
INTRODUCTION
Hepatocellular carcinoma (HCC) accounts for > 90% of all primary liver cancers and generally arises in the context of chronic liver diseases [1, 2]. Recent studies suggest that abnormal lipid metabolism may play an essential role in the development of nonalcoholic steatohepatitis (NASH) and its progression to HCC [3]. Excess dietary cholesterol has been shown to cause development of experimental NASH in different animal models, especially in the setting of a concurrent high-fat diet [4–7]. A high-fat and high-cholesterol (HFHC) diet is even shown to cause progression from simple steatosis to fibrosing steatohepatitis and HCC [7]. Furthermore, extensive dysregulation of hepatic cholesterol homeostasis leading to increased hepatic cholesterol levels has also been documented in NASH and HCC [3, 4, 8]. Dysregulation likely occurs at multiple levels, including increased hepatic levels of active sterol response element-binding protein-2 and elevated cholesterol biosynthesis, coupled with increased uptake of cholesterol-rich lipoproteins and decreased cholesterol excretion in bile as cholesterol. An aberrant feedback response to sterols appears to be a common phenomenon in cancer cells [9]. Free cholesterol accumulation in the liver is toxic and may be a trigger for progression of NASH and subsequent HCC development [3]. Excess cholesterol accumulation in cells is shown to trigger ER stress, oxidative stress, apoptosis, and inflammation, which aggravate NASH [10–13].
Our recent demonstration that an HFHC diet induces hepatic protein kinase C beta (PKCβ) expression, whereas PKCβ deficiency sensitizes mice to hepatic cholesterol accumulation, suggests that PKCβ has the potential to modulate inflammation, nonalcoholic steatohepatitis (NASH), and HCC [14–16]. It is interesting to note in this regard that loss of heterozygosity of human chromosome 16p region associated with the progression of HCC includes the PKCβ locus [17]. Recent studies have also indicated involvement of PKCβ and downstream ERK-1/2 in regulating lipid metabolism, oxidative stress, inflammation, and apoptosis, key aspects of the pathophysiology of HCC [18, 19]. PKCβ is reported to phosphorylate retinoblastoma (RB) in vitro [20]. The retinoblastoma (RB) gene, located at human chromosome 13q14, is also associated with development or progression of carcinomas in a large spectrum of tissues because this gene is often found to have been deleted in a variety of tumor types, including HCC [21–23]. In fact, 25–67% loss of heterozygosity on chromosome 13q was reported in HCC [24].
It is well established that the liver plays a critical role in regulating cholesterol homeostasis and is targeted by both dietary fat and cholesterol [25]. We previously assessed whether metabolic adaptation to an HFHC diet is associated with changes in hepatic PKCβ expression and also studied the consequence of PKCβ deficiency on diet-induced hepatic cholesterol homeostasis. We proposed that diet-induced PKCβ activation and dependent changes in the liver may prevent toxicity and maintains normal liver function despite repeated exposure to a toxic dose. The underlying mechanism involved upregulation of cholesterol conversion to bile salts coupled with reduction in the uptake of dietary cholesterol and biosynthesis. These results support the possibility that PKCβ is a physiological transducer of dietary lipids and plays a critical role in hepatic adaptation to dietary fats. In order to explore the potential role of the PKCβ isoform in diet-induced HCC, we examined the effects of a PKCβ deficiency on the retinoblastoma protein and family members, as well as compared PKCβ expression in HCC versus normal tissue. Identification of PKCβ as a cellular relay in diet-mediated changes in RB function can define the signaling events that could participate in oncogenesis.
RESULTS
To test if PKCβ and RB are functionally linked, we compared kinetics of hepatic PKCβ induction relative to appearance of phospho-RB and total RB expression levels. Establishing the temporal relationship between these variables will not only allow us to more accurately define the role of PKCβ in a physiological setting but, more importantly, will help to determine whether alteration in PKCβ expression and downstream signaling is an early event in the development of tumorigenesis.
WT and PKCβ-/- mice fed an HFHC diet for varying time periods were analyzed for hepatic PKCβ expression relative to phospho-RB and total RB. As shown in Figure 1A, PKCβ protein expression was markedly increased in a time-dependent manner; induction was observed at week one, peaked at two weeks, and remained elevated for at least three weeks (the last point measured). Notably, phospho-RB (Ser780) levels also showed a time-dependent increase, whereas this increase was absent in the livers of PKCβ-/- mice (Figure 1B). Surprisingly, PKCβ-/- mice demonstrated a marked reduction in the level of hepatic RB protein content. Total RB protein levels were decreased approximately four folds in the livers of these mice compared to controls. In contrast, expression levels of p38MAPK, PRMT7 and ATG7, and another family member, RBL-1, remained unaltered, whereas RBL-2 also showed a decrease in total levels in diet-fed PKCβ-/- livers. A transient decrease in expression of both RBL-1 and RBL-2 was also observed in both genotypes in livers of mice fed HFHC diet for 2 weeks.
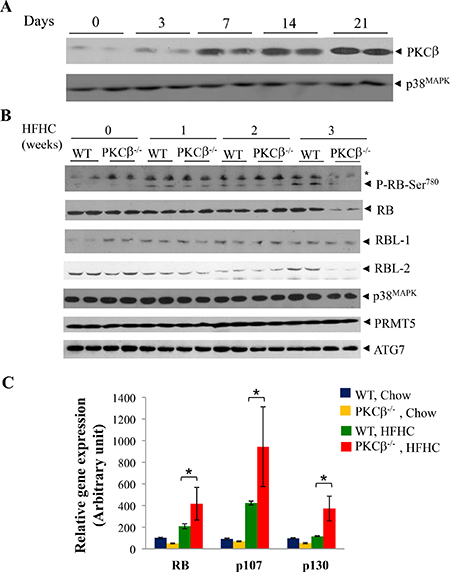
Figure 1: HFHC diet intake reduces RB protein content post-transcriptionally in PKCβ-/- liver compared to normal WT liver. (A) Eight-week-old WT and PKCβ-/- mice (n = 4) were fed an HFHC diet for indicated periods and expression levels of PKCβ isoform in the pooled liver extracts (2 livers per lane) were determined by immunoblotting. (B) Comparison of phospho-RB, RB, p107, and p130 levels in the livers of WT and PKCβ-/- mice fed an HFHC diet for the indicated periods. Equal amounts of protein were loaded per well and immunoblots were probed with antibody specific for PKCβ, RB, p107, p130, P-RB (Ser780), ATG7, PRMT5, and p38MAPK. (C) Q-RT-PCR analysis comparing relative mRNAs of RB, p107, and p130 in the pooled liver extracts of the above mice (16). Individual RNAs from the above livers were run in triplicate, and average values were calculated. Results are expressed as the mean ± SD. *, <0.05. The experiment was repeated twice with similar outcomes.
We also used quantitative RT-PCR to determine whether decreased levels of RB correlated with changes in transcript levels. When compared to control livers from mice fed an HFHC diet, RB transcript levels were increased by ~2-fold. Likewise, an increase in mRNA levels of RBL-1 (> 2.5-fold) and RBL-2 (~2-fold) were also noticed, possibly due to negative self-regulation and suppression of other family members by RB (Figure 1C) [26].
To attribute increased RB phosphorylation specifically to PKCβ, we transiently transfected PKCβ-/- hepatocytes, and reintroduction of PKCβ specifically resulted in increased in phospho-RB (Ser780) levels, further supporting that PKCβ phosphorylates RB specifically at Ser780 without affecting phosphorylation at other residues (Ser807 and Ser811) (Figure 2). Notably, an increase in Ser780 phosphorylation is not accompanied by an increase in RB protein levels in cultured hepatocytes, suggesting that participation of additional diet-responsive signaling pathways dictates RB protein level.

Figure 2: Reintroduction of human PKCβ in cultured cells restores RB (Ser780) phosphorylation with no significant effect on its abundance. PKCβ-/- hepatocytes were isolated from twelve-week-old C57BL/6J male mice as described previously (15). The recombinant adenovirus expressing green fluorescent protein GFP and GFP-PKCβ were used to transiently overexpress PKCβ as described previously (15, 29). After 36 h infection with 1 to 100 multiplicity of infection (MOI), cells were harvested with ice-cold PBS. Immunoblotting were used to compare phospho- and total RB levels. Quantitation of bands is shown in the right panel. The results shown are representative of at least three separate experiments. The detection of a weak band in the first two lanes possibly reflects slight cross-reactivity of the PKCβ antibody with other endogenous PKC isoforms, particularly PKCδ.
The global conformational changes that occur in RB upon phosphorylation are site-specific and remarkably diverse, providing a mechanism through which varying phosphorylation events can code for different functional outputs [27]. The role of S780 is presently not clear, but a previous study has reported that RB deficiency in adult mice for five to six months led to an increase in ploidy of hepatocytes [28]. In this study, loss of RB led to decreased numbers of 4n hepatocytes and a concomitant increase of 8n and 16n cells. To investigate the potential effects of PKCβ deficiency on the DNA content of hepatocytes in preneoplastic livers, we conducted flow cytometric analysis on hepatocytes obtained from WT and PKCβ-/-mice fed an HFHC diet for three weeks. This analysis showed that deficiency of PKCβ had no impact on ploidy in hepatocytes (Figure 3). These observations suggest that loss of RB for a shorter period may be less influential in perturbing DNA content in adult hepatocytes, and a longer RB deficiency is needed to see any measurable effects.
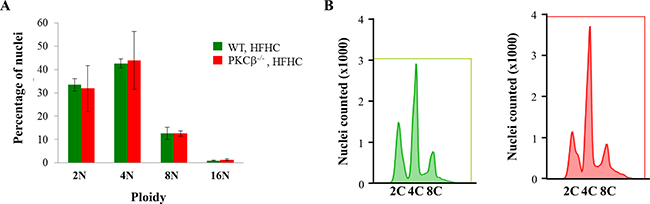
Figure 3: PKCβ deficiency does not increase ploidy in hepatocytes. (A) Quantification of ploidy by FACS of hepatocyte populations from control and PKCβ-/- livers (n = 5) after feeding an HFHC diet for three weeks. Results are expressed as the mean ± SD. * < 0.05. (B) Representative fluorescence-activated cell sorting profiles of propidium iodide-stained liver nuclei from the above mice. This experiment was repeated twice with similar outcomes
Autophagy is another protective mechanism that renders cells viable under stressful conditions. Emerging evidence suggests that this cellular process is also a tumor suppressor pathway [29]. We have previously shown that PKCβ deficiency, like RB, triggers autophagy [30, 31]. We therefore measured conversion of microtubule-associated protein LC3-I to LC3-II, a biochemical marker of autophagy that correlates with the formation of autophagosomes. As expected from our previous study, immunoblotting revealed a greater accumulation of LC3-II in the liver of PKCβ-/- mice compared to WT fed a chow diet, but upon feeding an HFHC diet, the observed differences in autophagy disappeared (Figure 4). Likewise, expression of Beclin-1, a key activator of the pathway, is higher in PKCβ-/- livers compared to control livers of chow-fed mice, and there is no difference in its protein levels between genotypes under dietary HFHC conditions. These results reinforce the concept that nutrient status and metabolic stressors can influence the autophagic response via signaling networks that often, but not exclusively, converge at PKCβ and RB, and participation of additional pathways potentially influence overall dietary modulation of autophagy induction. It is not surprising because autophagy is a complex process that requires a major degree of coordination among several sensors interacting with the autophagic machinery to detect fluctuations in key metabolic parameters [31].
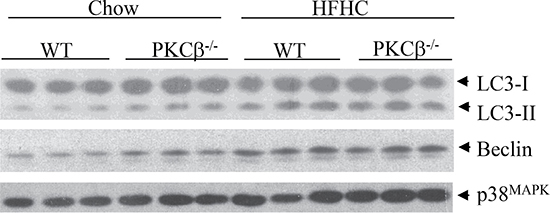
Figure 4: Comparison of hepatic autophagy levels between genotypes. WT and PKCβ-/- mice (n = 3) fed an HFHC diet for two weeks were sacrificed, and liver protein extracts were analyzed for the levels of autophagy markers. LC3-II accumulation was quantified by LC3-II/LC3-I ratio.
We also analyzed expression of both human splicing variants PKCβI and PKCβII in fifteen pairs of HCC and their paired nearby non-cancerous livers. The results are shown in Figure 5. The normal liver showed a homogenous expression pattern in comparison with the heterogeneous expression feature of HCC, which includes increased or decreased PKCβ mRNA in HCCs compared to control livers. We found that reduction and elevation of PKCβ was detected in 87% and 13% of HCCs examined, respectively. Interestingly, unlike PKCβI, decrease in PKCβII mRNA levels in HCCs compared to normal tissue was statistically significant (Figure 5). In addition, we compared the protein expression of PKCβII in the above samples. Again, expression was significantly reduced in the HCC livers (p < 0.028) when compared with normal distant and normal adjacent liver tissue (results not shown). Our data indicate that there is a downregulation of PKCβII in HCC.
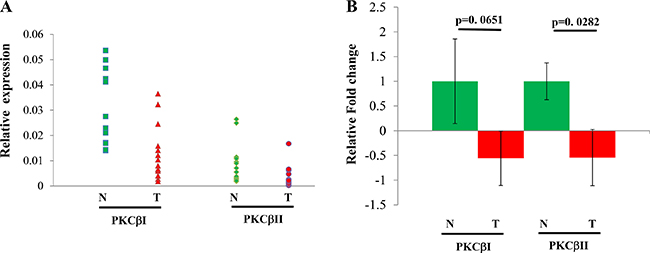
Figure 5: Individual (A) and combined (B) expression analyses of PKCβ isoforms in fifteen pairs of HCC and normal liver tissues by q-PCR. Total RNA from individual samples were subjected to real-time RT-PCR analysis to detect the mRNA expression of PKCβ-I and PKCβ-II. b-Actin was used as an internal control to normalize the samples. Data was analyzed using the delta delta ct method. The results were shown as mean ± SEM. All statistical analyses were performed by Student’s t-test or ANOVA in Excel; p < 0.05 was considered statistically significant. *< 0.05.
DISCUSSION
Dietary lipids are important regulators of cellular proliferation, not just as fuel substrates but also as specific modulators of the production and function of cell cycle-associated proteins. Results presented here support a role for the PKCβ regulatory network in hepatic adaptiveness to dietary fat and cholesterol. The loss of this protein may lead to the development or progression of diet-induced HCC, thus linking dietary intake of lipids with oncogenesis.
The molecular pathogenesis of HCC is complex and involves abnormal clonal expansion of dysplastic hepatocytes, anti-apoptotic signaling, and stimulation of cholesterol biosynthesis and uptake of dietary cholesterol [1, 32]. Following acute or chronic liver damage, quiescent hepatocytes enter the cell cycle and divide to restore the functional capacity of the liver. Precise control over hepatocyte proliferation is critical for the suppression of tumorigenesis in the liver, as chronic liver damage and corresponding cycles of regeneration are known to fuel the development of HCC [23]. There is clear evidence that RB pathways are involved in regulating hepatocyte proliferation. Correspondingly, the RB pathway is disrupted with high frequency in HCC [24]. Our observations suggest a role for PKCβ in the regulatory network controlling phosphorylation and stability of hepatic RB and another family member. This finding suggests that PKCβ may have some interesting biological consequences that contribute to liver tumorigenesis. Reduced PKCβ expression in a majority of our HCC samples supports a key role for this kinase as a tumor suppressor protein whose loss may predispose to tumor development. It is interesting to note in this regard that genetic alterations suppressing PKCβ activity or expression have recently been reported in a variety of cancer cell lines and in primary tumors [33, 34]. On the other hand, PKCβ overexpression observed in few HCC samples may be a programmed protective response of the organism to uncontrolled proliferation. This hypothesis is supported by a similar finding where overexpression of other tumor suppressor genes, such as RB and P73, has been detected in HCCs [35–37]. It is also possible that PKCβ, like P53 and P73, can increase its own expression in response to oncogenic activation or hypoxia or growth factor depletion/overexpression [38–40]. Individual genetic variations cannot account for heterogeneity of PKCβ expression patterns because cancer and noncancerous tissues from the same patients were examined in this study. However, it is possible that the heterogeneity of PKCβ expression is a direct result of the complexity of hepatocarcinogenesis in HCCs [41]. An important issue that might contribute to above results is the heterogeneity of HCC due to different etiologies because HCC generally arises in the context of chronic liver diseases, including chronic viral hepatitis, alcohol-induced liver injury, or other metabolic, dietary, or toxic factors such as fatty liver disease or aflatoxin ingestion. Although most of the patients had metabolic syndrome, more studies will be required to confirm and extend these observations; nevertheless, alteration in PKCβ expression implies that pharmacologic intervention of PKCβ may have broad therapeutic utility.
The exact mechanism by which PKCβ degrades RB is unknown. Multisite phosphorylation modulates the function of RB, and it has been proposed to act as a code in which discrete phosphorylation control specific activities [27]. Although the regulation of RB by phosphorylation has been extensively studied by multiple kinases, the role of phosphorylation in dictating proteasome-mediated RB protein degradation is largely unknown. Several lines of evidence indicate that RB phosphorylation and degradation are functionally linked [42, 43]. For example, gankyrin is shown to interact and stimulate RB degradation by increasing phosphorylation of specific residues in RB protein [44, 45]. Likewise, hypophosphorylated RB interacts with lamin A/C, and in the absence of lamin A/C, RB is degraded in a proteasome-dependent manner, suggesting that A-type lamins protect RB from degradation [46–48]. Given that RB undergoes enhanced degradation in a diet-fed PKCβ-/- liver, it will be interesting to examine whether PKCβ directly or through downstream signal modulates RB interaction and degradation [49]. S780, implicated in cell cycle regulation, is located in a C-terminal tail of the pocket domain [50]. One possibility is that S780 phosphorylation of RB regulates phosphorylation/acetylation/ubiquitinylation/ methylation/SUMOlyation at other sites and modulates intra- and/or intermolecular association. Gaining further insight into the role of specific nutrients and PKCβ in the control of RB post-translational modifications may improve our ability to manipulate cellular proliferation.
In summary, our results uncover a unique and novel metabolic regulatory axis that couples dietary fat/cholesterol to oncogenesis, and PKCβ activation may be the initial event in delaying dietary fat-induced HCC development and therefore may be an attractive therapeutic target for preventing its development.
MATERIALS AND METHODS
Animals and diets
HFHC diet studies with PKCβ-/- mice have been described previously [14, 15]. All mice were housed in a temperature-controlled room (22oC) with a 12 h light-12 h dark cycle, and maintained on a standard rodent diet (7912 rodent chow; Harland Tekland, WI). Eight-week-old WT and PKCβ-/- mice were fed an HFHC diet (Teklad TD-10014; Harland Tekland, WI) containing (by weight) 15% anhydrous milk fat, 1% cholesterol, and 0.5% cholate for indicated period. Mice were fed ad libitum with free access to water. Unless indicated, all experiments were performed on non-fasted male mice and mice were euthanized in the morning. All procedures and experiments on mice were approved by Institution Animal Care and Use Committee of the Ohio State University.
Western blotting
Liver were homogenized at 4oC by pestle in the buffer as previously described [14, 15]. Equal amounts of protein were run in 10% or 12% SDS-PAGE, transferred to nitrocellulose membranes and blocked in Tris-buffered saline with 0.1% Tween 20 containing 5% nonfat dry milk or BSA for 1 h at room temperature. Blots were incubated overnight at 4oC with following primary antibodies: PKCβ, total p38MAPK and total ERK (Santa Cruz Biotechnology); β-Actin, p106, p130, LC3 (Sigma, MO); RB, P-RB, PRMT5, P-ERK (Cell Signaling Biotechnology).
Isolation and culture of mouse hepatocytes
Mouse hepatocytes were isolated from 12 week-old C57BL/6J male mice as described previously [15]. Briefly, the liver was removed, and livers capsule were peeled off. Hepatocytes were dispersed by mechanical dissociation, and filtered through sterile100 mM-mesh nylon. The hepatocytes were washed twice in ice-cold Dulbecco’s modified Eagle’s medium (DMEM, Cellgro, Mediatech) supplemented with 10% FBS (Atlanta Biologicals, GA), penicillin/streptomycin (BioWhittaker, Lonza), insulin at 10 nM and Dexamethasone at 100 nM and resuspended in the same medium, then seeded at a density of 0.3 × 106 cells per well in collagen-coated (collagen I, rat tail) 6-well plate. After 2 h incubation, the culture medium was changed without insulin. The cell culture was continued for additional 4 h before the indicated experiments.
Infection of primary mouse hepatocytes with Ad-GFP or Ad-PKCβ
The recombinant adenovirus expressing green fluorescent protein GFP and GFP-PKCβ were used to overexpress PKCβ as described previously [15]. After 36 h infection with 1 to 100 multiplicity of infection (MOI), cells were harvested with ice-cold PBS. After spin, cells pellet were resuspended in cell lysis buffer (20 mM Tris buffer, 2 mM EDTA, 2mM EGTA, 0.1% SDS, proteinase and phosphatase inhibitors) for 30 min on ice, and sonicated briefly. The supernatant was collected after centrifugation. The protein assay was carried out before loading onto SDS-PAGE.
Gene expression profiles
Total RNA preparation, first-strand cDNA synthesis, and the transcript levels for indicated genes were quantified as described previously [15]. Reactions were normalized to Gadph expression using the DDCt method. Relative mRNA expression was expressed as fold change over the control mice. All samples were run in triplicate, and average values were calculated.
Flow cytometric analysis
Nuclei suspensions were obtained from frozen liver tissue as described previously [51]. Total DNA content of a minimum of 40,000 nuclei per liver sample was analyzed by the OSUCCC Analytical Cytometry Shared Resource using an LSRII (BD Biosciences). Cell cycle profiles were generated using FlowJo (Tree Star).
Human HCC specimens
Primary human hepatocellular tumor and normal liver samples were obtained from the Cooperative Human Tissue Network at the Ohio State University James Cancer Hospital. Tissue specimens were procured in accordance with The Ohio State University Cancer Internal Review Board guidelines. The detailed information of patients is described previously [52].
Statistical analysis
Data are presented as the mean+SE or mean+SD. To determine p values, two-tailed Student’s t tests were performed (unless otherwise indicated). P values indicate the level of statistical significance and P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We thank Dr. Gustavo Leone for Flow Cytometry analysis of liver samples from WT and PKCβ-/- mice.
CONFLICTS OF INTEREST
“The authors declare no conflicts of interest”.
FINANCIAL SUPPORT
This work was supported by HL079091 and the Ohio State University Dean fund (to KDM) and R01CA086978 (to STJ and KG).
REFERENCES
1. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012; 379:1245–1255.
2. Ioannou GN, Morrow OB, Connole ML, Lee SP. Association between dietary nutrient composition and the incidence of cirrhosis or liver cancer in the United States population. Hepatology. 2009; 50:175–184.
3. Ioannou GN. The role of cholesterol in the pathogenesis of NASH. Trends Endocrinol Metab. 2016; 27:84–95.
4. Lu M, Hu XH, Li Q, Xiong Y, Hu GJ, Xu JJ, Zhao XN, Wei XX, Chang CC, Liu YK, Nan FJ, Li J, Chang TY, et al. A specific cholesterol metabolic pathway is established in a subset of HCCs for tumor growth. J Mol Cell Biol. 2013; 5:404–15.
5. Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, van Bilsen M, Shiri-Sverdlov R, Hofker MH. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008; 48:474–486.
6. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, Chait A, Yeh MM, Quinn LS, Ioannou GN. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. 2013; 57:81–92.
7. Ichimura M, Kawase M, Masuzumi M, Sakaki M, Nagata Y, Tanaka K, Suruga K, Tamaru S, Kato S, Tsuneyama K, Omagari K. High-fat and high-cholesterol diet rapidly induces non-alcoholic steatohepatitis with advanced fibrosis in Sprague-Dawley rats. Hepatol Res. 2015; 45:458–69.
8. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. 2016; 76:2063–2070.
9. Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007; 46:1081–90.
10. Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003; 5:781–92.
11. Attia A, Ragheb A, Sylwestrowicz T, Shoker A. Attenuation of high cholesterol-induced oxidative stress in rabbit liver by thymoquinone. Eur J Gastroenterol Hepatol. 2010; 22:826–834.
12. Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am J Pathol. 2006; 168:1107–1118.
13. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010; 5:e11765.
14. Huang W, Bansode RR, Xie Y, Rowland L, Mehta M, Davidson NO, Mehta KD. Disruption of the murine protein kinase Cbeta gene promotes gallstone formation and alters biliary lipid and hepatic cholesterol metabolism. J Biol Chem. 2011; 286:22795–805.
15. Huang W, Mehta KD. Modulation of hepatic PKCβ in metabolic adaptation to a lithogenic diet. Cell Mol Gastroenter Hepatol. 2015; 1:395–405.
16. Mehta D, Mehta KD. PKCβ: expanding role in hepatic adaptation of cholesterol homeostasis to dietary fat/cholesterol. Am J Physiol- Gastroint Liver Physiol. 2017; In Press.
17. Sakai K, Nagahara H, Abe K, Obata H. Loss of heterozygosity on chromosome 16 in hepatocellular carcinoma. J Gastroenterol Hepatol. 1992; 7:288–292.
18. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004; 23:2838–2849.
19. Saba NS, Levy LS. Protein kinase C-beta inhibition induces apoptosis and inhibits cell cycle progression in AIDS-related Non-Hodgkin lymphoma cells. J Investig Med. 2012; 60:29–38.
20. Suzuma K, Takahara N, Suzuma I, Isshiki K, Ueki K, Leitges M, Aiello LP, King GL. Characterization of protein kinase C beta isoform’s action on retinoblastoma protein phosphorylation, vascular endothelial growth factor-induced endothelial cell proliferation, and retinal neovascularization. Proc Natl Acad Sci USA. 2002; 99:721–26.
21. Ashida K, Kishimoto Y, Nakamoto K, Wada K, Shiota G, Hirooka Y, Kamisaki Y, Itoh T, Kawasaki H. Loss of heterozygosity of the retinoblastoma gene in liver cirrhosis accompanying hepatocellular carcinoma. J Cancer Res Clin Oncol. 1997; 123:489–95.
22. Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006; 25:3778–86.
23. Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006; 6:674–87.
24. Murakami Y, Hayashi K, Hirohashi S, Sekiya T. Aberrations of the tumor suppressor p53 and retinoblastoma genes in human hepatocellular carcinomas. Cancer Res. 1991; 51:5520–25.
25. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990; 343:425–30.
26. Burkhart DL, Ngai LK, Roake CM, Viatour P, Thangavel C, Ho VM, Knudsen ES, Sage J. Regulation of RB transcription in vivo by RB family members. Mol Cell Biol. 2010; 30:1729–1745.
27. Rubin SM. Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci. 2013; 38:12–19.
28. Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ, Babcock GF, Lentsch AB, Fukasawa K, Knudsen ES. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res. 2005; 65:4568–4577.
29. Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016; 85:685–713.
30. Patergnani S, Marchi S, Rimessi A, Bonora M, Giorgi C, Mehta KD, Pinton P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy. 2013; 9:1367–85.
31. Jiang H, Martin V, Gomez-Manzano C, Johnson DG, Alonso M, White E, McDonnell JJ, Shinojima N, Fueyo J. The RB-E2F1 pathway regulates autophagy. Cancer Res. 2010; 70:7882–7893.
32. Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997; 276:60–66.
33. Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015; 160:489–502.
34. Dowling CM, Phelan J, Callender JA, Cathcart MC, Mehigan B, McCormick P, Dalton T, Coffey JC, Newton AC, O’Sullivan J, Kiely PA. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget. 2016; 7:20919–33. https://doi.org/10.18632/oncotarget.8062.
35. Huynh H. Overexpression of tumour suppressor retinoblastoma 2 protein (pRb2/p130) in hepatocellular carcinoma. Carcinogenesis. 2004; 25:1485–1494.
36. Stiewe T, Pützer BM. Role of p73 in malignancy: tumor suppressor or oncogene? Cell Death Differ. 2002; 9:237–45.
37. Tannapfel A, Wasner M, Krause K, Geissler F, Katalinic A, Hauss J, Mössner J, Engeland K, Wittekind C. Expression of p73 and its relation to histopathology and prognosis in hepatocellular carcinoma. J Natl Cancer Inst. 1999; 91:1154–58.
38. Scivittaro V, Ganz MB, Weiss MF. AGEs induce oxidative stress and activate protein kinase C-beta (II) in neonatal mesangial cells. Am J Physiol Renal Physiol. 2000; 278:F676–683.
39. Abrams ST, Brown BR, Zuzel M, Slupsky JR. Vascular endothelial growth factor stimulates protein kinase CbetaII expression in chronic lymphocytic leukemia cells. Blood. 2010; 115:4447–54.
40. Obeid LM, Blobe GC, Karolak LA, Hannun YA. Cloning and characterization of the major promoter of the human protein kinase C beta gene. Regulation by phorbol esters. J Biol Chem. 1992; 267:20804–10.
41. Fransvea E, Paradiso A, Antonaci S, Giannelli G. HCC heterogeneity: Molecular pathogenesis and clinical implications. Cell Oncol. 2009; 31:227–233.
42. Naetar N, Hutter S, Dorner D, Dechat T, Korbei B, Gotzmann J, Beug H, Foisner R. LAP2{alpha}-binding protein LINT-25 is a novel chromatin-associated protein involved in cell cycle exit. J Cell Sci. 2007; 120:737–747.
43. Barbie DA, Conlan LA, Kennedy BK. Nuclear tumor suppressors in space and time. Trends Cell Biol. 2005; 15:378–85.
44. Higashitsuji H, Itoh K, Nagao T, Dawson S, Nonoguchi K, Kido T, Mayer RJ, Arii S, Fujita J. Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat Med. 2000; 6:96–99.
45. Li J, Tsai MD. Novel insights into the INK4-CDK4/6-Rb pathway: counter action of gankyrin against INK4 protein regulates the CDK4-mediated phosphorylation of Rb. Biochemistry. 2002; 41:3977–3983.
46. Ozaki T, Saijo M, Murakami K, Enomoto H, Taya Y, Sakiyama S. Complex formation between lamin A and the retinoblastoma gene product: identification of the domain on lamin A required for its interaction. Oncogene. 1994; 9:2649–2653.
47. Mittnacht S, Hinds PW, Dowdy SF, Weinberg RA. Modulation of retinoblastoma protein activity during the cell cycle. Cold Spring Harb Symp Quant Biol. 1991; 56:197–209.
48. Markiewicz E, Dechat T, Foisner R, Quinlan RA, Hutchison CJ. Lamin A/C binding protein LAP2alpha is required for nuclear anchorage of retinoblastoma protein. Mol Biol Cell. 2002; 13:4401–13.
49. Nitta RT, Smith CL, Kennedy BK. Evidence that proteasome-dependent degradation of the retinoblastoma protein in cells lacking A-type lamins occurs independently of gankyrin and MDM2. PLoS One. 2007; 2:e963.
50. Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998; 391:859–865.
51. Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT, Tang X, Bae S, Srivastava A, Senapati S, Koivisto C, Martin CK, Cuitino MC, et al. E2f8 mediates tumor suppression in postnatal liver development. J Clin Invest. 2016; 126:2955–69.
52. Wang B, Hsu S, Frankel W, Ghoshal K, Jacob ST. Stat3-mediated activation of miR-23a suppresses gluconeogenesis in hepatocellular carcinoma by downregulating G6PC and PGC-1a. Hepatology. 2012; 56:186–197.