
INTRODUCTION
Neuroblastoma (NB) is a common extracranial pediatric solid tumor in the sympathetic nervous system, and its incidence rate is approximately 10.54 cases per one million per year in children younger than 15 years [1]. The most important clinical hallmark of NB is biological heterogeneity, and this hallmark is represented by its wide range of clinical behaviors and diverse treatment responses [2], which are closely related to their individual genetic or molecular features. Therefore, NB patients should be subjected to reasonable and accurate stratification depending on genetic or molecular features to improve treatment and prognosis. Studies on Children’s Oncology Group (COG) NB have focused on defining the risk groups for patient stratification and protocol assignment based on prognostic factors [3], including age at diagnosis [4], International Neuroblastoma Staging System (INSS) [5–7], International Neuroblastoma Pathology Classification (INPC), MYCN status, DNA index [8], 1p loss of heterogeneity (LOH) [9], and 11q LOH [10]. Despite these estimation systems, stratification is poorly implemented and thus results in overtreatment or undertreatment of patients with NB. Among these prognostic genes or genomes, MYCN amplification (MNA), associated with rapid disease progression in patients of all ages and stages, is the most characterized and unfavorable prognostic biomarker of NB [4, 11, 12]. However, MNA accounts for approximately 20% of genetic modifications in NB, and over 60% of patients with high-risk NB lack MNA [13]. The markers of these tumors, especially those in children with stage 4 diseases and poor prognosis, are weak. Thus, key factors related to NB should be identified to help effectively manage this disease.
High-risk aggressive tumors are typically characterized by various genomic alterations, including point mutations and copy number variations (CNVs) [14, 15]. Compared with adult tumors, mutation frequencies of tumor cells of children is relatively lower. Somatic chromosomal imbalance variation is a relevant feature of poor prognosis of NB [10, 16–18]. CNVs, amplifications, or deletions of genes of more than 1 kb are classified as chromosomal imbalance variation. In addition to MNA, the CNV of other single gene yields a low occurrence rate. As such, its importance is often disregarded, but the frequency of recurrent CNVs is relatively higher than that of point mutations [19, 20]. Thus far, none of the published CNV-based classifiers, except MYCN and 1p and 11q LOH, have been incorporated in clinical classification systems. Therefore, the relevance of CNVs in NB should be further investigated.
Different analytical techniques, including fluorescence in situ hybridization (FISH) and comparative genomic hybridization microarrays, have been used to detect CNVs. Next-generation sequencing (NGS) has been developed for clinical applications and future CNV assessment. One study compared the genomic profiles generated through exome sequencing data with those obtained from high-resolution Affymetrix single nucleotide polymorphism (SNP) microarrays and confirmed that several smaller genes can be identified through exome profiling but not through SNP microarray analyses [21]. They concluded that exome sequencing is a useful diagnostic tool to detect CNVs in NBs. Therefore, target-capture NGS was suitable for this study, and its application was extended on the basis of a comprehensive panel of regions where CNVs and point mutations occurred to identify important CNVs other than MNA and to improve the prognostic evaluation of patients without MNA.
RESULTS
Patient characteristics
A total of 96 patients were included and evaluated through FISH: 14 MNA(+) patients (4 in non-stage 4 and 10 in stage 4) and 82 MNA(−) patients (31 in non-stage 4 and 51 in stage 4). The three-year event-free survival (EFS) rates of 10 MNA(+) patients and 51 MNA(−) patients in stage 4 were 21% and 47%, respectively. These results indicated that 83.6% of the patients in stage 4 lacked MNA, and their prognosis remained poor. As such, these patients should be examined with other molecular tests to enhance stratification. Finally, 33 patients were evaluated (Supplementary Table 1). The average age of these patients was 38.546 ± 26.126 months, and of these patients, 12 aged ≤ 18 months and 21 aged > 18 months. Of the 33 patients, 16 were males and 17 were females. Histological classification revealed 16 exhibited favorable histology (FH) and 17 manifested unfavorable histology (UH). A total of 54.5% (18/33) patients suffered from distant metastasis, and the mean of serum NSE was 324.700 ± 391.227 ng/mL. Furthermore, 16 patients were in stage 4 (Table 1), 15 patients aged > 18 months, 13 patients were in the UH group, none showed MNA, and all of these patients received similar treatments.
Table 1: Clinical data and follow-up results of 16 patients with stage 4 NB
Patient NO. |
Age (month) /Age group |
INPC |
INSS |
CNV |
Event/ time of occurrence (month) |
VGPR |
|---|---|---|---|---|---|---|
2 |
72 (2) |
UH |
4 |
+ |
progress/15 |
NO |
6 |
53 (2) |
UH |
4 |
+ |
progress /16 |
NO |
8 |
40 (2) |
UH |
4 |
− |
progress /18 |
NO |
10 |
27 (2) |
UH |
4 |
+ |
none/35 |
YES |
11 |
12 (1) |
UH |
4 |
+ |
death/17 |
NO |
15 |
47 (2) |
UH |
4 |
− |
none/19 |
YES |
16 |
81 (2) |
UH |
4 |
− |
progress /20 |
YES |
17 |
71 (2) |
UH |
4 |
− |
none/22 |
YES |
19 |
67 (2) |
UH |
4 |
− |
none/19 |
YES |
20 |
57 (2) |
FH |
4 |
− |
none/23 |
YES |
22 |
39 (2) |
UH |
4 |
− |
relapse /18 |
NO |
26 |
52 (2) |
UH |
4 |
− |
none/17 |
YES |
30 |
62 (2) |
UH |
4 |
+ |
progress/13 |
NO |
31 |
40 (2) |
FH |
4 |
− |
progress/17 |
NO |
32 |
42 (2) |
FH |
4 |
− |
none/17 |
YES |
33 |
36 (2) |
UH |
4 |
+ |
relapse /10 |
NO |
Panel design and NGS data quality assessment
Whole genome sequencing analysis revealed that our panel, which was mainly based on CNVs, covered most of the chromosomal regions and candidate genes associated with NB. The whole genome data of more than 200 NB patients in foreign countries were analyzed and CADD, PROVEAN, and mutation assessor were utilized to filter mutant genes, which were defined as benign mutations. Some of the major CNV regions closely related to NB prognosis were selected because these regions were involved in the deletions or amplifications of genomic chromosomal imbalance and influential to the adverse prognosis of NB. Proto-oncogenes or tumor suppressor genes were also mainly located in these areas. Overall, our NB panel comprised 53 genes and 3 large chromosome regions (Supplementary Table 2). Moreover, 33 biopsies and matched leukocyte samples were subjected to capture-based deep sequencing with our panel to determine and quantify somatic mutations. The detailed quality control data of these samples are shown in Supplementary Table 3. All of the 33 tissue and leukocyte samples sequenced were then subjected to stringent QC allegations. The mean coverage depth in all of the target regions in all of the samples was 887×, and the mapped read percentage was over 99%. The imputed insert size and library complexity statistics shown in Supplementary Figure 1 revealed a mean insert size of 212 bp, and these findings indicated a high capture efficiency of the probes.
Mutation spectrum
The 33 biopsies and matched leukocyte cells were subjected to capture-based targeted sequencing. With the obtained high-depth sequencing data, the mutations in all of the cases were accurately quantified. Overall, 17 unique somatic aberrations, including 14 genes, were identified in 11 patients, and 13 CNVs were detected in 8 patients: 2 amplifications were observed in CDK4 of 12q14 and CCND1 of 11q13, 1 amplification was found in OS9 of 12q13, MYCN, and DDX1 of 2p24, and 1 deletion was located in 11q, cyclin-dependent kinase inhibitor 1C (CDKN1C) of 11p15.4, CDKN2A of 9p21, H19 of 11p15.5, RBMS3 of 3p24.1, and T-cell lymphoma invasion and metastasis 1 (TIAM1) of 21q22.11 (Figure 1).
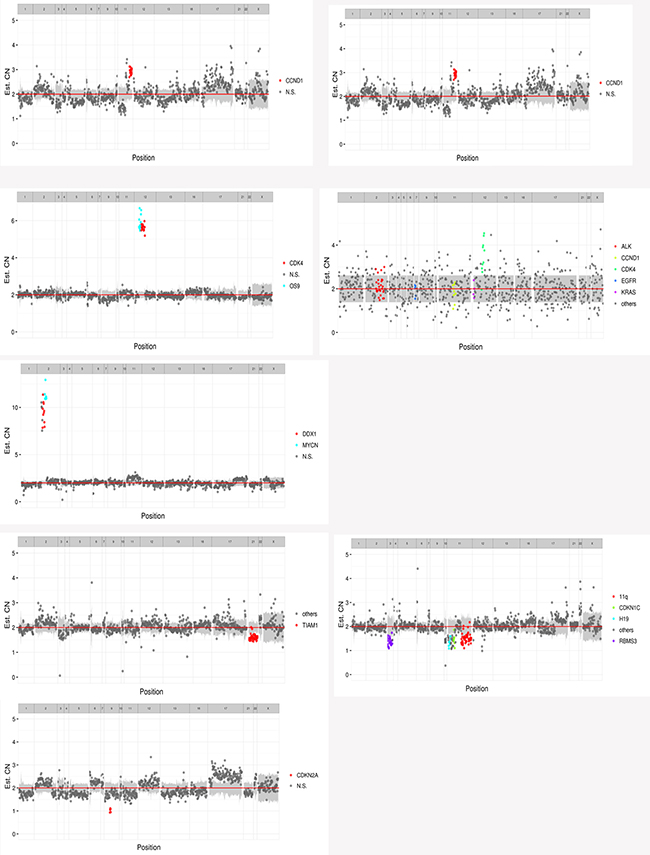
Figure 1: 13 CNVs of 8 patients. (CN_amp: copy number amplification; CN_del: copy number deletion).
TIAM1 gene deletion in one patient was a newly found variation, and the differentiated degree of neuroblast cells was poor. The reviewed pathological hematoxylin and eosin (H&E) staining results of the flat pieces of the tumor sample in microscopic 200 × and 400 × forms are shown in Figure 2.
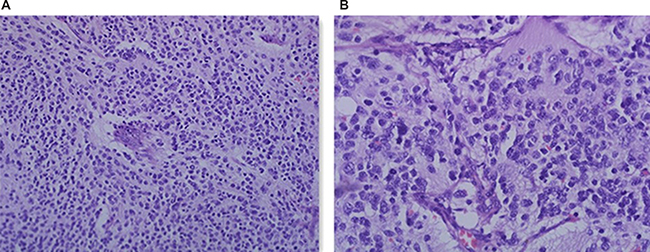
Figure 2: H&E staining of the patient with TIAM1 deletion. (A) HE 200 ×; (B) HE 400 ×.
In addition to CNVs, three missense variants were identified in three patients (ALK:2*F1174L, BRCA2: K2392N) and one frameshift variant was detected in one patient (PHOX2B: F86fs) in this cohort (Figure 3). ALK was previously reported as a frequently mutated gene in NB cases in terms of somatic mutations. All of the ALK mutations occurred in the kinase domain, and this observation suggested their pathogenic nature.
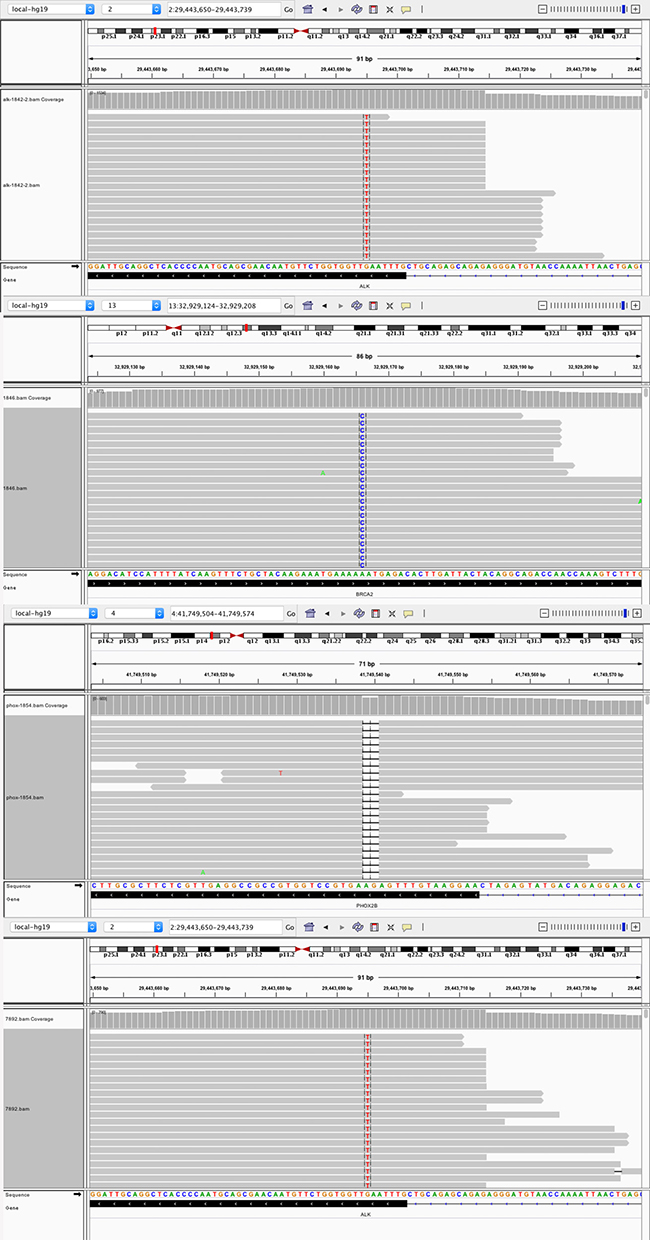
Figure 3: Four point mutations in four patients. (2 ALK: F1174L; BRCA2: K2392N; PHOX2B: F86fs).
All of the changes, including CNVs, accounted for 76.5% (13/17)in comparison to 23.5% (4/17) of point mutations were detected at a high frequency. The results of somatic mutations are illustrated in Figure 4 and Supplementary Table 4.
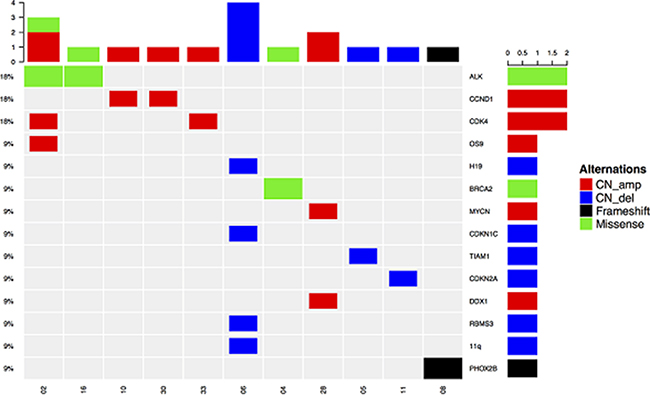
Figure 4: Oncoprint outcome of 11 patients (detected somatic mutations were mostly CNVs, including CN amp and CN del). CDK4, CCND1, and ALK variations appeared most frequently, and they were present in two patients).
Identification of CNVs associated with other prognostic factors
In this study, 11 CNVs among 32 patients, in which one case of amplification of MYCN and DDX1 was excluded, were associated with other significant prognostic effects, namely, age at diagnosis, INSS, INPC, and distant metastasis. In Table 2, CNVs were also associated with UH (P = 0.003) and more likely to occur in stage 4 (P = 0.041). Only one deletion occurred in TIAM1 in stage 1. All amplifications occurred in patients in stage 4 and in the UH group.
Table 2: Clinical and tumor characteristics of the discovery cohort (n = 32)
Characteristic |
Number (%) |
CNV (%) |
P-value |
|---|---|---|---|
Age |
0.465 |
||
≤ 18 months |
12 (37.5) |
2 (6.3) |
|
> 18 months |
20 (62.5) |
5 (15.6) |
|
Gender |
0.500 |
||
boy |
16 (50) |
3 (9.4) |
|
girl |
16 (50) |
4 (12.5) |
|
INSS |
0.041 |
||
non–stage 4 |
16 (50) |
1 (3.1) |
|
4 |
16 (50) |
6 (18.8) |
|
Histology (INPC) |
0.003 |
||
Favorable (FH) |
16 (50) |
0 (0) |
|
Unfavorable (UH) |
16 (50) |
7 (21.9) |
|
Distant Metastatic |
0.061 |
||
+ |
17 (53.1) |
6 (18.8) |
|
− |
15 (46.9) |
1 (3.1) |
|
Serum NSE |
0.389 |
||
> 100 ng/mL |
19(59.4) |
5 (15.6) |
|
≤ 100 ng/mL |
13(39.4) |
2 (6.3) |
Identification of CNVs associated with prognosis
16 patients in Stage 4 received the same chemotherapy and surgery, lacked MNA, and belonged to nearly the same age group and INPC group to avoid the interference of other prognostic factors. The three-year EFS of patients without CNV (10 patients) was 60.0% in comparison to the EFS (16.7%) of patients with CNV (6 patients) (HR = 0.1344; 95%, CI = 0.027 to 0.678; P = 0.015, log-rank test; Figure 5). Of the 16 patients, 9 experienced adverse events: 1 patient died, 2 patients underwent relapse, and 6 patients exhibited progression. Of the 9 patients, 7 manifested definite somatic events, mostly CNVs (Table 1).
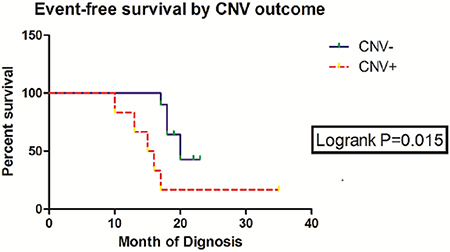
Figure 5: Summary of three-year EFS rates and log-rank test comparisons by CNV outcome.
Identification of CNVs associated with treatment effects
The differences between CNV(+) and CNV(−) of the 16 patients were analyzed in terms of their sensitivity to chemotherapy and dependence on very good partial response (VGPR) after four cycles of chemotherapy. The results showed that VGPR in the patients with CNVs were generally difficult to achieve (P = 0.059; Figure 6 and Table 1).
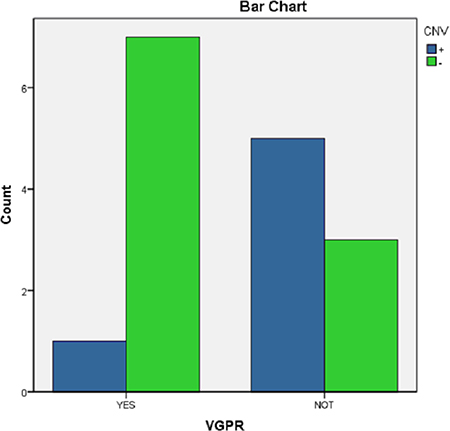
Figure 6: Distribution of obtaining a VGPR after four cycles of chemotherapy between CNV(+) and CNV (−).
Value of four point mutations
Three missense variants were found in three patients (ALK:2*F1174L, BRCA2: K2392N) and one frameshift variant in one patient (PHOX2B: F86fs) in this cohort, and these conditions accounted for 23.5% of all the results. BRCA2 was found in the patients with stage 1 and in the FH group. Other point mutations were observed in the patients with stage 4 and in the UH group. The patients with ALK or PHOX2B progressed eventually.
DISCUSSION
The amplification of MYCN gene (MNA) or the v-myc avian myelocytomatosis viral oncogene NB-derived homolog is a strong independent adverse prognostic factor associated with other adverse prognostic factors. For example, MNA is related to UH, old age, and INSS stage 4 [7, 11]. MNA is also found in a high proportion of adrenal primary tumors [22], and its incidence may differ according to the involvement of specific metastatic sites [23]. However, most patients lack MNA [13], and their prognosis remains poor. A comprehensive and definitive analysis of clinical and other genetic predictors in a large cohort of patients without MNA has yet to be performed. Most amplifications, such as ALK, DDX1, and ODC1, often occur concomitantly with MNA. Other CNVs, such as the amplifications of CDK4, CCND1,and CDK6 and deletions of CDKN2A and RBMS3, which are nonsyntenic with the MYCN locus, likely yield a low recurrence to the amplicon of MYCN, but these findings have yet to be investigated [24–29]. However, the frequency of recurrent CNVs is relatively higher than that of point mutations [19, 20], which are essential for patients without MNA.
To investigate the significance of other CNVs in these samples without MNA and screen other mutations, we applied capture-based NGS, integrated key CNV and point mutation regions related to NB, and deeply sequenced 33 patients without MNA defined by FISH. Our NB panel comprised 53 genes and 3 large chromosome regions. All of the 33 tissue and leukocyte samples sequenced in this study were subjected to stringent QC allegations. A mean coverage depth of 887× was found in all of the target regions in all of the samples. The mapped read percentage was over 99%. Finally, 13 CNVs were detected in 33 patients and 4 significant point mutations were screened. The reliability of our approach was validated, and this approach was compared with traditional CNV detection method (FISH) in terms of MNA evaluation. Our results indicated that specificity, negative predictive value, and false-positive rate were 97%, 100%, and 3%, respectively. Furthermore, 1 case of MNA was detected through NGS possibly because this method could capture smaller amplified fragments.
Our sequencing results revealed that CNVs were commonly detected. These CNVs were related to other prognostic factors, and they were detected mostly in patients in stage 4 and UH group. These CNVs could also suggest poor prognosis in stage 4 patients who received the same treatment regimen and presented poor responses to conventional chemotherapy. These CNVs showed important clinical values in the predicting prognosis and guiding the targeted therapy of NB in patients without MNA. Our conclusion is consistent with previous findings, which revealed that somatic chromosomal imbalance variation is an important feature of the poor prognosis of NB. For example, the deletions of chromosomes 1p, 3p, 4p, and 11q, the gain of chromosomes 1q, 2p, and 17q, and MYCN amplifications were associated with aggressive clinical features, and this finding implied poor prognosis [10, 16–18]. The LOH of chromosome 1p36 and the gain of 17q in NB were closely related to other high-risk clinical features, such as old age, MYCN amplification, and distant metastasis [10, 30–33]. In the presence of MYCN-amplified NB, the deletion fragment of 1p is often larger [25, 27]. Furthermore, 11q deletion is frequently found in tumors without MYCN and associated with poor prognosis in NB [10]. In our study, three large chromosomal regions captured in our assay panel were selected. Finally, 1 case of 11q deletion was found in our study, and the patient developed disease progression after 16 months, and this finding fully confirmed that 11q deletion could be a poor prognostic indicator of NB. Although the relationship between these chromosomal fragments and NB has been clarified, the CNV values in NB clinical treatment, prognostic analysis, and pathogenetic studies remain unclear.
Our study on CNVs found that CDK4 and CCND1 amplifications were the most common, and 11q, CDKN2A, and RBMS3 deletions have been detected in NB [24–29, 31, 34, 35]. Other CNVs, such as deletions of H19 [36] and CDKN1C [37], have been rarely reported. 11q deletions and other CNVs also occur predominantly in tumors without MNA, and this observation is consistent with that in previous studies [27, 28]. CDKN2A encodes p16INK4a transcripts and functions as an inhibitor of the cell cycle activators CDK4 and CDK6, whose activity is required for cell cycle G1/S transition [38]. CCND1 or cyclin D1 forms a complex and serves as a regulatory subunit of CDK4 or CDK6. OS-9 gene, an endoplasmic reticulum lectin, is usually co-amplified with CDK4 [39]. H19, which is imprinted and maternally expressed transcript and non-protein coding, and CDKN1C in 11p15.5 regions are two imprinted genes implicated in cell cycle and associated with Beckwith–Wiedemann syndrome, which is a rare pediatric overgrowth disorder [40]. Most of these genes are involved in the cell cycle, and this finding suggests that NB may be susceptible to being addicted to individually activated oncogenes during cell cycle regulation. Cell cycle-related molecular inhibitors may be used to treat NB. For example, CDK4/6 inhibition has provided an effective strategy to treat a subset of NB tumors [41].
Among the 17 mutations observed in this study, TIAM1 deletion is a new finding. This gene encodes the protein of the GEF family members and activates the downstream Rac1 protein [42]. Once the neurotrophic factor NGF binds to the neurotrophin receptor TrkA, it activates TIAM1 through Ras and then converts Rac1-GDP into a biologically active Rac1-GTP to act on actin and promote neuronal outgrowth and Schwann cell migration [43]. By contrast, P75NGFR/RhoA pathway functions as a negative regulatory TrkA/TIAM1/Rac1 pathway and thus facilitates the outward retraction of protruding axons and the conversion of cells to become round [44, 45]. The balance of these signaling pathways regulate the growth of neuronal axons and the migration of Schwann cells, which is the process of nerve cell differentiation [46, 47]. NB is derived from primordial neural crest cells, and other immature neuronal cells, such as neuroblasts and gangminic neuroblast cells, are produced because of the differentiation disorder of neural stem cells into ganglion cells and Schwann cell and causes the occurrence of NB [48]. Hence, NB can be used as a good model to examine the differentiation mechanism of tumors [6]. The exploration of genes and molecules involved in NB differentiation is the key to understanding the pathogenesis of this condition in various genes and signaling pathways. However, the accurate differentiation mechanism has yet to be established because of the complexity of signal pathways and molecules participating in NB differentiation. Ras/MAPK signaling pathway is primarily involved in NB differentiation and mediated by neurotrophic factor receptor (NGFR) [49–51]. However, this signaling pathway is implicated in the occurrence and development of other adult epithelial tumors, and Ras/TIAM1/Rac1 pathway likely participates in this mechanism. The Trk/TIAM1/Rac1 pathway promotes the role of positive differentiation. P75NGFR/RhoA negatively regulates this process and participates in dedifferentiation. Therefore, the two signaling pathways modulate NB differentiation. TIAM1 deletion causes the dedifferentiation of nerve cells and thus leads to tumor occurrence. Molenaar JJ et al [52]. reported that three cases of TIAM1 inactive mutations are detected in 87 patients with NBs and the downstream signaling pathway is blocked, and this finding is consistent with our results. Either TIAM1 mutation or deletion blocks the Trk/TIAM1/Rac1 pathway, and this finding indicates the importance of gene and signaling pathway. To verify this observation, we reviewed the diagnosis of patients with TIAM1 deletion and to observe the morphological characteristics of tumor cells microscopically. Our results revealed that the differentiation of neural cells was poor, and the diagnosis of children was in the UH group. Through the NGS of the NB patients, TIAM1 was screened more scientifically, and this finding showed a basis for the analysis of NB pathogenesis. Therefore, target-capture NGS is an important tool to screen disease pathogenesis and provided insights into protein functional verification.
This study demonstrated that other CNVs, such as CDK4 and CCND1 amplifications and CDKN2A, CDKN1C, and RBMS3 deletions, could be the superior choice to identify patient stratification in MYCN-negative tumors. These CNVs are associated with NB prognosis and other risk factors. Patients with CNVs usually elicit poor responses to conventional chemotherapy. Comprehensive genetic/genomic profiling through in-depth NGS rather than focusing on single genetic markers could include prognostic information and improve targeted therapy. Further analysis through clinical trials and sample expansion are required to enhance the risk-group stratification in patients without MNA. TIAM1 deletion is a new mutation that may be involved in NB differentiation and may provide a basis for NB pathogenesis by using NGS screening method.
MATERIALS AND METHODS
Patient selection and analysis
The electronic medical records of 127 pediatric patients who underwent diagnostic workup for NB in our medical center between 2012 and 2015 were retrospectively reviewed. Of these patients, 96 suffered from NB and underwent MYCN tested by FISH, and these patients were enrolled in this study. Their clinical stages were determined according to the INSS [9] and histologic classification from INPC [7, 53]. Tumor groups were reviewed by the Department of Pathology in Tianjin Medical University Cancer Institute and Hospital. The patients were treated according to international treatment protocols. Distant metastasis refers to the metastasis of tumors in sites that the primary tumor is not connected. Efficacy evaluation and VGPR were determined as follows [54]: primary tumor volume was reduced from 99% to 90%. All of the measurable metastases disappeared, catecholamines and metabolites were recovered to normal levels, and 99Tc scan bone lesions must be positive because bone metastases are not healed, but all of the lesions should be negative in the MIBG examination. MNA was defined by international criteria with FISH [12].
To explore the CNV-guided risk stratification, we enrolled 33 patients from those MNA(−) patients: 16 patients in non-stage 4 were randomly selected, and the 17 other patients were carefully selected(The set of conditions was as follows: (a) stage 4, (b) only received the same chemotherapy and surgery, and (c) age, and the INPC group was as far as possible consistent. This set ruled out the effect of stage, treatment differences, age, and INPC on prognosis). All tumor tissue samples were confirmed by pathology assessment to contain tumor cells more than 60%.
The objects for survival analysis were defined as MNA(−) showed by NGS and Stage 4 of neuroblastoma to avoid the interference of other prognostic factors. Survival was statistically analyzed with SPSS and GraphPad Prism. The EFS event was defined as the first occurrence of relapse, progression, secondary malignancy, or death from any cause. In the absence of an event, the survival time was censored in the last known time. The time to the event was determined from the date of diagnosis. Two-sided log-rank tests were conducted to examine the differences between survival curves. Survival curves were evaluated according to Kaplan–Meier method and compared via a log-rank test with P < 0.05, which was considered significant. Clinical analysis was performed in chi-square test. In all of the analyses, P-values less than 5% were considered significant.
Tumor tissue collection and DNA extraction
For each patient enrolled in the study, tissue biopsy was obtained from our hospital tissue bank with agreement. DNA was extracted using DNeasy Blood & Tissue Kit according to the manufacturer’s instructions. DNA concentration was measured by Qubit dsDNA assay.
Preparation of white blood cells
At time of biopsy, 10 ml of peripheral blood was obtained, stored in ethylenediaminetetraacetic (EDTA) acid tubes and let it stand at room temperature for 72 hours. The blood cells was stratified by centrifuging for 10 min at 2000 g at 4°C. The the white blood cells was then removed to a new tube and stored at −20°C until further analysis. gDNA was recovered from 4 to 5 ml 200ul of nucleated cell layer using the DNeasy Blood & Tissue Kit (Qiagen). Quantification of gDNA was performed using the Qubit 2.0 Fluorimeter with the dsDNA HS assay kits (Life Technologies, Carlsbad, CA). A minimum of 50 ng of gDNA is required for NGS library construction.
NGS library preparation
DNA shearing was performed using transposase, synchronized fragmentation and adaptor ligation. Fragments of size 200–400 bp were selected by bead (Agencourt AMPure XP Kit), followed by hybridization with capture probes baits, hybrid selection with magnetic beads and PCR amplification. A bioanalyzer high-sensitivity DNA assay was then performed to assess the quality and size of the fragments and indexed samples were sequenced on Nextseq500 sequencer (Illumina, Inc., USA) with pair-end reads.
Capture-based targeted DNA sequencing
DNA concentration and genomic DNA quality were measured by Qubit dsDNA assay and 260 nm/280 nm absorption ratio, respectively. DNA was hybridized with the capture probes baits, selected with magnetic beads and PCR amplified. Quality and size range of amplified fragments were then assessed by bioanalyzer high-sensitivity DNA assay, and indexed samples sequenced on a NextSeq 500 sequencer with pair-end reads.
Sequence data analysis
Sequence data were mapped to the human genome (hg19) using BWA aligner 0.7.10. Local alignment optimization, variant calling and annotation were performed using GATK 3.2, MuTect, and VarScan. Tumor sample was compared against paired white blood cells to identify somatic variants. Variants were filtered using the VarScan fpfilter pipeline, with loci with depth less than 100 filtered out. Minimal of 5 supporting reads were required for INDELs and 8 supporting reads were required for SNV calling. Variants were annotated with ANNOVAR and SnpEff v3.6. Annotation database including the ExAC, 1000 Genomes, dbSNP, ESP6500SI-V2, and COSMIC. DNA translocation analysis was performed using both Tophat2 and Factera 1.4.3. CNV were identified using in-house R scripts based on the coverage ratio of the capture intervals in tumor and normal samples. Coverage depth data were corrected for the sequencing bias due to GC content and probe density. The mean centers and variation levels of coverage depth were then calculated from normal white blood cells as reference. The difference of adjusted coverage depth for each gene from tumor sample and reference was evaluated by t-test. Genes with p-value < 1e-4 in the t-test and CN > 2.5 or < 1.5 were considered as amplification or deletion.
ACKNOWLEDGMENTS AND FUNDING
Thanks are due to the National Key Clinical Specialist Construction Programs of China (No.2013-544), Medical University of Tianjin Science Foundation(2014KYM07), Molecular Diagnostic Platform Construction Project and Foster Projects (Category C) of National Cancer Clinical Medicine Research Center for funding support, and to Professor Kexin Chen and Fengju Song, and partner Jiayi Liu for valuable discussion. The same gratitude is also given to Dr. Shaokun Chuai for technical guidance and support.
CONFLICTS OF INTEREST
The authors declared no potential financial conflicts of interest.
REFERENCES
1. London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, Thorner P, Brodeur G, Maris JM, Reynolds CP, Cohn SL. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. Journal of clinical oncology. 2005; 23:6459–6465.
2. Matthay KK, George RE, Yu AL. Promising therapeutic targets in neuroblastoma. Clinical cancer research. 2012; 18:2740–2753.
3. Shimada H, Chatten J, Newton WA Jr, Sachs N, Hamoudi AB, Chiba T, Marsden HB, Misugi K. Histopathologic prognostic factors in neuroblastic tumors: definition of subtypes of ganglioneuroblastoma and an age-linked classification of neuroblastomas. Journal of the National Cancer Institute. 1984; 73:405–416.
4. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. The New England journal of medicine. 1985; 313:1111–1116.
5. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B. Terminology and morphologic criteria of neuroblastic tumors: recommendations by the International Neuroblastoma Pathology Committee. Cancer. 1999; 86:349–363.
6. Shimada H, Umehara S, Monobe Y, Hachitanda Y, Nakagawa A, Goto S, Gerbing RB, Stram DO, Lukens JN, Matthay KK. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children’s Cancer Group. Cancer. 2001; 92:2451–2461.
7. Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, Stram DO, Gerbing RB, Lukens JN, Matthay KK, Castleberry RP. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999; 86:364–372.
8. Goto S, Umehara S, Gerbing RB, Stram DO, Brodeur GM, Seeger RC, Lukens JN, Matthay KK, Shimada H. Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children’s Cancer Group. Cancer. 2001; 92:2699–2708.
9. Evans AE, D’Angio GJ, Randolph J. A proposed staging for children with neuroblastoma. Children’s cancer study group A. Cancer. 1971; 27:374–378.
10. Attiyeh EF, London WB, Mosse YP, Wang Q, Winter C, Khazi D, McGrady PW, Seeger RC, Look AT, Shimada H, Brodeur GM, Cohn SL, Matthay KK, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. The New England journal of medicine. 2005; 353:2243–2253.
11. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science (New York, NY). 1984; 224:1121–1124.
12. Ambros PF, Ambros IM, Brodeur GM, Haber M, Khan J, Nakagawara A, Schleiermacher G, Speleman F, Spitz R, London WB, Cohn SL, Pearson AD, Maris JM. International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. British journal of cancer. 2009; 100:1471–1482.
13. Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Current opinion in pediatrics. 2005; 17:7–13.
14. Caldas C. Cancer sequencing unravels clonal evolution. Nature biotechnology. 2012; 30:408–410.
15. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481:506–510.
16. Janoueix-Lerosey I, Schleiermacher G, Michels E, Mosseri V, Ribeiro A, Lequin D, Vermeulen J, Couturier J, Peuchmaur M, Valent A, Plantaz D, Rubie H, Valteau-Couanet D, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. Journal of clinical oncology. 2009; 27:1026–1033.
17. Deyell RJ, Attiyeh EF. Advances in the understanding of constitutional and somatic genomic alterations in neuroblastoma. Cancer genetics. 2011; 204:113–121.
18. Caron H, van Sluis P, van Hoeve M, de Kraker J, Bras J, Slater R, Mannens M, Voute PA, Westerveld A, Versteeg R. Allelic loss of chromosome 1p36 in neuroblastoma is of preferential maternal origin and correlates with N-myc amplification. Nature genetics. 1993; 4:187–190.
19. Shen J, Wu W, Gao C, Ochin H, Qu D, Xie J, Gao L, Zhou Y, Cui Y, Liu J. Chromosomal copy number analysis on chorionic villus samples from early spontaneous miscarriages by high throughput genetic technology. Molecular cytogenetics. 2016; 9:7.
20. Sausen M, Leary RJ, Jones S, Wu J, Reynolds CP, Liu X, Blackford A, Parmigiani G, Diaz LA Jr, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE, Hogarty MD. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nature genetics. 2013; 45:12–17.
21. Fransson S, Ostensson M, Djos A, Javanmardi N, Kogner P, Martinsson T. Estimation of copy number aberrations: Comparison of exome sequencing data with SNP microarrays identifies homozygous deletions of 19q13.2 and CIC in neuroblastoma. International journal of oncology. 2016; 48:1103–1116.
22. Vo KT, Matthay KK, Neuhaus J, London WB, Hero B, Ambros PF, Nakagawara A, Miniati D, Wheeler K, Pearson AD, Cohn SL, DuBois SG. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. Journal of clinical oncology. 2014; 32:3169–3176.
23. Morgenstern DA, London WB, Stephens D, Volchenboum SL, Hero B, Di Cataldo A, Nakagawara A, Shimada H, Ambros PF, Matthay KK, Cohn SL, Pearson AD, Irwin MS. Metastatic neuroblastoma confined to distant lymph nodes (stage 4N) predicts outcome in patients with stage 4 disease: A study from the International Neuroblastoma Risk Group Database. Journal of clinical oncology. 2014; 32:1228–1235.
24. Mosse YP, Diskin SJ, Wasserman N, Rinaldi K, Attiyeh EF, Cole K, Jagannathan J, Bhambhani K, Winter C, Maris JM. Neuroblastomas have distinct genomic DNA profiles that predict clinical phenotype and regional gene expression. Genes, chromosomes & cancer. 2007; 46:936–949.
25. Mosse YP, Greshock J, Margolin A, Naylor T, Cole K, Khazi D, Hii G, Winter C, Shahzad S, Asziz MU, Biegel JA, Weber BL, Maris JM. High-resolution detection and mapping of genomic DNA alterations in neuroblastoma. Genes, chromosomes & cancer. 2005; 43:390–403.
26. Carr J, Bown NP, Case MC, Hall AG, Lunec J, Tweddle DA. High-resolution analysis of allelic imbalance in neuroblastoma cell lines by single nucleotide polymorphism arrays. Cancer genetics and cytogenetics. 2007; 172:127–138.
27. Fix A, Peter M, Pierron G, Aurias A, Delattre O, Janoueix-Lerosey I. High-resolution mapping of amplicons of the short arm of chromosome 1 in two neuroblastoma tumors by microarray-based comparative genomic hybridization. Genes, chromosomes & cancer. 2004; 40:266–270.
28. Molenaar JJ, van Sluis P, Boon K, Versteeg R, Caron HN. Rearrangements and increased expression of cyclin D1 (CCND1) in neuroblastoma. Genes, chromosomes & cancer. 2003; 36:242–249.
29. Su WT, Alaminos M, Mora J, Cheung NK, La Quaglia MP, Gerald WL. Positional gene expression analysis identifies 12q overexpression and amplification in a subset of neuroblastomas. Cancer genetics and cytogenetics. 2004; 154:131–137.
30. Maris JM, Weiss MJ, Guo C, Gerbing RB, Stram DO, White PS, Hogarty MD, Sulman EP, Thompson PM, Lukens JN, Matthay KK, Seeger RC, Brodeur GM. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: a Children’s Cancer Group study. Journal of clinical oncology. 2000; 18:1888–1899.
31. Caren H, Erichsen J, Olsson L, Enerback C, Sjoberg RM, Abrahamsson J, Kogner P, Martinsson T. High-resolution array copy number analyses for detection of deletion, gain, amplification and copy-neutral LOH in primary neuroblastoma tumors: four cases of homozygous deletions of the CDKN2A gene. BMC genomics. 2008; 9:353.
32. Bown N, Lastowska M, Cotterill S, O’Neill S, Ellershaw C, Roberts P, Lewis I, Pearson AD. 17q gain in neuroblastoma predicts adverse clinical outcome. U.K. Cancer Cytogenetics Group and the U.K. Children’s Cancer Study Group. Medical and pediatric oncology. 2001; 36:14–19.
33. Bown N, Cotterill S, Lastowska M, O’Neill S, Pearson AD, Plantaz D, Meddeb M, Danglot G, Brinkschmidt C, Christiansen H, Laureys G, Speleman F, Nicholson J, et al. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. The New England journal of medicine. 1999; 340:1954–1961.
34. Spitz R, Oberthuer A, Zapatka M, Brors B, Hero B, Ernestus K, Oestreich J, Fischer M, Simon T, Berthold F. Oligonucleotide array-based comparative genomic hybridization (aCGH) of 90 neuroblastomas reveals aberration patterns closely associated with relapse pattern and outcome. Genes, chromosomes & cancer. 2006; 45:1130–1142.
35. Fix A, Lucchesi C, Ribeiro A, Lequin D, Pierron G, Schleiermacher G, Delattre O, Janoueix-Lerosey I. Characterization of amplicons in neuroblastoma: high-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes, chromosomes & cancer. 2008; 47:819–834.
36. Astuti D, Latif F, Wagner K, Gentle D, Cooper WN, Catchpoole D, Grundy R, Ferguson-Smith AC, Maher ER. Epigenetic alteration at the DLK1-GTL2 imprinted domain in human neoplasia: analysis of neuroblastoma, phaeochromocytoma and Wilms’ tumour. British journal of cancer. 2005; 92:1574–1580.
37. Brioude F, Netchine I, Praz F, Le Jule M, Calmel C, Lacombe D, Edery P, Catala M, Odent S, Isidor B, Lyonnet S, Sigaudy S, Leheup B, et al. Mutations of the Imprinted CDKN1C Gene as a Cause of the Overgrowth Beckwith-Wiedemann Syndrome: Clinical Spectrum and Functional Characterization. Human mutation. 2015; 36:894–902.
38. Sharpless NE. INK4a/ARF: a multifunctional tumor suppressor locus. Mutation research. 2005; 576:22–38.
39. Su YA, Hutter CM, Trent JM, Meltzer PS. Complete sequence analysis of a gene (OS-9) ubiquitously expressed in human tissues and amplified in sarcomas. Molecular carcinogenesis. 1996; 15:270–275.
40. Fryssira H, Amenta S, Kanber D, Sofocleous C, Lykopoulou E, Kanaka-Gantenbein C, Cerrato F, Ludecke HJ, Bens S, Riccio A, Buiting K. A novel large deletion of the ICR1 region including H19 and putative enhancer elements. BMC medical genetics. 2015; 16:30.
41. Rihani A, Vandesompele J, Speleman F, Van Maerken T. Inhibition of CDK4/6 as a novel therapeutic option for neuroblastoma. Cancer cell international. 2015; 15:76.
42. Engers R, Mueller M, Walter A, Collard JG, Willers R, Gabbert HE. Prognostic relevance of Tiam1 protein expression in prostate carcinomas. British journal of cancer. 2006; 95:1081–1086.
43. Habets GG, Scholtes EH, Zuydgeest D, van der Kammen RA, Stam JC, Berns A, Collard JG. Identification of an invasion-inducing gene, Tiam-1, that encodes a protein with homology to GDP-GTP exchangers for Rho-like proteins. Cell. 1994; 77:537–549.
44. Leeuwen FN, Kain HE, Kammen RA, Michiels F, Kranenburg OW, Collard JG. The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. The Journal of cell biology. 1997; 139:797–807.
45. Boissier P, Huynh-Do U. The guanine nucleotide exchange factor Tiam1: a Janus-faced molecule in cellular signaling. Cellular signalling. 2014; 26:483–491.
46. Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes & development. 2002; 16:1587–1609.
47. Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A. Evidence for the involvement of Tiam1 in axon formation. The Journal of neuroscience. 2001; 21:2361–2372.
48. Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nature Reviews Cancer. 2013; 13:397–411.
49. Brodeur GM, Minturn JE, Ho R, Simpson AM, Iyer R, Varela CR, Light JE, Kolla V, Evans AE. Trk receptor expression and inhibition in neuroblastomas. Clinical cancer research. 2009; 15:3244–3250.
50. Shimada H, Nakagawa A, Peters J, Wang H, Wakamatsu PK, Lukens JN, Matthay KK, Siegel SE, Seeger RC. TrkA expression in peripheral neuroblastic tumors: prognostic significance and biological relevance. Cancer. 2004; 101:1873–1881.
51. Kiessling MK, Rogler G. Targeting the RAS pathway by mitogen-activated protein kinase inhibitors. Swiss medical weekly. 2015; 145:w14207.
52. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012; 483:589–593.
53. Peuchmaur M, d’Amore ES, Joshi VV, Hata J, Roald B, Dehner LP, Gerbing RB, Stram DO, Lukens JN, Matthay KK, Shimada H. Revision of the International Neuroblastoma Pathology Classification: confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer. 2003; 98:2274–2281.
54. Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. Journal of clinical oncology. 1993; 11:1466–1477.