
INTRODUCTION
Intracellular growth-promoting signals are transduced through the activation of the small G protein RAS, which activates RAF and then activates mitogen-activated protein kinase (MAPK) kinase (MEK), leading to the phosphorylation and activation of MAPK (originally called extracellular signal-regulated kinase [ERK]). MEK1 and MEK2 show a unique specificity for ERK1/2 and the ERKs have wide spectrum substrates, many of which play a role in promoting cell proliferation and survival [1]. Small-molecule inhibitors of one or more components of the RAS-MAPK signaling pathway have potential therapeutic properties for some cancers, particularly those that have a dysregulated MAPK pathway (for example, B-RAF-mutated melanoma and K-RAS/B-RAF-mutated colorectal cancer) [2]. MEK inhibitors inhibit MEK1 and/or MEK2, homologous dual specificity kinases that share ERK as their only known catalytic substrate [3]. PD098059 was the first MEK inhibitor to be described [4] and several other MEK inhibitors have since been developed in clinical use for various cancers [5]. The Food and Drug Administration (FDA) of the United States recently approved one of these agents, trametinib for the treatment of patients with unrespectable or metastatic melanomas harboring B-RAFV600E or B-RAFV600K mutations, as detected by an FDA-approved test (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm481951.pdf). AZD6244 (Selumetinib) is a non-ATP competitive inhibitor and is highly specific for MEK1/2. Multiple phase 2 studies of this drug have been conducted in patients with a wide range of cancers [5].
Although recent studies have demonstrated impressive antitumor activities of MEK inhibitors, many tumors show intrinsic resistance of these agents. Reliable biomarkers of susceptibility or resistance to MEK inhibitors are thus needed to allow tailoring of individualized treatments with these compounds and to reduce the risk of unnecessary drug toxicities. It has been suggested that activating mutations in the B-RAF gene represent the most important predictive biomarker for sensitivity to MEK inhibitors [6, 7]. However, early clinical data have revealed that B-RAF mutant tumors were not uniformly responsive to MEK inhibitors although tumors with these mutations seemed to be more sensitive. [8, 9] Furthermore, the duration of the responses to MEK inhibitors is reported to be relatively short (median ≤ 5 months) in melanoma [10]. Other biomarkers such as RAS mutations or ERK activation correlate poorly with the sensitivity of cells to MEK inhibitors [11, 12].
The NCI-60 is a set of 60 human cancer cell lines derived from diverse tissues, including brain, blood and bone marrow, breast, colon, kidney, lung, ovary, prostate, and skin. These cell lines have now been subjected to a battery of experiments including extensive pharmacological characterization analyses via treatment with over 100,000 chemical compounds, chromosome karyotyping, and gene expression analysis using various DNA microarray platforms [13]. The current approaches to studying the genetic basis of cancer are exploring substantial components of the expressed genome rather than focusing on individual genes [14]. Researchers can now use standards-based repositories such as the Stanford Microarray Database and the Gene Expression Omnibus, which have developed to store and disseminate the results of microarray experiments [15].
The proto-oncogene β-catenin has been linked to the pathogenesis of hepatocellular carcinoma, colorectal carcinoma, lung cancer, malignant breast tumors, and leukemia through the Wnt-signal transduction pathway [16] Immunoglobulin Transcription Factor-2 (ITF-2) is one of several specific target genes of T-cell factor-dependent transcription upon translocation of β-catenin to the nucleus [17].
In our current study we aimed to screen for novel response predictive markers for the AZD6244, MEK inhibitor by analyzing published microarray data from AZD6244 sensitivity profiling of the NCI-60 cell lines and also aimed to validate the candidate markers in both primary and acquired resistance models.
RESULTS
Screening of cancer cell lines for their response to a MEK inhibitor, AZD6244
We performed assays for the growth response of a panel of NCI-60 cell lines to 10 μM or less AZD6244 by proliferation assay. On the basis of the response to AZD6244, we selected 6 sensitive (IC50 ≤ 0.5 μM) and 6 resistant (IC50 > 5 μM) cell lines (Supplementary Figure 1). The IC50 values for the 6 resistant cell lines ranged from 6.032 to 125.9 μM, while the 6 sensitive cell lines showed IC50 values of less than 0.5 μM (range, 0.02923 to 0.4870 μM). One resistant and 5 sensitive cell lines harbored a B-RAF mutation; and 1 resistant and 1 sensitive line contained a K-RAS mutation. One resistant cell line had an N-RAS mutation (Table 1).
Table 1: Characteristics of 12 cell lines: 6 cell lines were sensitive to a MEK inhibitor, AZD6244, and 6 cell lines were resistant to the agent
Cell line |
Origin |
Gene mutation |
IC50 for AZD6244 (μM) |
|||
|---|---|---|---|---|---|---|
B-RAF |
K-RAS |
N-RAS |
||||
Resistant |
SF-295 |
Glioblastoma |
- |
- |
- |
7.383 |
SNB-19 |
Glioblastoma |
- |
- |
- |
15.13 |
|
LOX-IMVI |
Melanoma |
V600E |
- |
- |
6.032 |
|
K-562 |
CML |
- |
- |
- |
82.07 |
|
CCRF-CEM |
ALL |
- |
G12D |
wt |
125.9 |
|
HL-60 |
APL |
- |
- |
Q61L |
20.95 |
|
Sensitive |
UACC-62 |
Melanoma |
V600E |
- |
- |
0.4870 |
UACC-257 |
Melanoma |
V600E |
- |
- |
0.4650 |
|
M14 |
Melanoma |
V600E |
- |
- |
0.06103 |
|
SK-MEL-28 |
Melanoma |
V600E |
- |
- |
0.2098 |
|
COLO-205 |
Colon cancer |
V600E |
- |
- |
0.02923 |
|
SW-620 |
Colon cancer |
- |
G12V |
- |
0.05538 |
|
CML, chronic myeloid leukemia; ALL, acute lymphoblastic leukemia; APL, acute promyelocytic leukemia.
Selection of ITF-2 as a potential AZD6244 resistance marker from public microarray data
Following public microarray data acquisition and analysis, a total of 62 differentially expressed genes (DEGs) were selected from 12 cell lines (6 AZD6244 resistant; CCRF-CEM, HL-60, K-562, LOX-IMVI, SF-295 and SNB-19 vs. 6 AZD6244 sensitive; COLO-205, SW-620, M14, SK-MEL-28, UACC-257 and UACC-62) (Supplementary Figure 2). Of these selected genes, 18 were up-regulated and 44 were down-regulated in AZD6244 resistant cell lines (Supplementary Table 1). Table 2 lists the top 10 AZD6244 resistant signature genes and we confirmed the expression using quantitative real-time PCR for these 10 genes (data not shown). We selected the Immunoglobulin transcription factor-2 (ITF-2) gene because it showed the strongest upregulation in resistant lines and previous studies have suggested that this gene is a downstream target of the Wnt/β-catenin/T-cell factor pathway [18].
Table 2: Top 10 resistant signature genes for AZD6244
Symbol |
Description |
Entrez ID |
Fold-change* |
|---|---|---|---|
ITF-2 |
Immunoglobulin transcription factor 2 |
6925 |
5.91 |
ZNF22 |
zinc finger protein 22 |
7570 |
5.66 |
TGFB1 |
transforming growth factor, beta 1 |
7040 |
5.62 |
ZEB1 |
zinc finger E-box binding homeobox 1 |
6935 |
5.58 |
ASF1A |
ASF1 anti-silencing function 1 homolog A |
25842 |
3.58 |
BCAT1 |
branched chain amino-acid transaminase 1, cytosolic |
586 |
3.57 |
RANBP6 |
RAN binding protein 6 |
26953 |
2.78 |
SACS |
spastic ataxia of Charlevoix-Saguenay |
26278 |
2.63 |
MAP3K4 |
mitogen-activated protein kinase kinasekinase 4 |
4216 |
2.45 |
NAA15 |
N(alpha)-acetyltransferase 15, NatA auxiliary subunit |
80155 |
2.39 |
*Fold-change of mean expression intensities in resistant group/sensitive group.
Increased expression of ITF-2 in cell lines having primary or acquired resistance to AZD6244
The mRNA levels of ITF-2 in AZD6244 resistant cancer cell lines were found to be significantly higher than in cell lines sensitive to this inhibitor (Figure 1A). Moreover, western blot using an anti-ITF-2 monoclonal antibody (mAb) showed that the ITF-2 protein level was elevated in 5 of 6 AZD6244 resistant cell lines but in only one sensitive cell line. The p-ERK levels were not significantly associated with the sensitivity to AZD6244 (Figure 1B).
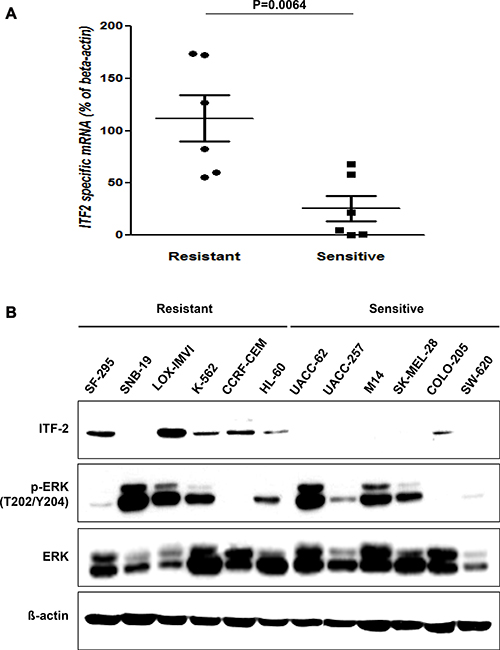
Figure 1: Basal levels of ITF-2 in AZD6244 resistant and sensitive cell lines. (A) Relative expression of endogenous ITF-2 mRNA in each indicated cell line based on the expression in SW620. Each sample was tested in triplicate, and gene expression levels were normalized to those of β-actin mRNA. The mRNA levels of ITF-2 in AZD6244 resistant cell lines were significantly higher than those in sensitive cell lines. Statistical significances are calculated by the Mann-Whitney U-test. (B) Equal amounts of total cellular proteins were subjected to western blot analysis for ITF-2, and phospho-specific and total ERK1/2. ITF-2 was elevated in AZD6244 resistant cell lines except SNB-19, but the p-ERK levels were not significantly different according to the sensitivity to AZD6244. Beta-actin was included as a loading control.
M14/AZD-3 is 33.4-fold more resistant to AZD6244 compared to parent M14 (Figure 2A). Direct sequencing demonstrated that both M14 and M14/AZD-3 had B-RAFV600E mutation, but did not have mutations in MEK1 exon 3 and exon 6 which encompass the validated resistant alleles (Supplementary Figure 3) [51]. M14/AZD-3 needed higher concentration of AZD6244 to decrease the p-ERK level and did not induce poly (ADP-ribose) polymerase (PARP) cleavages following treatment of AZD6244 (Figure 2B). Western blot demonstrated that the ITF-2 protein level was elevated in M14/AZD-3 as well as LOX-IMVI, while it was not in parent M14 cell line (Figure 2C). ITF-2 protein overexpression in both primary (LOX-IMVI) and acquired (M14/AZD-3) resistant cell lines suggested the possible role of ITF-2 in resistance to AZD6244. In contrast, the levels of RAS/RAF/MEK/ERK signaling proteins were not significantly changed in resistant cell lines (M14/AZD-1, M14/AZD-3, and M14/AZD-4) compared to M14 (Supplementary Figure 4).
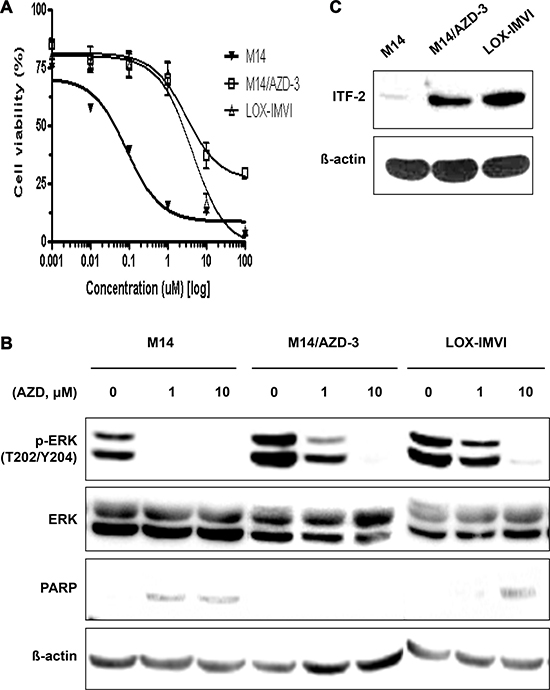
Figure 2: Establishment of an acquired AZD6244 resistant cell line, M14/AZD-3 and overexpression of ITF-2 in M14/AZD-3. (A) M14/AZD-3 cell line was established by long-term treatment of an AZD6244 sensitive melanoma cell line, M14, with increasing dose of AZD6244. (B) With 1 μM of AZD6244, suppression of phosphorylated ERK (p-ERK) and cleavage of poly (ADP-ribose) polymerase (PARP) did occur in M14, but not in M13/AZD-3 (acquired AZD6244 resistant cell line) and LOX-IMVI (primary AZD6244 resistant cell line). (C) Overexpression of ITF-2 protein in M14/AZD-3 and LOX-IMVI.
Suppression of ITF-2 by siRNA in primary and acquired resistant cell lines
To exam whether ITF-2 overexpression is associated with the resistance to AZD6244, M14/AZD-3 and LOX-IMVI cells were transfected with either 50 nM of control siRNAs or siRNA against ITF-2 (siITF-2) (Figure 3). Transfection of siITF-2 into M14/AZD-3 and LOX-IMVI effectively reduced ITF-2 protein expression, but transfection alone did not induce cleavage of PARP (Figure 3A) or did not inhibit cell proliferation (Figure 3B).
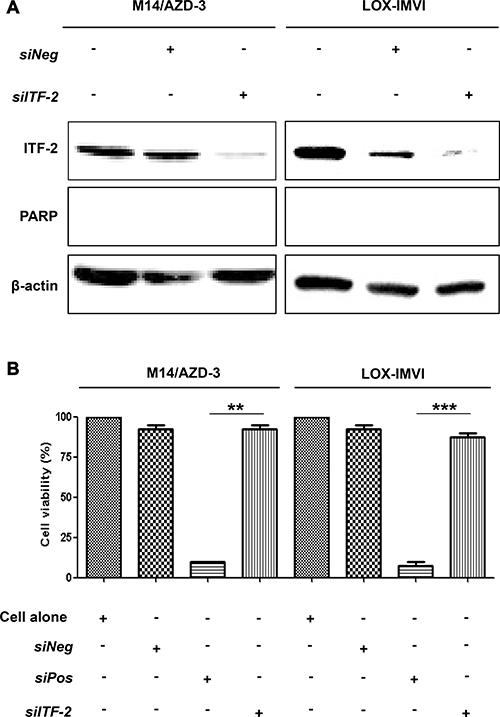
Figure 3: Suppression of ITF-2 by siRNA in primary and acquired AZD6244 resistant cell lines. M14/AZD-3 and LOX-IMVI cells were transfected with ITF-2 siRNA (siITF-2), non-targeting control siRNA (siNeg) and cell death control siRNA (siPos). All siRNAs were used as 50 nM concentrations for 24 h. (A) Endogenous ITF-2 mRNA levels decreased by the transfection of siITF-2, but transfection alone did not induce cleavage of poly (ADP-ribose) polymerase (PARP). (B) The transfection of siITF-2 did not influence on the cell viability in resistant cell lines. **P = 0.001, ***P < 0.001.
Although the transfection of siITF-2 alone did not induce the death of AZD6244 resistant cells, it might alter response of the resistant cells to AZD6244. To test this hypothesis, we treated AZD6244 resistant cells with 10 μM of AZD6244 for 24 h following transfection of siITF-2. The Live-Dead cell staining showed that ITF-2 suppression by transfection of siITF-2 dramatically increased the proportion of dead cells with treatment of AZD6244 (Figure 4A). Western blot analysis also demonstrated increased cleavage of procaspase-3 and PARP with treatment of AZD6244 following transfection of siITF-2 (Figure 4B).
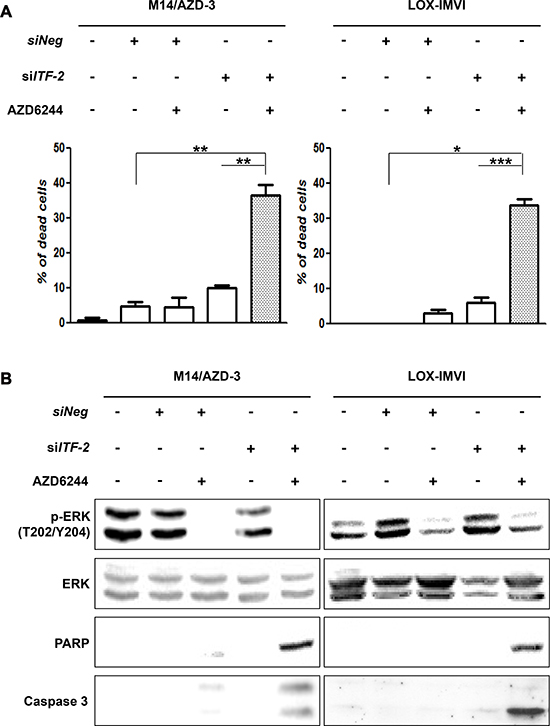
Figure 4: Combination of AZD6244 and siRNA against ITF-2. The cytotoxic and apoptotic effects were assessed after treatment of AZD6244 resistant cells with 10 μM of AZD6244 for 24 h following transfection of siRNA against ITF-2 (siITF-2). (A) Live-Dead cell staining showed dramatically increased proportion of dead cells by combination of AZD6244 and siITF-2. *P = 0.002, **P = 0.001, ***P < 0.001. (B) Western blot analysis also demonstrated increased cleavage of procaspase-3 and poly (ADP-ribose) polymerase (PARP) with treatment of AZD6244 following transfection of siITF-2 (Figure 4B).
This phenomenon could also be found in other primary AZD6244 resistant cell line, human glioma-derived SF-295. The cell line had increased ITF-2 level, which was effectively suppressed by two different siRNAs against ITF-2 (siITF-2 #1 and siITF-2 #2) (Supplementary Figure 5A). The apoptotic and cytotoxic effects of AZD6244 on SF-295 cells were enhanced by transfection of both siRNAs as evidenced by inhibition of phosphorylated ERK (p-ERK) and increased cleavage of PARP in western blot (Supplementary Figure 5B) and increase of the proportion of dead cells in Live-Dead cell staining (Supplementary Figure 5C).
These results suggest that the ITF-2 suppression could enhance the susceptibility to AZD6244 in melanoma cells with primary or acquired resistance.
Activation of Wnt signaling pathway in primary and acquired resistant cell lines
Because ITF-2 is known as a downstream target of Wnt signaling pathway, we examined the pathway in AZD6244 sensitive cell line (M14) and resistant cell lines (M14/AZD-3 and LOX-IMVI). Translocation of dephosphorylated β-catenin to the nucleus and accumulation of phosphorylated Ser9 of GSK3β in cytosol fraction were found in AZD6244 resistant cell lines (M14/AZD-3 and LOX-IMVI), but not in AZD6244 sensitive cell line, M14 (Figure 5A). The same findings were observed in SF-295 (Supplementary Figure 6). These findings suggest that activation of Wnt signaling pathway due to inactivation of GSK3β leads to increased transcription of ITF-2 in AZD6244 resistant cells. Because GSK3β is known to be phosphorylated at Ser9 by the most downstream kinase of the classical MAPK cascade, called ribosome S6 kinase (RSK), [19] we assayed total and phosphorylated p90RSK (Tyr573). Interestingly, we found that p90RSK was downregulated in AZD6244 resistant cell lines (Figure 5B).
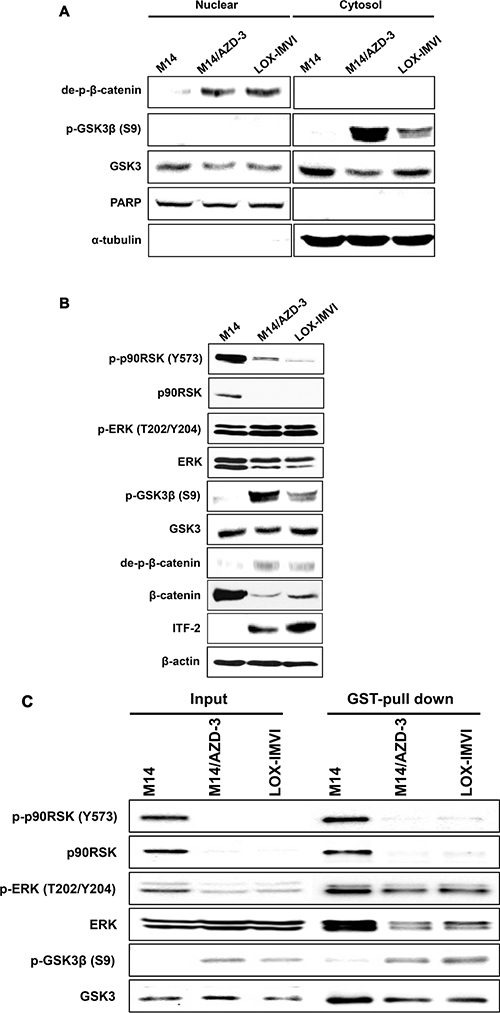
Figure 5: ITF-2 induction by activation of Wnt signaling pathway. (A) Western blot analysis for Wnt/β-catenin signaling proteins demonstrated that dephosphorylated β-catenin was translocated to the nucleus and phosphorylated Ser9 of GSK3β was accumulated in cytosol fraction in AZD6244 resistant cell lines (M14/AZD-3 and LOX-IMVI). (B) Western blot analysis showed that the p90RSK was downregulated in AZD6244 resistant cell lines. (C) GST pull-down assay using pGEX-GSK3β and Glutathione-Sepharose 4B (GST) beads demonstrated the direct interaction of p-ERK and GSK3β and phosphorylation of GSK3β at Ser9 in AZD6244 resistant cell lines (M14/AZD-3 and LOX-IMVI).
We assumed that p-ERK might directly interact with GSK3β and we performed GST pull-down assay using pGEX-GSK3β and Glutathione-Sepharose 4B (GST) beads. GSK3β fused GST bead was interacted with phosphorylated p90RSK (Tyr573) and ERK (T202/Y204) in M14 but the interaction did not activate phosphorylation of GSK3β (S9). In contrast, the bead was interacted with phosphorylated ERK (T202/Y204) and phosphorylation of GSK3β (S9) was induced in both resistant cell lines without p90RSK (Figure 5C). The findings demonstrated that p-ERK was directly bound to GSK3β, which was phosphorylated at Ser9, in AZD6244 resistant cell lines (M14/AZD-3 and LOX-IMVI).
DISCUSSION
Drug resistance, either primary or acquired, is one of the most challenging aspects of anticancer agents. A critical component of cancer drug development is the identification of biomarkers that can be used to predict resistance to these agents. Biomarker identification is also important for minimizing patient exposure to potentially toxic agents for little clinical benefit and to develop rational approaches for overcoming drug resistance. Many studies have been under taken to gain an improved understanding of the mechanisms of cancer drug resistance and have identified factors that seemed to be associated with resistance to MEK inhibitors, [20] including MEK1 mutations, [21] over-expression of microphthalmia-associated transcription factor (MITF), [22] aberrant expression of musculoaponeurotic fibrosarcoma (MAF), [23] concurrent activation of the phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway [24, 25] or the signal transducer and activator of transcription 3 (STAT3) pathway, [26] and an ‘invasive phenotype’ of transcriptional pathway signatures [27]. In our current study, we obtained evidence that the over-expression of ITF-2 might be a novel biomarker of cellular resistance to MEK inhibitors because the mRNA and protein levels of this gene in AZD6244 resistant cell lines were significantly elevated and suppression of ITF-2 by siRNA enhanced the susceptibility of AZD6244 resistant cells to this inhibitor. These results were observed with both primary AZD6244 resistance cell line (LOX-IMVI) and acquired resistance cell line (M14/AZD), which was established as a model for acquired resistance using a sensitive cell line harboring B-RAF mutation. Our study results might provide a clue to prevent or overcome the development of acquired resistance to MEK inhibitors, which is a common and serious problem in real clinical settings.
The ITF-2 gene (also known as TCF4, E2-2, ME2, or SEF2) is located on human chromosome 18q21, consists of 20 exons (exons 1 and 20 are noncoding), and spans 360 kb. ITF-2 belongs to the class A subfamily of basic helix-loop-helix (bHLH) transcriptional regulators [28, 29]. This gene is different from T-cell transcription factor 4 on human chromosome 10q25.3, which was previously termed TCF4, but is now designated as TCF7L2. The ITF-2 gene encodes two splice variants, ITF-2A and ITF-2B. ITF-2A is a truncated splice variant with a low transcriptional potency. ITF-2B produces a larger protein, which is ubiquitously expressed and dimerizes with other classes of bHLH proteins to regulate tissue-specific gene expression through E-box sites in various cell types including myocytes, osteoblasts, B and T lymphocytes, and neuronal cells [30]. Previous studies have demonstrated that ITF-2 is a downstream target of the Wnt/β-catenin/ TCF7L2 pathway, which is frequently disrupted in human cancers [17, 18, 31]. It is also well known that nuclear transcriptional activity of β-catenin is enhanced upon sustained oncogenic stimulation by the RAS-MAPK signaling pathway [32]. Like c-MYC and cyclin D1, ITF-2 functions as an oncogene when deregulated in human colon cancer or ovarian cancer, [18, 31] although other studies have reported that the inactivation of ITF-2 may play a role in early gastric carcinoma progression or in the colonic adenoma to carcinoma transition [33, 34]. In another earlier study aiming to identify predictive biomarkers of the response to AZD6244 in colorectal cancer, members of the Wnt signaling pathway were found to be highly over-expressed in response to AZD6244 and shRNA knockdown experiments indicated the functional involvement of these factors in mediating resistance to this inhibitor [35]. In our study, we observed increase of inactive form of GSK3β and translocation of β-catenin to nucleus in AZD6244 resistant cell lines (M14/AZD3 and LOX-IMVI), leading to increased transcription of ITF-2. It has been known that GSK3β is phosphorylated at Ser9 by stress-activated p90RSK [36]. In our study, however, p90RSK was downregulated in AZD6244 resistant cell lines and GST pull-down assay showed that p-ERK was bound to GSK3β, which was phosphorylated at Ser9. Thus, it is reasonably assumed that p-ERK directly interacts with GSK3β leading to phosphorylation at Ser9, and activation of Wnt pathway results in the transcription of ITF-2 gene in AZD6244 resistant cells (Figure 6). Downstream signals of ITF-2 related to resistance to MEK inhibitor remain to be studied.
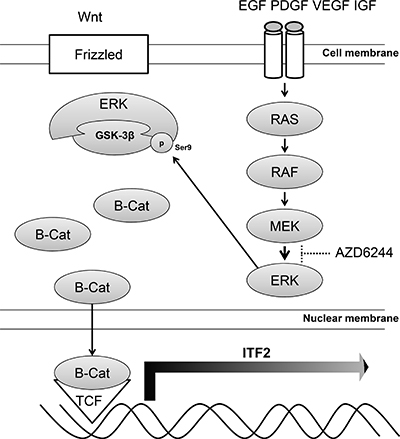
Figure 6: Model of the ITF-2 induction via ERK/GSK3β/β-catenin. In AZD6244 resistant cells, p-ERK directly interacts with GSK3β, leading to phosphorylation at Ser9 and dephosphorylation of β-catenin. The dephosphorylated β-catenin is translocated to the nucleus and the association of β-catenin and T-cell factor results in the transcription of ITF-2 gene, one of Wnt target genes.
MEK inhibitors have generally shown limited effectiveness as monotherapeutic agents and combination regimens are a possible treatment approach to overcome MEK inhibitor resistance. Indeed, simultaneously inhibition of the MAPK and PI3K/AKT/mTOR pathways has been proposed for melanoma [37]. Moreover, in a K-RAS mutant lung cancer mouse model, a combination of a MEK inhibitor at doses below the maximum tolerated dose with a PI3K/AKT/mTOR inhibitor was found to suppress the growth of the tumors [38]. In another previous in vitro study using melanoma cell lines, a high degree of cross-resistance to a B-RAF inhibitor (vemurafenib) and a MEK inhibitor (AZD6244) was reported, and genetic or pharmacological inhibition of the PI3K/AKT/mTOR pathway was found to partially or completely reverse this resistance [39]. Furthermore, a combination of AZD6244 and an BCL2 inhibitor (ABT-263) was shown to be synergistic in causing a cell death response in colon cancer or melanoma cells harboring B-RAF or RAS mutations, and also to delay the onset of acquired resistance and in some cases overcome acquired resistance to AZD6244 [40]. The MAPK pathway is activated by BCR-ABL in nilotinib-resistant chronic myeloid leukemia (CML) cells and nilotinib acts synergistically with MEK inhibitors to kill drug-resistant CML cells both in vitro and in vivo [41]. The available evidence suggests that cytotoxic chemotherapeutic agents can activate the MAPK pathway through diverse processes and that MEK inhibitors may effectively blocked the activation [42]. It has also been demonstrated that there are synergistic or additive effects of MEK inhibitors in combination with various cytotoxic chemotherapeutic agents. In vitro studies using leukemia cell lines have reported that MEK inhibitors enhance the cytotoxic effects of cytarabine or doxorubicin [43, 44]. Similar enhanced effects of MEK inhibitors in combination with cytotoxic drugs have been observed in xenograft tumor models of solid cancers including melanoma, [45] and biliary, [46] colon, [47, 48] lung, [48] pancreatic [48] or hepatocellular carcinoma [49]. When MEK inhibitors are combined with traditional chemotherapeutic agents, drug scheduling may have a critical disease management role because MEK inhibitors cause cell cycle arrest whereas many standard cytotoxic agents kill actively dividing cancer cells [46, 48]. In our current study, a combination of AZD6244 and ITF-2 siRNA induced cell death in AZD6244 resistant cell lines, although the magnitude of enhanced susceptibility to AZD6244 was not adequate for clinical application, probably due to the only partial suppression of ITF-2 expression by siRNA. Wnt/ β-catenin pathway inhibitors might suppress ITF-2 more effectively and may provide a novel combination approach for MEK inhibitors.
In summary, we have identified the ITF-2 gene as a novel resistance biomarker for an MEK inhibitor, AZD6244. The mRNA and protein levels of ITF-2 were found to be significantly elevated in cell lines with primary or acquired resistance to AZD6244 and a targeted silencing of ITF-2 by siRNA significantly enhanced the susceptibility of primary resistant cells (LOX-IMVI) and acquired resistant cells (M14/AZD-3) to AZD6244. Activation of Wnt signaling pathway through direct interaction of p-ERK and GSK3β seems to be a mechanism for increase of ITF-2.
MATERIALS AND METHODS
Cancer cell lines
The NCI-60 cell lines were purchased from the Jackson Laboratory (Bar Harbor, ME) and were cultured at 37°C in 5% CO2 in RPMI-1640 medium containing 5% or 10% (v/v) fetal bovine serum and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA). Information regarding the mutation status of the B-RAF, K-RAS, and N-RAS genes of each cell line was obtained from the Wellcome Trust Sanger institute website (http://cancer.sanger.ac.uk/cosmic/sample/overview?id=1295340, http://cancer.sanger.ac.uk/cosmic/sample/overview?id=1238117).
Establishment of an AZD6244 resistant melanoma cell line
Human melanoma M14 cell line was sensitive to AZD6244 in vitro. M14 cells were continuously exposed to gradually increasing concentration of AZD6244 up to 500 nM. An AZD6244 resistant subclone was isolated using limiting dilution method and AZD6244 resistant cell lines (M14/AZD-1, M14/AZD-3, and M14/AZD-4) could be established. In most experiments, M14/AZD-3 was used.
Cell proliferation assay
Cell viability was assessed by the luminescent-based CellTiter-Glo system (Promega, Madison, WI) and Cell counting kit -SK (Dojindo Molecular Technologies, Inc. Rockville, MD) in accordance with the manufacturer’s instructions. Briefly, cells were plated at 1,000–3,000 per well in a 96-well opaque plate and were incubated in complete growth medium. Cells were treated with various concentrations of MEK1/2 inhibitor (AZD6244), which was synthesized by Chemizon (Seongnam, Korea) and prepared as a 50 mmol/L stock solution in DMSO. After 48 h, cell viability was determined by measuring luminescent signals with a VICTOR™ X Light luminescence plate reader (PerkinElmer, Waltham, MA). The concentration of AZD6244 required for 50% growth inhibition was scored as the half maximal inhibitory concentration (IC50) values.
Public microarray data acquisition and data analysis
To identify candidate genes associated with resistant or sensitive responses to AZD6244 genome-wide gene expression profiling data from Affymetrix HG-U133A chips were downloaded from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo). The chip data were analyzed with RNA-eXpress (www.rnaexpress.org) using its default analysis settings and a quintile normalization method. Analyses of preprocessed data were performed using BRB-ArrayTools (linus.nci.nih.gov/BRB-ArrayTools.html). A two-sample t-test was performed between sensitive and resistant groups at P < 0.005 to select for DEGs between the two groups.
Genomic DNA PCR and direct sequencing
Genomic DNA was purified using the QIAamp DNA mini kit (Qiagen, Valencia, CA). All PCR amplifications were performed with HotStar HiFidelity Polymerase kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. B-RAF codon V600 was PCR amplified using following primers: 5′GGCCAAAAATTTAATCAGTGGA3′ and 5′ AGCCTCAATTCTTACCATCC3′. 50 μl PCR reactions were 35 cycles at 95°C for 30 sec, 56°C for 30 sec, 72°C 45 sec. The MEK1 genetic mutation was accomplished using published methods [51]. The sequencing primers were the same as the PCR forward primers and the sequencing reaction was performed using standard-seq single (Macrogen Inc. Seoul. Kr).
Quantitative real-time RT-PCR
Total RNAs were extracted using TRIzol® Reagent (Invitrogen) and converted into cDNA using a QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA). The transcript levels were quantified via the SYBR Green real time PCR method using LightCycler® 480 SYBR Green I Master Mix (Roche Applied Science) on a Roche-480 system using primer sets (Supplementary Table 2). After an initial denaturation step at 95°C for 5 min, amplification occurred over 45 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 30 s. Each sample was tested in triplicate, and gene expression levels were normalized to those of beta-actin.
Inhibition of gene expression by siRNA transfection
The siRNA was used for targeted silencing of ITF-2. Because off-target activity of siRNA may complicate the interpretation of phenotypic effects in gene silencing, two kinds of ITF-2 siRNA’s were used for the experiments: siITF-2 #1 (Integrated DNA technologies, Inc., Coralville, IA) and siITF-2 #2 (Santa Cruz Biotechnology, Inc., Dallas, TX). The level of off-target effects was assessed by western blot analysis using an anti-ITF-2 mAb in M14/AZD-3 and both ITF-2 siRNA’s significantly reduced ITF-2 expression levels (Supplementary Figure 7). A non-targeting control siRNA (negative control) and a cell death control siRNA (positive control) were purchased from Qiagen. Cells were seeded at 2 × 105 per well in a 6-well plate and were transfected with 50 μM of siRNA using jetPRIME® in accordance with the manufacturer’s instructions (Polyplus, Illkirch, France). Cells were then incubated at 37°C for 24 h.
Western blot analysis
Cells were lysed with lysis buffer (cell signaling technology, Beverly, MA) and protein samples (50 μg) were separated by SDS-PAGE and were blotted onto polyvinylidene difluoride membranes (Millipore, Bedford, MA). After blocking with 5% (w/v) non-fat dry milk powder for 1 h, the membranes were incubated with primary antibodies overnight at 4°C, then with secondary antibody conjugated with horseradish peroxidase (Enzo Life Sciences, Farmingdale, NY). Specific antigen-antibody complexes were detected by enhanced chemiluminescence using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Waltham, MA). Specific antibodies were as follows: ITF-2 (Abcam, Cambridge, MA), phospho-beta-catenin (p-beta-catenin), beta-catenin, phospho-ERK (p-ERK), ERK, phospho-GSK3 beta (Y216 and S9), GSK-3 alpha and beta, phospho-90RSK (Y573), 90RSK, PARP, cleaved caspase-3 (Cell Signaling Technology, Beverly, MA), dephospho-beta-catenin (de-p-beta-catenin) (Enzo bioscience Inc. France), KRAS, B-RAF, phosphor-MEK, MEK, and beta-actin (Sigma-Aldrich, St. Louis, MO).
To isolate cytoplasmic and nuclear fractions, cells were incubated using NE-PER® Nuclear and cytoplasmic extraction reagents (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s protocol. For comparison, one equivalent of cytoplasmic proteins and two equivalents of nuclear proteins were analyzed side-by-side by western blot analysis with different antibodies.
Immunofluorescence assay
Cells (2 × 105) grown on sterile glass coverslips were transfected with either non-targeting control siRNA or ITF-2 siRNA. These transfected cells were then incubated with 1:50 anti-ITF-2 (Santa Cruz Biothchnology, Dallas, TX) at room temperature for 1 h after fixation with 4% paraformaldehyde and permeabilization with triton X-100. The cells were then incubated with secondary anti-goat antibody conjugated with fluorescein isothiocyanate (Bethyl, Montgomery, TX) and counterstained with a 4 μg/mL DAPI solution. After the coverslips were mounted with fluorescence mounting medium (Dako, Carpinteria, CA), cells were viewed using a fluorescence microscope (Olympus-IX71 Olympus, Tokyo, Japan).
Flow cytometric apoptosis analysis
Cells (1 × 106) were treated with AZD6244 for 24 h, washed twice with cold PBS, and resuspended in 1× Binding Buffer at a concentration of 1 × 106 cells/mL. A 100 μL aliquot of this solution (1 × 105 cells) was then transferred to a 5 mL culture tube to which 5 μL of FITC Annexin V was added. These cells were incubated for 15 min at room temperature in the dark before assaying with an Annexin V–FITC Apoptosis Detection Kit I (BD Biosciences, San Jose, CA), in accordance with the manufacturer’s protocol. The percentage of apoptotic cells was analyzed by flow cytometry within 1 h (Becton Dickinson, San Jose, CA).
Assessment of cell death by Live-Dead cell staining
Cell death after siRNA transfection with or without AZD6244 treatment was assessed using Live-Dead Cell Staining Kit (Enzo Life Sciences). This kit utilizes a cell permeable green fluorescent dye to stain live cells. Dead cells were stained by propidium iodide, a red fluorescent dye, which is actively pumped out of the cytoplasm in viable cells. Stained cells were visualized by fluorescence microscopy (Olympus, Tokyo, Japan).
GST pull-down assay
pGEX-GSK3β (Addgene, Cambridge, MA) was introduced into Escherichia coli BL21(DE3) cells (Stratagene, La Jolla, CA) by transformation and the cells were cultured in 5 ml LB medium at 37°C to the mid-log phase. Protein expression was induced by 1mM isopropyl-1-thio-beta-D-galactopyranoside (IPTG). After culturing for 3 h, cells were pelleted by centrifugation and then suspended in 100 μL of a lysis buffer (Cell Signaling Technology, Beverly, MA). Recombinant GST-GSK3β was purified using Glutathione- Sepharose 4B beads (GE Healthcare Life Sciences. UK). Protein lysates of M14, M14/AZD-3, and LOX-IMVI cell lines were respectively combined with 50 μL pre-cleaned Glutathione- Sepharose 4B beads and the mixture was incubated under shaking for 16 h at 4°C. The incubated beads were washed three times with lysis buffer (Cell Signaling Technology, Beverly, MA). After washing, 2× SDS-PAGE loading buffer was added to the washed beads and proteins were extracted from the beads by boiled for 5 min. Proteins were analyzed by 10% SDS-PAGE and western blot analysis.
Statistical analysis
Statistical differences in cell viability, presented as mean ± standard error of the mean (SEM), were evaluated by the Student t test, while those in relative expression of endogenous ITF-2 mRNA were evaluated by the Mann-Whitney U test. Death rates of two cell lines in assessment of Live-Dead cell staining were compared by a Fisher’s exact test. Statistical analyses were performed using SPSS version 21.0 software. For all analyses, the P values were 2-tailed, and P < 0.05 was considered as statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This research were supported by Basic Science Research Program though the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (NRF-2011-0012625).
CONFLICTS OF INTEREST
All authors have no conflicts of interests to declare.
REFERENCES
1. Yoon S, Seger R. The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth factors. 2006; 24:21–44.
2. Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nature reviews Cancer. 2004; 4:937–947.
3. Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991; 65:663–675.
4. Watts SW. Serotonin activates the mitogen-activated protein kinase pathway in vascular smooth muscle: use of the mitogen-activated protein kinase kinase inhibitor PD098059. The Journal of pharmacology and experimental therapeutics. 1996; 279:1541–1550.
5. Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. Journal of hematology & oncology. 2013; 6:27.
6. Garon EB, Finn RS, Hosmer W, Dering J, Ginther C, Adhami S, Kamranpour N, Pitts S, Desai A, Elashoff D, French T, Smith P, Slamon DJ. Identification of common predictive markers of in vitro response to the Mek inhibitor selumetinib (AZD6244; ARRY-142886) in human breast cancer and non-small cell lung cancer cell lines. Molecular cancer therapeutics. 2010; 9:1985–1994.
7. Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, Golub TR, Sebolt-Leopold J, Sellers WR, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006; 439:358–362.
8. Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004; 22:4456–4462.
9. Sznol M. Molecular markers of response to treatment for melanoma. Cancer J. 2011; 17:127–133.
10. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367:107–114.
11. Wang Y, Van Becelaere K, Jiang P, Przybranowski S, Omer C, Sebolt-Leopold J. A role for K-ras in conferring resistance to the MEK inhibitor, CI-1040. Neoplasia. 2005; 7:336–347.
12. Yeh JJ, Routh ED, Rubinas T, Peacock J, Martin TD, Shen XJ, Sandler RS, Kim HJ, Keku TO, Der CJ. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol Cancer Ther. 2009; 8:834–843.
13. Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006; 6:813–823.
14. Hanauer DA, Rhodes DR, Sinha-Kumar C, Chinnaiyan AM. Bioinformatics approaches in the study of cancer. Curr Mol Med. 2007; 7:133–141.
15. Kim HS, Kim SC, Kim SJ, Park CH, Jeung HC, Kim YB, Ahn JB, Chung HC, Rha SY. Identification of a radiosensitivity signature using integrative metaanalysis of published microarray data for NCI-60 cancer cells. BMC Genomics. 2012; 13:348.
16. Morin PJ. beta-catenin signaling and cancer. Bioessays. 1999; 21:1021–1030.
17. Kolligs FT, Bommer G, Goke B. Wnt/beta-catenin/tcf signaling: a critical pathway in gastrointestinal tumorigenesis. Digestion. 2002; 66:131–144.
18. Kolligs FT, Nieman MT, Winer I, Hu G, Van Mater D, Feng Y, Smith IM, Wu R, Zhai Y, Cho KR, Fearon ER. ITF-2, a downstream target of the Wnt/TCF pathway, is activated in human cancers with beta-catenin defects and promotes neoplastic transformation. Cancer Cell. 2002; 1:145–155.
19. Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, Bartholomeusz G, Li Y, Pan Y, Li Z, Bargou RC, Qin J, Lai CC, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005; 19:159–170.
20. Davies MA, Kopetz S. Overcoming resistance to MAPK pathway inhibitors. J Natl Cancer Inst. 2013; 105:9–10.
21. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, MacConaill LE, Zhang J, Gray NS, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009; 106:20411–20416.
22. Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, Beroukhim R, Milner DA, Granter SR, Du J, Lee C, Wagner SN, Li C, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005; 436:117–122.
23. Annunziata CM, Hernandez L, Davis RE, Zingone A, Lamy L, Lam LT, Hurt EM, Shaffer AL, Kuehl WM, Staudt LM. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood. 2011; 117:2396–2404.
24. Gopal YN, Deng W, Woodman SE, Komurov K, RamP, Smith PD, Davies MA. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010; 70:8736–8747.
25. Won JK, Yang HW, Shin SY, Lee JH, Heo WD, Cho KH. The crossregulation between ERK and PI3K signaling pathways determines the tumoricidal efficacy of MEK inhibitor. J Mol Cell Biol. 2012; 4:153–163.
26. Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L, Minna JD, Fang B, Roth JA. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011; 71:3658–3668.
27. Dry JR, Pavey S, Pratilas CA, Harbron C, Runswick S, Hodgson D, Chresta C, McCormack R, Byrne N, Cockerill M, Graham A, Beran G, Cassidy A, et al. Transcriptional pathway signatures predict MEK addiction and response to selumetinib (AZD6244). Cancer Res. 2010; 70:2264–2273.
28. Lassar AB, Davis RL, Wright WE, Kadesch T, Murre C, Voronova A, Baltimore D, Weintraub H. Functional activity of myogenic HLH proteins requires hetero-oligomerization with E12/E47-like proteins in vivo. Cell. 1991; 66:305–315.
29. Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000; 20:429–440.
30. Kim SK, Jang HR, Kim JH, Kim M, Noh SM, Song KS, Kang GH, Kim HJ, Kim SY, Yoo HS, Kim YS. CpG methylation in exon 1 of transcription factor 4 increases with age in normal gastric mucosa and is associated with gene silencing in intestinal-type gastric cancers. Carcinogenesis. 2008; 29:1623–1631.
31. Zhai Y, Wu R, Schwartz DR, Darrah D, Reed H, Kolligs FT, Nieman MT, Fearon ER, Cho KR. Role of beta-catenin/T-cell factor-regulated genes in ovarian endometrioid adenocarcinomas. Am J Pathol. 2002; 160:1229–1238.
32. Lemieux E, Cagnol S, Beaudry K, Carrier J, Rivard N. Oncogenic KRAS signalling promotes the Wnt/beta-catenin pathway through LRP6 in colorectal cancer. Oncogene. 2014; 34:4914–27.
33. Herbst A, Bommer GT, Kriegl L, Jung A, Behrens A, Csanadi E, Gerhard M, Bolz C, Riesenberg R, Zimmermann W, Dietmaier W, Wolf I, Brabletz T, et al. ITF-2 is disrupted via allelic loss of chromosome 18q21, and ITF-2B expression is lost at the adenoma-carcinoma transition. Gastroenterology. 2009; 137:639–648, 648 e631–639.
34. Joo JK, Kim SH, Kim HG, Kim DY, Ryu SY, Lee KH, Lee JH. CpG methylation of transcription factor 4 in gastric carcinoma. Ann Surg Oncol. 2010; 17:3344–3353.
35. Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, Selby HM, Kachaeva MI, Flanigan SA, Kulikowski GN, Leong S, Arcaroli JJ, Messersmith WA, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther. 2010; 9:3351–3362.
36. Vene R, Cardinali B, Arena G, Ferrari N, Benelli R, Minghelli S, Poggi A, Noonan DM, Albini A, Tosetti F. Glycogen synthase kinase 3 regulates cell death and survival signaling in tumor cells under redox stress. Neoplasia. 2014; 16:710–722.
37. Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006; 5:1136–1144.
38. Simmons BH, Lee JH, Lalwani K, Giddabasappa A, Snider BA, Wong A, Lappin PB, Eswaraka J, Kan JL, Christensen JG, Shojaei F. Combination of a MEK inhibitor at sub-MTD with a PI3K/mTOR inhibitor significantly suppresses growth of lung adenocarcinoma tumors in Kras(G12D-LSL) mice. Cancer Chemother Pharmacol. 2012; 70:213–220.
39. Atefi M, von Euw E, Attar N, Ng C, Chu C, Guo D, Nazarian R, Chmielowski B, Glaspy JA, Comin-Anduix B, Mischel PS, Lo RS, Ribas A. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One. 2011; 6:e28973.
40. Sale MJ, Cook SJ. The BH3 mimetic ABT-263 synergizes with the MEK1/2 inhibitor selumetinib/AZD6244 to promote BIM-dependent tumour cell death and inhibit acquired resistance. Biochem J. 2013; 450:285–294.
41. Packer LM, Rana S, Hayward R, O’Hare T, Eide CA, Rebocho A, Heidorn S, Zabriskie MS, Niculescu-Duvaz I, Druker BJ, Springer C, Marais R. Nilotinib and MEK inhibitors induce synthetic lethality through paradoxical activation of RAF in drug-resistant chronic myeloid leukemia. Cancer Cell. 2011; 20:715–727.
42. Chang-Yew Leow C, Gerondakis S, Spencer A. MEK inhibitors as a chemotherapeutic intervention in multiple myeloma. Blood Cancer J. 2013; 3:e105.
43. McCubrey JA, Abrams SL, Ligresti G, Misaghian N, Wong EW, Steelman LS, Basecke J, Troppmair J, Libra M, Nicoletti F, Molton S, McMahon M, Evangelisti C, et al. Involvement of p53 and Raf/MEK/ERK pathways in hematopoietic drug resistance. Leukemia. 2008; 22:2080–2090.
44. Nishioka C, Ikezoe T, Yang J, Yokoyama A. Inhibition of MEK signaling enhances the ability of cytarabine to induce growth arrest and apoptosis of acute myelogenous leukemia cells. Apoptosis. 2009; 14:1108–1120.
45. Haass NK, Sproesser K, Nguyen TK, Contractor R, Medina CA, Nathanson KL, Herlyn M, Smalley KS. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008; 14:230–239.
46. Xu J, Knox JJ, Ibrahimov E, Chen E, Serra S, Tsao M, Cao P, Vines D, Green DE, Metran-Nascente C, McNamara MG, Hedley DW. Sequence dependence of MEK inhibitor AZD6244 combined with gemcitabine for the treatment of biliary cancer. Clin Cancer Res. 2013; 19:118–127.
47. Davies BR, Logie A, McKay JS, Martin P, Steele S, Jenkins R, Cockerill M, Cartlidge S, Smith PD. AZD6244 (ARRY-142886), a potent inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase 1/2 kinases: mechanism of action in vivo, pharmacokinetic/pharmacodynamic relationship, and potential for combination in preclinical models. Mol Cancer Ther. 2007; 6:2209–2219.
48. Holt SV, Logie A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer. 2012; 106:858–866.
49. Huynh H, Chow PK, Soo KC. AZD6244 and doxorubicin induce growth suppression and apoptosis in mouse models of hepatocellular carcinoma. Molecular cancer therapeutics. 2007; 6:2468–2476.
50. Loes IM, Immervoll H, Angelsen JH, Horn A, Geisler J, Busch C, Lonning PE, Knappskog S. Performance comparison of three BRAF V600E detection methods in malignant melanoma and colorectal cancer specimens. Tumour Biol. 2015; 36:1003–1013.
51. Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, MacConaill LE, Zhang J, Gray NS, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci USA. 2009; 106:20411–6.