubicin (0.05μM) for 24h. C, Western blots showing the increased p53 and p27KIP1 in miR-17/20 transduced MCF-7 cells. GDI served as loading control. D, Western blots showing the increased p53 expression in miR-17/20 transduced MCF-7 cells in the presence (lane 1 and 2) or absence (lane 3 and 4) of doxorubicin treatment. E, Western blot showing decreased p53 in anti-miR-17 and anti-miR-20a- transduced MCF-7 cells. F, miR-17/20 sensitizes NAFA cells to doxorubicin-induced apoptosis. G, Western blot showing increased p27KIP1 BAX, p-γ/H2A, PARP and Caspase 9 in miR-17/20 transduced NAFA cells. β-Tubulin served as loading control.
miR-17/20 increased MCF-7 cell sensitivity to tamoxifen.
The anti-estrogen tamoxifen is the most commonly used treatment for patients with estrogen-receptor α (ER)-positive breast cancer. Tamoxifen resistance occurs in ERα+ breast cancer cells including MCF-7. miR-17/20 transduced MCF-7 cells and control were treated with 15 uM tamoxifen up to 36 hours. The relative cell survival was determined using the MTT assay. As shown in Figure 2B, miR-17/20 overexpression attenuated relative cell survival in the presence of tamoxifen (Fig. 2B, C). MCF-7 control cells showed sensitivity to 15 uM tamoxifen after 36h treatment while miR-17/20 transduced MCF-7 cells showed sensitivity after 24 hours treatment when compared to cells without tamoxifen treatment (Fig. 2C). At both 24h and 35h timepoints, control MCF-7 cells showed more resistance to tamoxifen than miR-17/20 transduced cells (Fig. 2C).
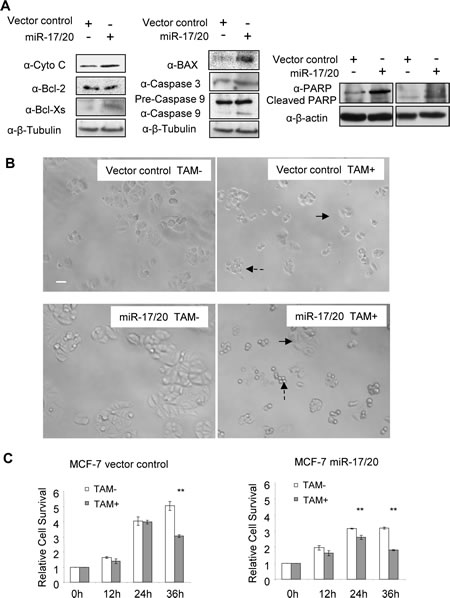
Figure 2: miR-17/20 increases tamoxifen sensitivity of MCF-7 cells. A, Western blot showing the regulation of apoptosis pathway-related genes in miR-17/20 transduced MCF-7 cells. B, Phase contrast images of miR-17/20 or control transduced MCF-7 cells after 36h treatment (15uM tamoxifen). Survived cells are indicated with solid arrow, apoptotic cells are indicated with dashed arrow. C, MTT assays measuring the relative survival rate of MCF-7 cells after treatment with tamoxifen (15uM) for the indicated times. The data are mean +SEM (n=5), **p<0.01.
miR-17/20 attenuated doxorubicin resistance in MCF-7 cell.
MCF-7 cells were treated with different concentrations (0-500 nM) of doxorubicin for 48h and 72h, followed by quantitative analysis of cell survival. Compared to control cells, miR-17/20 transduced MCF-7 showed increased sensitivity to 400 nM and 500 nM doxorubicin. The reduction in cell survival of miR-17/20 transduced cells was most pronounced (~20% vs. ~35%) after 72h of treatment (Fig. 3A). miR-17/20 overexpression decreased the IC50 of MCF-7 cells to doxorubicin (48h treatment). The MCF-7 cell growth curve with 500 nM doxorubicin treatment demonstrated significantly enhanced sensitivity to doxorubicin to doxorubicin after 24h of treatment (Fig. 3B).
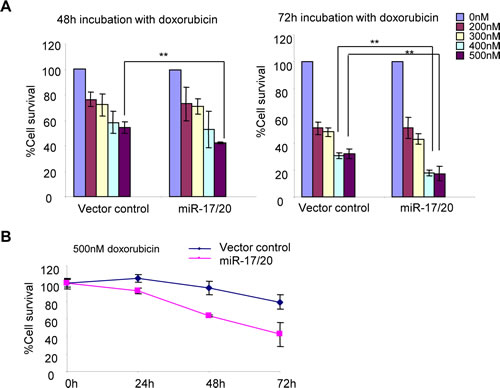
Figure 3: miR-17/20 increases the sensitivity of MCF-7 cells to doxorubicin. A, Cell survival of miR-17/20 or control transduced MCF-7 cells after treatment with doxorubicin at indicated concentrations for 48h and 72h. B, Cell survival curves of miR-17/20 and control transduced MCF-7 treated with 0.5 μM doxorubicin for 0, 24, 48, and 72 hours. The data represent mean + SEM (n=5).
Akt1 is required for miR-17/20 sensitization of breast tumor cells to doxorubicin-induced apoptosis.
Akt and p53 both are important regulators of stress signal-induced cellular apoptosis (36). miR-17/20 sensitized the ErbB2-induced NAFA cell line to apoptosis. Akt1 is required for the progression of ErbB2-induced breast tumorigenesis in transgenic mice (37). We investigated whether Akt1 is involved in the miR-17/20 sensitized cellular apoptosis. We therefore derived breast tumor cell lines from MMTV-ErbB2/Akt1-/- and MMTV-ErbB2/Akt1+/+ litter mate control mice and thereby assessed the role of Akt1 in miR-17/20 mediated apoptosis. The mammary tumor cell lines were transduced with either miR-17/20 or control. Apoptosis assays indicated more apoptotic cells in Akt1-/- tumor cells than that in Akt1+/+ cells after doxorubicin treatment (Fig. 4A). In addition to doxorubicin, puromycin was applied to Akt1+/+ and Akt1-/- breast tumor cells to analyze the induced cellular apoptosis. As shown in supplementary Figure S3, Akt1-/- cells were more sensitive to puromycin than Akt1+/+ cells at the indicated concentrations.
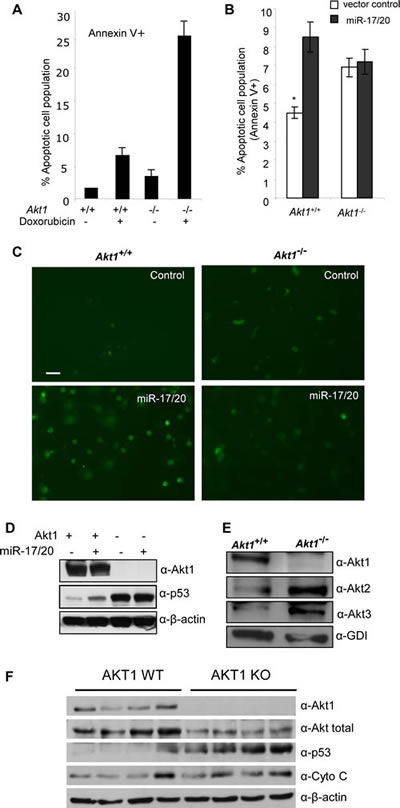
Figure 4: Akt1 is required for miR-17/20 sensitization of breast tumor cells to doxorubicin-induced apoptosis. A, Annexin V staining showing the increased apoptosis in Akt1-/- murine ErbB2 breast tumor cells treated with doxorubicin at 0.5μM for 24h. B, Annexin V staining as a marker of apoptosis in miR-17/20 transduced Akt1+/+ or Akt1-/- murine ErbB2 breast tumor cells, **p<0.01. C, Tunnel assays for apoptosis in Akt1-/ - vs. Akt1+/+ murine ErbB2 breast tumor cells. D, Western blot of miR-17/20 transduced Akt1-/- and Akt+/+ murine ErbB2 breast tumor cells for p53. β-Tubulin is a loading control. E, Western blot of Akt1, Akt2 and Akt3 expression in Akt1-/- murine ErbB2 mammary tumor cells. GDI served as loading control. F: Western blot of Akt1, total Akt, Cyto C and p53 expression in multiple WT and Akt1-/- murine ErbB2 mammary tumors.
miR-17/20 transduction was able to sensitize the Akt1+/+ cells to doxorubicin treatment, but not Akt1-/- cells (Fig. 4B, C). Western analysis showed that miR-17/20 overexpression promoted p53 expression in Akt1+/+ cells rather than Akt1-/- cells (Fig. 4D). Notably, Akt1-/- cells showed much more p53 expression than Akt1+/+ cells (Fig. 4D), which is consistent with our observation of more apoptotic cells in Akt1-/- cells. Interestingly, increased Akt2 and Akt3 expression was detected in Akt1-/- cells compared to Akt1+/+ cells (Fig. 4E). Additional analysis on multiple breast tumor samples from Akt1-/- mice also showed increased expression of p53 and Cyto C compared with Akt1+/+ mice (Fig.4F).
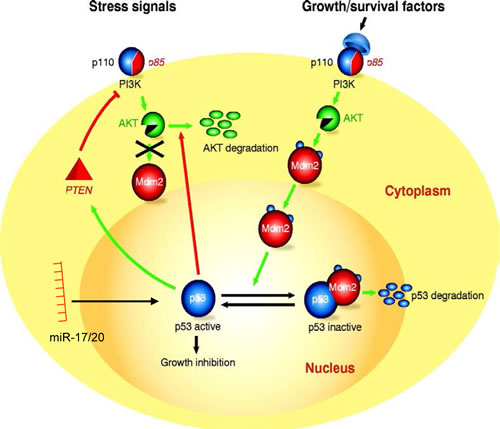
Figure 5: Schematic representation of the molecular mechanisms by which miR-17/20 and Akt1 regulate p53 abundance and thereby apoptosis.
DISCUSSION
The current studies demonstrated that miR-17/20 induces apoptosis in response to the DNA damaging agents doxorubicin and tamoxifen. MiR-17/20 transduction of MCF-7 cells induced p53 and apoptosis characterized by induction of BAX, release of cytochrome C and induction of Bcl-X(S). Using Akt1 knockout mammary epithelial cells, and breast cancer derived cell lines from transgenic mice, we demonstrated the induction of p53 by miR-17/20 required Akt1. Akt1 inhibited p53 abundance in cultured mammary epithelial cells. These studies identify a novel pathway by which the non-coding genome mediates apoptosis in response to DNA damaging agents, and thereby enhances sensitivity to drug treatment.
In the current studies, miR-17/20 enhanced tamoxifen-induced apoptosis. Addition of tamoxifen induced apoptosis with a 4-fold increase in cellular apoptosis at 24 hours. In prior studies, tamoxifen-induced apoptosis was shown to involve the estrogen receptor ERα and additional signaling kinases, including protein kinase C, calmodulin TGF-α, map kinases including JNK and p38 map kinase, and oxidative stress involving the mitochondrial permeability transition (38)(Reviewed in (39)). 10 uM tamoxifen treatment induces genotoxic stress in MCF-7 cells via a DNA damage response through free-radical production (40).
Consistent with the role of the miR-17/20 in growth inhibition, the human miR-17/20 cluster, which is located on chromosome 13q31, undergoes loss of heterozygosity in a number of malignancies, including breast cancer. In transgenic mice, miR-17/20 overexpression reduced overall tissue growth resulting in small organs (41). In breast cancer cells, the cell cycle is controlled through a cyclin D1-miR-17/20 auto-regulatory feedback loop (33). Consistent with a model in which miRNA-17 retards cellular growth in transgenic mice, these studies demonstrated miR-17/20 inhibits cyclin D1 gene expression via a 3’ UTR binding site. The cyclin D1 3’ UTR is spliced in the normal population (41). The miR-17/20-mediated inhibition of breast cancer cellular proliferation via cyclin D1 repression, together with the finding that miR-17/20 inhibits breast cancer cell invasiveness (42), is consistent with a model in which miR-17/20 is a negative growth regulator in breast cancer cells. In this regard, miR-17/20 levels were reduced in highly invasive breast cancer cell lines and node-positive breast cancer specimens (42). Furthermore, cell conditioned media from miR-17/20 overexpressing MCF-7 cells inhibited the invasiveness of MDA-MB-231 cells by inhibiting secretion of several cytokines and plasminogen activators. In the current work we found the apoptosis induction by miR-17/20 in breast cancer cells. miR-17/20 overexpression induced the expression of p53, Bax, Cyto C and caspases which are key components of p53-mediated apoptosis pathway (Figure 2A and Supplementary Figure S4).
In terms of same “seed” sequence shared by miR-17 and miR-20a,1,220 conserved target genes for both miR-17 and miR-20a are predicted by TargetScan tool, in which quite a few genes are involved in cellular apoptosis regulation including caspase 7, caspase 2, BCL2-like 11, et al. Ingenuity Pathway Analysis (IPA) on the 1,220 predicted target genes revealed a cell death network (Supplemental Figure S5) and a cellular apoptosis pathway (Supplemental Figure S6).
Deletion of Akt1 abrogated both the inhibition of cellular proliferation and the induction of apoptosis by miR-17/20. In the current studies, Akt1 was required for miR-17/20-mediated induction of p53 abundance. These findings are consistent with prior observations that Akt1 stabilized p53 in response to DNA damaging agents. Double-stranded DNA breaks induce cellular lesions that may lead to chromosomal rearrangements and instability. Defense mechanisms to maintain stability include cell cycle arrest, DNA repair, and apoptosis. The double-stranded DNA breaks induced by DNA damaging agents, including doxorubicin, as used in the current studies, are recognized by large phosphatidylinositol 3 protein kinases, including Ataxia Telangiectasia Mutated (ATM) and DNA-dependent Protein Kinases (DNA-PK) which sequentially implement the DNA damage response. In resting cells the levels of p53 protein are low due to the activity of E3 ubiquitin ligases (MDM2, Pirh2, and COP1). Akt2 was required for irradiation-induced p53 stabilization (43). In mouse embryo fibroblasts and lymphoblasts, this DNA-PKc/Akt/GSK3á/Mdm2 signaling pathway prevented Mdm2-mediated p53 degradation. In the current studies, the Akt1-/- MEC cells expressed both Akt2 and Akt3, indicating that Akt1 is necessary in mammary epithelial cells for this signaling pathway.
MATERIALS AND METHODS
Cell culture and reagents.
Murine mammary epithelial tumor cells (MMTV-ErbB2/Akt1+/+ or MMTV-ErbB2/Akt1-/-) were isolated from MMTV-ErbB2 mammary gland tumors generated in bi-transgenic mice, and maintained as previously described (37). The NAFA cell line derived from the MMTV-NeuT transgenic mouse was cultured in DMEM containing penicillin and streptomycin (100 mg of each/L) and supplemented with 10% fetal bovine serum (FBS). Tamoxifen (MP Bio), doxorubicin (Sigma), were used at doses and for the times indicated in the individual figure legends.
Retrovirus infection, small RNA/plasmid transfection.
miR-17/20Retroviral production and infection methods were described in detail before (40). The ecotropic packaging vector, pSV-![]() –E-MLV which providesecotropic packaging helper function, was used with retrovirus to infect cells. F For cellular transfection with anti-miRNA (Ambion), RNAiMax (Invitrogen) was used following the manufacturer’s instructions.
–E-MLV which providesecotropic packaging helper function, was used with retrovirus to infect cells. F For cellular transfection with anti-miRNA (Ambion), RNAiMax (Invitrogen) was used following the manufacturer’s instructions.
Western blot analysis.
Whole-cell lysates (50 μg) were separated by 10% SDS-PAGE, and the proteins were transferred to nitrocellulose membrane. The following antibodies were used for Western blotting: anti-cyclin D1 from Neomarker (Fremont, CA); anti-Bcl-Xs from Calbiochem (Darmstadt, Germany); anti-β-Tubulin (sc-9104), anti-β-actin (sc-47778), anti-p53 (sc-6243G), anti-p57 (sc-1040), anti-p27 (sc-528), anti-Cytochrome C (SC-7159), anti-Bax (sc-7480), anti-BCL-2 (sc-7382), anti PARP (sc-7150), anti-Caspase 9 (sc-8355), anti-Caspase 3 (sc-7148) were from Santa Cruz Biotechnology (Santa Crus, CA); anti-GDI from RTG Solutions (Gaithersburg, MD); anti-Akt1 (#2967 ), anti-Akt2 ( #5239 ), anti-Akt3 (#4059 ), anti-p-y-H2A ( #9718s ) were from Cell Signaling.
Cell proliferation assays.
MCF-7 Cells were infected with the pMSCVpuro-miR-17/20 cluster or the pMSCVpuro empty vector. After puromycin selection, 4x104 cells were seeded into each well of a 6-well plate in triplicate and cell number was counted daily for 3 to 4 days under a microscope using a hemocytometer. For the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium(MTT) assay, 4x103 cells/well were seeded into a 96-well plate in triplicate and cell growth was measured after 24 hours’ culture.
TUNEL assay.
miR-17/20 transduced cells and control were cultured in medium containing doxorubicin (0.05μM). After 24-48 hours cells were plated into 96-well plate in triplicate. Apoptosis assays were performed using the In Situ Cell Death Detection Kit, TMR red (Roche Diagnostics, Mannheim, Germany) following the manufacturer’s instructions.
Annex V staining.
Akt1+/+ and Akt1-/ - murine ErbB2 breast tumor cells were incubated with fluorochrome-conjugated Annexin V followed by Propidium Iodide staining solution treatment. Flow cytometry analysis was applied to measure apoptotic cells.
miRNA target gene prediction and pathway analysis.
The target gene prediction of hsa-miR-17 and hsa-miR-20 was performed using TargetScan Human 6.2 version (released June 2012) (http://www.targetscan.org/). The predicted target genes were further analyzed for pathway analysis and network analysis using Ingenuity Pathway Analysis (IPA).
Statistical analysis.
Data are presented as mean + SEM. The standard two-tailed student’s t-test was used for analysis and P<0.05 was considered significant.
ACKNOWLEDGEMENTS
This work was supported by grants 2012CB966800 from National Basic Research Program of China and 81172515 from NSFC (to Yu. Z), partly by grants 13JC1401702 and 124119a7100 from Science and Technology Commission of Shanghai Municipality (Z. Yu); partly by the Foundation for Innovative Research Groups of the NSFC (81221001); partly by grants R01CA070896, R01CA075503, R01CA132115, R01CA107382, R01CA086072 (R.G.P.), the Kimmel Cancer Center NIH Cancer Center Core grant P30CA056036 (R.G.P.), generous grants from the Dr. Ralph and Marian C. Falk Medical Research Trust and the Margaret Q. Landenberger Research Foundation, and a grant from Pennsylvania Department of Health (R.G.P.). The Department specifically disclaims responsibility for an analysis, interpretations or conclusions. There are no conflicts of interest associated with this manuscript.
REFERENCES
1. Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol 1999; 19: 5800-10.
2. Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J. Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo J 1997; 16: 2783-93.
3. Dudek H, Datta SR, Franke TF, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 1997; 275: 661-5.
4. Majewski N, Nogueira V, Bhaskar P, et al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell 2004; 16: 819-30.
5. Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res 2001; 264: 29-41.
6. Cheng JQ, Ruggeri B, Klein WM, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci U S A 1996; 93: 3636-41.
7. Downward J. Mechanisms and consequences of activation of protein kinase B/Akt. Curr Opin Cell Biol 1998; 10: 262-7.
8. Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev 1999; 13: 2905-27.
9. Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005; 8: 179-83.
10. Lee RJ, Albanese C, Fu M, et al. Cyclin D1 is required for transformation by activated Neu and is induced through an E2F-dependent signaling pathway. Mol Cell Biol 2000; 20: 672-83.
11. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001; 3: 245-52.
12. Hutchinson J, Jin J, Cardiff RD, Woodgett JR, Muller WJ. Activation of Akt (protein kinase B) in mammary epithelium provides a critical cell survival signal required for tumor progression. Mol Cell Biol 2001; 21: 2203-12.
13. Wilbur DC, Barrows GH. Estrogen and progesterone receptor and c-erbB-2 oncoprotein analysis in pure in situ breast carcinoma: an immunohistochemical study. Mod Pathol 1993; 6: 114-20.
14. Lund E, Guttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science 2004; 303: 95-8.
15. Yi R, Qin Y, Macara IG, Cullen BR. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev 2003; 17: 3011-6.
16. Zhang H, Kolb FA, Jaskiewicz L, Westhof E, Filipowicz W. Single processing center models for human Dicer and bacterial RNase III. Cell 2004; 118: 57-68.
17. Ambros V. The functions of animal microRNAs. Nature 2004; 431: 350-5.
18. Cullen BR. Transcription and processing of human microRNA precursors. Mol Cell 2004; 16: 861-5.
19. Lai EC. Micro RNAs are complementary to 3’ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat Genet 2002; 30: 363-4.
20. Krek A, Grun D, Poy MN, et al. Combinatorial microRNA target predictions. Nat Genet 2005; 37: 495-500.
21. Calin GA, Ferracin M, Cimmino A, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med 2005; 353: 1793-801.
22. He L, Thomson JM, Hemann MT, et al. A microRNA polycistron as a potential human oncogene. Nature 2005; 435: 828-33.
23. Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell 2005; 122: 6-7.
24. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6: 857-66.
25. He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447: 1130-4.
26. Calin GA, Sevignani C, Dumitru CD, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 2004; 101: 2999-3004.
27. Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004; 64: 3753-6.
28. Johnson SM, Grosshans H, Shingara J, et al. RAS is regulated by the let-7 microRNA family. Cell 2005; 120: 635-47.
29. Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A 2002; 99: 15524-9.
30. Cimmino A, Calin GA, Fabbri M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 2005; 102: 13944-9.
31. Eiriksdottir G, Johannesdottir G, Ingvarsson S, et al. Mapping loss of heterozygosity at chromosome 13q: loss at 13q12-q13 is associated with breast tumour progression and poor prognosis. Eur J Cancer 1998; 34: 2076-81.
32. Lin YW, Sheu JC, Liu LY, et al. Loss of heterozygosity at chromosome 13q in hepatocellular carcinoma: identification of three independent regions. Eur J Cancer 1999; 35: 1730-4.
33. Yu Z, Wang C, Wang M, et al. A cyclin D1/microRNA 17/20 regulatory feedback loop in control of breast cancer cell proliferation. J Cell Biol 2008; 182: 509-17.
34. Hossain A, Kuo MT, Saunders GF. Mir-17-5p regulates breast cancer cell proliferation by inhibiting translation of AIB1 mRNA. Mol Cell Biol 2006; 26: 8191-201.
35. Hayashita Y, Osada H, Tatematsu Y, et al. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res 2005; 65: 9628-32.
36. Manfe V, Biskup E, Rosbjerg A, et al. miR-122 regulates p53/Akt signalling and the chemotherapy-induced apoptosis in cutaneous T-cell lymphoma. PLoS One; 7: e29541.
37. Ju X, Katiyar S, Wang C, et al. Akt1 governs breast cancer progression in vivo. Proc Natl Acad Sci USA 2007; 104: 7438-43.
38. Zheng A, Kallio A, Harkonen P. Tamoxifen-induced rapid death of MCF-7 breast cancer cells is mediated via extracellularly signal-regulated kinase signaling and can be abrogated by estrogen. Endocrinology 2007; 148: 2764-77.
39. Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis 2001; 6: 469-77.
40. Li Z, Wang C, Jiao X, et al. Cyclin D1 regulates cellular migration through the inhibition of thrombospondin 1 and ROCK signaling. Mol Cell Biol 2006; 26: 4240-56.
41. Shan SW, Lee DY, Deng Z, et al. MicroRNA MiR-17 retards tissue growth and represses fibronectin expression. Nat Cell Biol 2009; 11: 1031-8.
42. Yu Z, Willmarth NE, Zhou J, et al. microRNA 17/20 inhibits cellular invasion and tumor metastasis in breast cancer by heterotypic signaling. Proc Natl Acad Sci USA 2010; 107: 8231-6.
43. Boehme KA, Kulikov R, Blattner C. p53 stabilization in response to DNA damage requires Akt/PKB and DNA-PK. Proc Natl Acad Sci U S A 2008; 105: 7785-90.