
Introduction
Sunlight that reaches our skin consists of infrared, visible, ultraviolet A (UVA) and ultraviolet B (UVB) light. Ultraviolet B targets epidermis and damages DNA in keratinocytes and melanocytes, thus initiating carcinogenesis [1-6].
Long wavelength ultraviolet light UVA, which is less energetic than UVB, is a main component of UV radiation we exposed to. UVA deeply penetrates into the skin (the dermal layer), targeting dermal fibroblasts. In vitro, UVA can induce senescence in fibroblasts [7-10]. Senescent fibroblasts can promote carcinogenesis and also contribute to photo-aging [11].
In cell culture, induction of senescence includes two steps: cell cycle arrest followed by geroconversion [12, 13]. During geroconversion cells become hypertrophic (large cell morphology) and hyper-functional (hyper-secretory phenotype or SASP), acquire β- galactosidase (β-Gal) staining, a marker of lysosomal hyperfunction, and lose re-proliferative potential [12-17]. Geroconversion is in part mTORC1-dependent and is partially suppressed by rapamycin and other rapalogs [18-34]. Pan-mTOR inhibitors, which inhibit both rapamycin-sensitive and -insensitive activities of mTORC1 [35,36], further suppress geroconversion [31-33, 37].
Here we showed that UVA caused senescence in normal human WI38t fibroblasts and primary adult dermal murine fibroblasts. Rapamycin and pan-mTOR inhibitors prevented UVA-induced senescent morphology. mTOR inhibitors did not abrogate cell cycle arrest and in contrast reinforced it. Thus, mTOR inhibitors suppressed geroconversion of UVA-arrested fibroblasts.
Results
Torins and rapamycin potentiate cell cycle arrest caused by UVA
In pilot experiments, we selected 8-10 J/cm2 of UVA irradiation to induce senescence in human WI38t fibroblasts. As shown in Figure 1, irradiation caused predominately G2 arrest, decreasing the number of cells in S-phase. We next investigated effects of low concentrations of mTOR inhibitors on UVA-induced arrest: 5 nM rapamycin and 30 nM Torin 1 and Torin 2, an optimal non-toxic gerosuppressive concentration of Torins [37]. mTOR inhibitors further decreased S phase fraction in UVA-arrested cells (Figure 1). mTOR inhibitors shifted arrest from G2 to G1, so cells were arrested equally in G1 and G2 with rapamycin and predominantly in G1 with Torins.
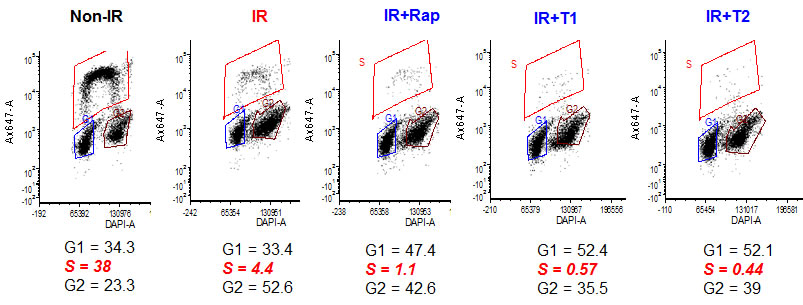
Figure 1: mTOR inhibitors re-enforce arrest caused by UVA in WI38t fibroblasts. Fibroblasts were irradiated with 8 J/cm2 of UVA (IR) and were left untreated or treated with either rapamycin (Rap, 5 nM), Torin 1 (T1, 30 nM) or Torin 2 (T2, 30 nM) immediately after irradiation. Non-irradiated (non-IR) cells serve as proliferation control. 24 h after irradiation cells were pulsed with 10 µM Edu for 1 h, trypsinized, fixed in 4% paraformaldehyde, stained for Edu and DAPI as described in Methods and analyzed by flow cytometry. Note: percentage of EDU-positive (S phase) cells is measured to estimate S phase. G1 and G2 were estimated by DAPI staining, so % of G1+G2+S is not necessarily 100.
Torins and rapamycin prevent UVA-induced senescence in WI38t fibroblasts
Irradiated WI38t cells developed senescent phenotype: enlarged cell morphology and SA-β-gal staining (Figure 2A). Rapamycin and Torins 1 and 2 prevented hypertrophic morphology , when drugs were added immediately after irradiation (Figure 2) or when cells were additionally pre-treated for 1 day before irradiation and treated after irradiation too (Supplementary Figure S1). UVA-irradiated fibroblasts treated with mTOR inhibitors remained smaller/thinner in size compared to irradiated cells (Figure 2A and Supplementary Figure S1). For all further studies, we chose to treat cells after irradiation (as depicted in schemas in Figure 2B and 3B).
Since one of the features of senescent cells is hyperfunction, we compared metabolic rate of irradiated senescent WI38t fibroblasts and control non-irradiated cells by measuring reduction of resazurin to fluorescent resorufin. Resazurin was added to cultures 6-7 days after irradiation and fluorescence of resorufin was measured (Figure 2B, 2C and Supplementary Figure S2). Then, cells were trypsinized and counted in hemocytometer and ratios of fluorescence units to cell numbers were calculated. In non-irradiated cultures, rapamycin and Torins did not affect ratios of fluorescence intensities to cell numbers (Supplementary Figure S2), indicating that metabolic rate per cell was constant. In irradiated cells, fluorescence intensity per cell was high (Figure 2C) and 5nM rapamycin and 30 nM Torin 1 and 2 decreased metabolic activity of irradiated cells (Figure 2C).
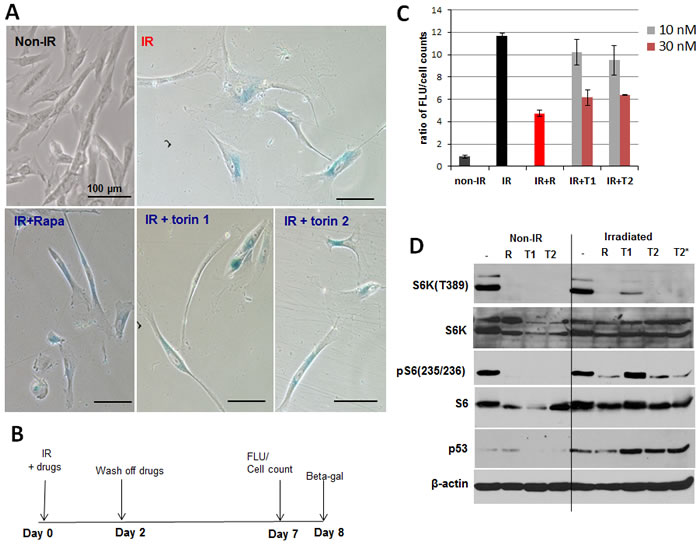
Figure 2: mTOR inhibitors suppress senescent phenotype induced by UVA in WI38t fibroblasts. A. SA-β-gal staining. WI38t cells were irradiated with 8 J/cm2 (IR) and treated with mTOR inhibitors as shown in schema B. 8 days after irradiation cells were stained for SA-β-gal and microphotographed. Non-IR – non-irradiated cells serve as control. C. mTOR inhibitors reduce metabolic activity of senescent WI38t fibroblasts. Cells were induced to senesce by UVA irradiation and treated as shown in schema (B). 7 days after irradiation cellular metabolic activity was measured using CellTiter Blue reagent as described in Methods, followed by cell counts. Data present a ratio of fluorescence units (FLU) to cell counts, i.e. fluorescence units per cell. Bars are mean ± SD. R – rapamycin 5 nM; T1 – Torin 1; T2 – Torin 2. D. Immunoblot analysis. WI38t cells were irradiated or not (non-IR) with 8 J/cm2 and drugs were added immediately after irradiation. 24 h later cells were lysed and immunoblotting was performed with indicated antibodies.
While decreasing mTOR activity, mTOR inhibitors did not decrease p53 in UVA-irradiated cells
UVA induced p53 (Figure 2D), which is known to mediate cell cycle arrest caused by irradiation. Rapamycin did not abrogate p53 induction and Torins even potentiated it. So suppression of senescent phenotype by mTOR inhibitors is not due to abrogation of the pathway leading to cell cycle arrest. The arrest pathway remained intact (Figure 2D), consistent with cell cycle distribution (Figure 1). We next investigated the geroconversion pathway. UVA did not inhibit phospho-S6K (T389) and phospho-S6(S235/236) (Figure 2D). Treatment with rapamycin and 30 nM Torin 2 resulted in sustained inhibition of phospho-S6K and S6 (S235/236). In agreement with previous results [37], Torin 1 was less potent than Torin 2 (Figure 2D). Curiously, mTOR inhibitors including rapamycin were less effective in inhibiting S6 phosphorylation in UVA-irradiated cells compared to non-irradiated control cells (Figure 2D).
Rapamycin and Torins suppressed geroconversion of UVA-irradiated WI38t fibroblasts even when treatment began 24 h after radiation (Figure 3B). Treated cells remained morphologically smaller (Figure 3A) and less hypertrophic as indicated by reduced amount of protein per cell (Figure 3C).
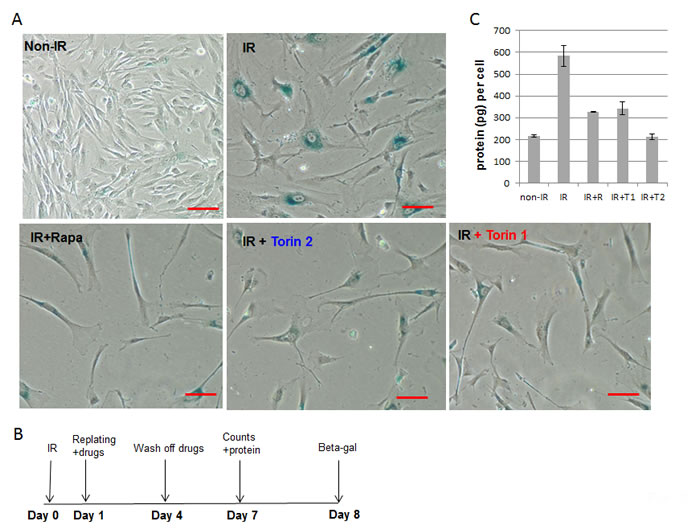
Figure 3: mTOR inhibitors suppress senescent morphology and hypertrophy in WI38t fibroblasts, when treatment was delayed by 24 h after irradiation. A. SA-β-gal staining. Cells were irradiated with 10 J/cm2 (IR) and treated as shown in schema B. 8 days after irradiation cells were stained for SA-β-gal and microphotographed; bar- 100 µm; C. Cells were treated as in (B). 7 days after irradiation, cells were counted and lysed followed by measuring protein amounts. Data present protein per cell. Non-IR – non-irradiated control cells.
Torins and rapamycin prevent UVA-induced senescence in primary adult mouse skin fibroblasts
We extended our study to primary adult mouse skin fibroblasts. Primary skin fibroblast cultures were isolated from murine skin and used at early passage (p.2) since these cells undergo replicative senescence when cultured in vitro by passage 5. Primary skin fibroblasts were pre-treated with mTOR inhibitors for 3 hours, then irradiated with UVA and treatment continued for 4 days followed by SA-β-gal staining (Figure 4A). SA-β-gal-positive cells were counted in 4-6 fields of each condition and percent of senescent cells was calculated (Figure 4B). Non-irradiated control cell culture had a noticeable number of senescent cells (~ 21%), which is not unusual for primary cultures. Four days after irradiation, number of senescent cells doubled in control irradiated cultures. mTOR inhibitors decreased fraction of SA-β-gal positive enlarged cells in irradiated cultures. As in the case of WI38t cells, these effects were associated with inhibition of mTORC1 pathway as evidenced by inhibited phosphorylation of S6K at T389 and S6 at S235/236 (Figure 5).
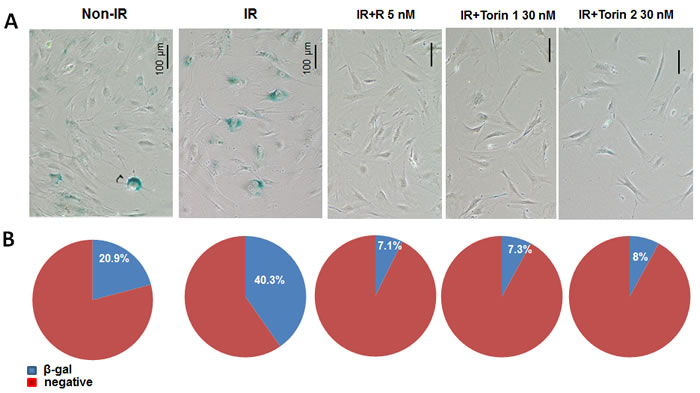
Figure 4: mTOR inhibitors suppress senescent morphology in UVA-irradiated primary adult mouse skin fibroblasts. A. Cells were pre-treated with mTOR inhibitors for 3h and then irradiated with 10 J/cm2 (IR). Drugs were re-added after irradiation. Four days after irradiation cells were stained for SA-β-gal. Non-IR – non-irradiated control; IR – irradiated, R – rapamycin ; T1 – Torin 1; T2 – Torin 2 . B. Numbers of β-gal positive and negative cells were counted in 4-6 fields for each sample. Counts were combined and percentage of β-gal positive cells was calculated.
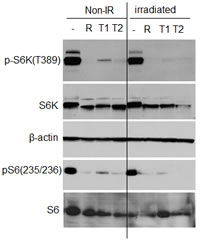
Figure 5: UVA irradiation does not inhibit mTOR pathway in primary adult mouse fibroblasts. Immunoblot analysis. Primary adult murine fibroblasts were pre-treated with mTOR inhibitors for 3 h and then irradiated with 10 J/cm2. Drugs were re-added and cells were lysed 24 h after irradiation. Non-IR – non-irradiated control; R – rapamycin (5 nM); T1 - Torin 1 (30 nM); T2 – Torin 2 (30 nM).
Discussion
Here we showed that UVA caused cell cycle arrest followed by mTOR-dependent geroconversion, which could be suppressed by rapamycin and Torin 1 and 2. mTOR inhibitors prevented only the second step of senescence program: geroconversion. Cell cycle arrest caused by UVA was not abrogated. Furthermore, the arrest was re-enforced. mTOR inhibitors by themselves slow down cell cycle progression. It is important to emphasize because of the common misunderstanding of the difference between cell cycle arrest and senescence [12, 13]. mTOR inhibitors arrest cell cycle, yet inhibit geroconversion in arrested (quiescent) cells. Cells remain quiescent, not senescent. Quiescent cells retain the ability to re-proliferate. So mTOR inhibitors inhibit proliferation but may preserve re-proliferative potential, which can be evident when cells are re-stimulated to proliferate [12,13, 31]. We emphasize again that mTOR inhibitors do not abrogate senescent arrest, do not re-activate cell cycle, do not stimulate proliferation. They preserve the potential to re-proliferate, when cell cycle is re-activated by removing CDK inhibition [12, 13, 31].
Suppression of geroconversion in UVA-treated fibroblasts has several implications. First, by inducing senescence in dermal fibroblasts, UVA may create pro-carcinogenic micro-environment to promote premalignant keratinocytes and melanocytes. In fact, hyper-functional senescent cells secrete tumor-promoting molecules and support carcinogenesis [38-43]. By suppressing development of UV-induced senescent phenotype in stromal fibroblasts, mTOR inhibitors may prevent UV-induced tumors. In fact, rapamycin suppress UVB-induced skin cancer in mice [44], decrease clusters of premalignant cells with mutant p53 after UVA+UVB-radiation [45]. Although not much is known about the effect of mTOR inhibitors on UV-induced carcinogenesis, it is recognized that rapamycin prevents cancer by other carcinogens [46] and spontaneous cancer in animals and humans [47-61]. Also, rapamycin prevents TPA-induced skin tumors [62]. Noteworthy, TPA can activate mTOR and induce cellular senescence in certain cell types [63]. Rapamycin prevents cancer in a wide variety of cancer-prone murine models [64-70]. Rapamycin and everolimus prevent skin cancer in humans: namely, in transplant patients receiving rapamycin (sirolimus) and everolimus [57-61]. mTOR inhibitors are very attractive chemopreventive modality, given their systemic anti-aging effects [54, 55].
Finally, by reducing cellular senescence, rapamycin may be considered to prevent photo aging. Rapalogs (rapamycin and everolimus) can be used not only systemically but also topically. Rapalog-based creams are expected not to interfere with sun tanning and vitamin D3 synthesis.
materials and Methods
Cell lines and reagents
WI38-tert (WI38t) fibroblasts were provided by Dr. Eugene Kendal (Roswell Park Cancer Institute, Buffalo, NY) and described previously [71]. WI38t cells were cultured in DMEM, supplemented with 10% FBS and pen/strep. Primary adult mouse skin fibroblasts were a kind gift from Dr. G. Paragh laboratory (Roswell Park Cancer Institute, Buffalo, NY). Primary fibroblasts were maintained in DMEM supplemented with 10% FBS, pen/strep and antibiotic-antimycotic (ANTI-ANTI; Thermo Fisher Scientific, Grand Island, NY).
Rapamycin was purchased from LC laboratories (Woburn, MA). Torin 1 and Torin 2 were from Selleckchem (Houston, TX). Stock solutions were prepared in DMSO.
Senescence induction
Cells were induced to senesce by exposure to UVA1 radiation, which makes up to 75% of UVA rays. UVA1 irradiation was produced by a UVP transilluminator with 5x8W Hitachi F8T5 UVA1 fluorescent light tubes. The spectral output was determined by a STS-UV-L-25-400-SMA STS Microspectrometer (Ocean Optics Inc). UVA1 dose was determined using an International light radiometer/photometer (IL1400A).
Immediately before irradiation complete medium was replaced with DMEM without phenol red/5%FBS. Proliferating cells were irradiated for an interval needed to deliver 8 or 10 J/cm2 of UVA1 rays; control non-irradiated (mock irradiated) cells were placed in the same hood for an identical interval but not being irradiated. Then cells were treated with mTOR inhibitors or left untreated and incubation was continued in complete growth medium.
Irradiated cells were treated with mTOR inhibitors as described in Figure legends.
After several days, cells were scored for senescence markers, such as SA-β-gal activity, hypertrophy (displayed as protein amount per cell) and increased metabolic activity (rate of reduction of resazurin to resorufin).
Cell cycle distribution and DNA synthesis
To determine rate of DNA synthesis, cells were pulsed with 10 µM EdU (5-ethynyl-2’-deoxyuridine; Thermo Fisher Scientific, Grand Island, NY) for 1 hour. Labeled cells were trypsinized, washed in PBS and fixed in 4% paraformaldehyde for 20 min at RT and washed in PBS. Fixed cells were further stained by incubating in the staining cocktail (4 mM CuSO4, 100 mM sodium ascorbate and 4.8 µM Alexa Flour 647 Azide triethylammonium (Thermo Fisher Scientific)) for 20 min at RT followed by washes in PBS. Cells were re-suspended in PBS and counterstained with 5 µg/ml of DAPI. Analyses were performed on flow cytometer Fortessa B at Roswell Park Cancer Institute FACS facility, using BD FACS Diva Software (BD Biosciences). Data were collected for 20000 cells and analyzed with FCS Express 4 (De Novo Software, Glendale, CA).
SA-β-gal staining was performed using Senescence-galactosidase staining kit (Cell Signaling Technology, Danver, MA) according to manufacturer’s protocol. Photos were taken under bright field microscope.
Immunoblot analysis
Whole cell lysates were prepared using boiling lysis buffer (1% SDS, 10 mM Tris.HCl, pH 74.). Protein concentrations were determined using BCA reagent (Thermo Scientific, Rockford, IL) and equal amounts of proteins were separated onto Criterion or mini gradient polyacrylamide gels (Bio-Rad, Hercules, CA) and transferred to PVDF membranes (Bio-Rad). The following primary antibodies were used: rabbit antibodies for phospho-S6 (Ser235/236), phospho-S6K(T389 ) and total S6K - were from Cell Signaling Biotechnology. Mouse anti-S6 antibody was from Cell Signaling Biotechnology. Anti-β-actin-HRP antibody was from Sigma-Aldrich (St. Louis, MO); mouse antibody for p53 (Ab-6) was from Oncogene Research products (La Jolla, CA). Secondary anti-rabbit and anti-mouse HRP-conjugated antibodies were from Cell Signaling Biotechnology as previously described [20] , [21] .
Metabolic activity
Test was performed using CellTiter Blue reagent (Promega, Madison, WI). The resazurin dye (CellTiter Blue) is reduced to resorufin by metabolically active cells, resulting in the generation of a fluorescent product. 200 µl of CellTiter Blue was added per 1 ml of medium to the cells followed by incubation at 37ºC, 5% CO2 for 3 h. Fluorescence was recorded at 560/590 nm. Then cells were counted and ratios of fluorescence units to cell numbers were calculated.
Author Contributions
MVB conceptualized the project, designed the experiments. OL performed the experiments. MVB and OL wrote the manuscript.
Acknowledgments AND FUNDING
Funding: Roswell Park Cancer Institute, Buffalo, NY.
Conflicts of interests
I am the Editor in Chief of the Journal Oncotarget, I did not participate in the acceptance, rejection or peer review process for this manuscript.
Other author has no conflicts to declare.
References
1. De Fabo EC, Noonan FP, Fears T, Merlino G. Ultraviolet B but not ultraviolet A radiation initiates melanoma. Cancer Res. 2004; 64: 6372-6376.
2. Lin SW, Wheeler DC, Park Y, Cahoon EK, Hollenbeck AR, Freedman DM, Abnet CC. Prospective study of ultraviolet radiation exposure and risk of cancer in the United States. Int J Cancer. 131: E1015-1023.
3. de Gruijl FR, Forbes PD. UV-induced skin cancer in a hairless mouse model. Bioessays. 1995; 17: 651-660.
4. van Kranen HJ, de Gruijl FR. Mutations in cancer genes of UV-induced skin tumors of hairless mice. J Epidemiol. 1999; 9: S58-65.
5. Zaidi MR, De Fabo EC, Noonan FP, Merlino G. Shedding light on melanocyte pathobiology in vivo. Cancer Res. 2012; 72: 1591-1595.
6. Zaidi MR, Davis S, Noonan FP, Graff-Cherry C, Hawley TS, Walker RL, Feigenbaum L, Fuchs E, Lyakh L, Young HA, Hornyak TJ, Arnheiter H, Trinchieri G, et al. Interferon-gamma links ultraviolet radiation to melanomagenesis in mice. Nature. 2011; 469: 548-553.
7. Herrmann G, Brenneisen P, Wlaschek M, Wenk J, Faisst K, Quel G, Hommel C, Goerz G, Ruzicka T, Krieg T, Sies H, Scharffetter-Kochanek K. Psoralen photoactivation promotes morphological and functional changes in fibroblasts in vitro reminiscent of cellular senescence. J Cell Sci. 1998; 111: 759-767.
8. Ma W, Wlaschek M, Hommel C, Schneider LA, Scharffetter-Kochanek K. Psoralen plus UVA (PUVA) induced premature senescence as a model for stress-induced premature senescence. Exp Gerontol. 2002; 37: 1197-1201.
9. Wlaschek M, Ma W, Jansen-Durr P, Scharffetter-Kochanek K. Photoaging as a consequence of natural and therapeutic ultraviolet irradiation—studies on PUVA-induced senescence-like growth arrest of human dermal fibroblasts. Exp Gerontol. 2003; 38: 1265-1270.
10. Borlon C, Debacq-Chainiaux F, Hinrichs C, Scharffetter-Kochanek K, Toussaint O, Wlaschek M. The gene expression profile of psoralen plus UVA-induced premature senescence in skin fibroblasts resembles a combined DNA-damage and stress-induced cellular senescence response phenotype. Exp Gerontol. 2007; 42: 911-923.
11. Campisi J. The role of cellular senescence in skin aging. J Investig Dermatol Symp Proc. 1998; 3: 1-5.
12. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4: 159-165. doi: 10.18632/aging.100443.
13. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014: 13: 3628-3635.
14. Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003; 4: 358-362.
15. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007; 371: 21-31.
16. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013; 123: 966-972.
17. Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy. 2013; 9: 260-262.
18. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008; 7: 3355-3361.
19. Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A. 2010; 107: 9660-9664.
20. Leontieva OV, Natarajan V, Demidenko ZN, Burdelya LG, Gudkov AV, Blagosklonny MV. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A. 2012; 109: 13314-13318.
21. Leontieva OV, Demidenko ZN, Blagosklonny MV. Contact inhibition and high cell density deactivate the mammalian target of rapamycin pathway, thus suppressing the senescence program. Proc Natl Acad Sci U S A. 2014; 111: 8832-8837.
22. Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY). 2010; 2: 924-935. doi: 10.18632/aging.100265.
23. Dulic V. Senescence regulation by mTOR. Methods Mol Biol. 2013; 965: 15-35.
24. Iglesias-Bartolome R, Patel V, Cotrim A, Leelahavanichkul K, Molinolo AA, Mitchell JB, Gutkind JS. mTOR inhibition prevents epithelial stem cell senescence and protects from radiation-induced mucositis. Cell Stem Cell. 2012; 11: 401-414.
25. Serrano M. Dissecting the role of mTOR complexes in cellular senescence. Cell Cycle. 2012; 11: 2231-2232.
26. Cho S, Hwang ES. Status of mTOR activity may phenotypically differentiate senescence and quiescence. Mol Cells. 2012; 33: 597-604.
27. Luo Y, Li L, Zou P, Wang J, Shao L, Zhou D, Liu L. Rapamycin enhances long-term hematopoietic reconstitution of ex vivo expanded mouse hematopoietic stem cells by inhibiting senescence. Transplantation. 2014; 97: 20-29.
28. Hinojosa CA, Mgbemena V, Van Roekel S, Austad SN, Miller RA, Bose S, Orihuela CJ. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp Gerontol. 2012; 47: 958-965.
29. Halicka HD, Zhao H, Li J, Lee YS, Hsieh TC, Wu JM, Darzynkiewicz Z. Potential anti-aging agents suppress the level of constitutive mTOR- and DNA damage- signaling. Aging (Albany NY). 2012; 4: 952-965. doi: 10.18632/aging.100521.
30. Selman C, Sinclair A, Pedroni SM, Irvine EE, Michie AM, Withers DJ. Evidence that hematopoietic stem cell function is preserved during aging in long-lived S6K1 mutant mice. Oncotarget. 2016; 7: 29937-29943. doi: 10.18632/oncotarget.8729.
31. Leontieva OV, Demidenko ZN, Blagosklonny MV. Dual mTORC1/C2 inhibitors suppress cellular geroconversion (a senescence program). Oncotarget. 2015; 6: 23238-23248. doi: 10.18632/oncotarget.4836.
32. Sousa-Victor P, Garcia-Prat L, Munoz-Canoves P. Dual mTORC1/C2 inhibitors: gerosuppressors with potential anti-aging effect. Oncotarget. 2015; 6: 23052-23054. doi: 10.18632/oncotarget.5563.
33. Walters HE, Deneka-Hannemann S, Cox LS. Reversal of phenotypes of cellular senescence by pan-mTOR inhibition. Aging (Albany NY). 2016; 8: 231-244. doi: 10.18632/aging.100872.
34. Gu Z, Tan W, Ji J, Feng G, Meng Y, Da Z, Guo G, Xia Y, Zhu X, Shi G, Cheng C. Rapamycin reverses the senescent phenotype and improves immunoregulation of mesenchymal stem cells from MRL/lpr mice and systemic lupus erythematosus patients through inhibition of the mTOR signaling pathway. Aging (Albany NY). 2016; 8:1102-1114. doi: 10.18632/aging.100925.
35. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009; 284: 8023-8032
36. Thoreen CC, Sabatini DM. Rapamycin inhibits mTORC1, but not completely. Autophagy. 2009; 5:725-726.
37. Leontieva OV, Blagosklonny MV. Gerosuppression by pan-mTOR inhibitors. Aging (Albany NY). 2016; 8: 3535-3551. doi: 10.18632/aging.101155.
38. Parrinello S, Coppe JP, Krtolica A, Campisi J. Stromal-epithelial interactions in aging and cancer: senescent fibroblasts alter epithelial cell differentiation. J Cell Sci. 2005; 118: 485-496.
39. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001; 98: 12072-12077.
40. Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002; 34: 1401-1414.
41. Mercier I, Camacho J, Titchen K, Gonzales DM, Quann K, Bryant KG, Molchansky A, Milliman JN, Whitaker-Menezes D, Sotgia F, Jasmin JF, Schwarting R, Pestell RG, et al. Caveolin-1 and accelerated host aging in the breast tumor microenvironment: chemoprevention with rapamycin, an mTOR inhibitor and anti-aging drug. Am J Pathol. 2012; 181: 278-293.
42. Pani G. From growing to secreting: new roles for mTOR in aging cells. Cell Cycle. 2011; 10: 2450-2453.
43. Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, Hong S, Berry LS, Reichelt S, Ferreira M, Tavare S, Inoki K, Shimizu S. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011; 332: 966-970.
44. Wulff BC, Kusewitt DF, VanBuskirk AM, Thomas-Ahner JM, Duncan FJ, Oberyszyn TM. Sirolimus reduces the incidence and progression of UVB-induced skin cancer in SKH mice even with co-administration of cyclosporine A. J Invest Dermatol. 2008; 128: 2467-2473.
45. Voskamp P, Bodmann CA, Rebel HG, Koehl GE, Tensen CP, Bouwes Bavinck JN, El Ghalbzouri A, Van Kranen HJ, Willemze R, Geissler EK, De Gruijl FR. Rapamycin impairs UV induction of mutant-p53 overexpressing cell clusters without affecting tumor onset. Int J Cancer. 2012; 131: 1267-1276.
46. Granville CA, Warfel N, Tsurutani J, Hollander MC, Robertson M, Fox SD, Veenstra TD, Issaq HJ, Linnoila RI, Dennis PA. Identification of a highly effective rapamycin schedule that markedly reduces the size, multiplicity, and phenotypic progression of tobacco carcinogen-induced murine lung tumors. Clin Cancer Res. 2007; 13: 2281-2289.
47. Kauffman HM, Cherikh WS, Cheng Y, Hanto DW, Kahan BD. Maintenance immunosuppression with target-of-rapamycin inhibitors is associated with a reduced incidence of de novo malignancies. Transplantation. 2005; 80: 883-889.
48. Cullis B, D’Souza R, McCullagh P, Harries S, Nicholls A, Lee R, Bingham C. Sirolimus-induced remission of posttransplantation lymphoproliferative disorder. Am J Kidney Dis. 2006; 47: e67-72.
49. Law BK. Rapamycin: an anti-cancer immunosuppressant? Crit Rev Oncol Hematol. 2005; 56: 47-60.
50. Campistol JM, Eris J, Oberbauer R, Friend P, Hutchison B, Morales JM, Claesson K, Stallone G, Russ G, Rostaing L, Kreis H, Burke JT, Brault Y, et al. Sirolimus Therapy after Early Cyclosporine Withdrawal Reduces the Risk for Cancer in Adult Renal Transplantation. J Am Soc Nephrol. 2006; 17: 581-589.
51. Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, Ranieri E, Gesualdo L, Schena FP, Grandaliano G. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005; 352: 1317-1323.
52. Knoll GA, Kokolo MB, Mallick R, Beck A, Buenaventura CD, Ducharme R, Barsoum R, Bernasconi C, Blydt-Hansen TD, Ekberg H, Felipe CR, Firth J, Gallon L, et al. Effect of sirolimus on malignancy and survival after kidney transplantation: systematic review and meta-analysis of individual patient data. Bmj. 2014; 349: g6679.
53. Fernandez A, Hu S, Kirsner RS. Sirolimus: a potential chemopreventive agent. J Invest Dermatol. 2008; 128: 2352.
54. Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008; 7: 1520-1524.
55. Blagosklonny MV. Rapalogs in cancer prevention: Anti-aging or anticancer? Cancer Biol Ther. 2012; 13: 1349-1354.
56. de Gruijl FR, Koehl GE, Voskamp P, Strik A, Rebel HG, Gaumann A, de Fijter JW, Tensen CP, Bavinck JN, Geissler EK. Early and late effects of the immunosuppressants rapamycin and mycophenolate mofetil on UV carcinogenesis. Int J Cancer. 2010; 127: 796-804.
57. Halleck F, Budde K. Transplantation: Sirolimus for secondary SCC prevention in renal transplantation. Nat Rev Nephrol. 2012; 8: 687-689.
58. Alberu J, Pascoe MD, Campistol JM, Schena FP, Rial Mdel C, Polinsky M, Neylan JF, Korth-Bradley J, Goldberg-Alberts R, Maller ES. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation. 2011; 92: 303-310.
59. Euvrard S, Morelon E, Rostaing L, Goffin E, Brocard A, Tromme I, Broeders N, del Marmol V, Chatelet V, Dompmartin A, Kessler M, Serra AL, Hofbauer GF, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. N Engl J Med. 2012; 367: 329-339.
60. Alter M, Satzger I, Schrem H, Kaltenborn A, Kapp A, Gutzmer R. Non-melanoma skin cancer is reduced after switch of immunosuppression to mTOR-inhibitors in organ transplant recipients. J Dtsch Dermatol Ges. 2014; 12: 480-488.
61. Karia PS, Azzi JR, Heher EC, Hills VM, Schmults CD. Association of Sirolimus Use With Risk for Skin Cancer in a Mixed-Organ Cohort of Solid-Organ Transplant Recipients With a History of Cancer. JAMA Dermatol. 2016; 152: 533-540.
62. Checkley LA, Rho O, Moore T, Hursting S, DiGiovanni J. Rapamycin is a potent inhibitor of skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Prev Res (Phila). 2011; 4: 1011-1020.
63. Leontieva OV, Blagosklonny MV. Tumor promoter-induced cellular senescence: cell cycle arrest followed by geroconversion. Oncotarget. 2015; 5: 12715-12727. doi: 10.18632/oncotarget.3011.
64. Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Antoch MP, Blagosklonny MV. Rapamycin extends maximal lifespan in cancer-prone mice. Am J Pathol. 2010; 176: 2092-2097.
65. Comas M, Toshkov I, Kuropatwinski KK, Chernova OB, Polinsky A, Blagosklonny MV, Gudkov AV, Antoch MP. New nanoformulation of rapamycin Rapatar extends lifespan in homozygous p53-/- mice by delaying carcinogenesis. Aging (Albany NY). 2012; 4: 715-722. doi: 10.18632/aging.100496.
66. Komarova EA, Antoch MP, Novototskaya LR, Chernova OB, Paszkiewicz G, Leontieva OV, Blagosklonny MV, Gudkov AV. Rapamycin extends lifespan and delays tumorigenesis in heterozygous p53+/- mice. Aging (Albany NY). 2012; 4: 709-714. doi: 10.18632/aging.100498.
67. Donehower LA. Rapamycin as longevity enhancer and cancer preventative agent in the context of p53 deficiency. Aging (Albany NY). 2012; 4: 660-661. doi: 10.18632/aging.100494.
68. Christy B, Demaria M, Campisi J, Huang J, Jones D, Dodds SG, Williams C, Hubbard G, Livi CB, Gao X, Weintraub S, Curiel T, Sharp ZD, Hasty P. p53 and rapamycin are additive. Oncotarget. 2015; 6: 15802-15813. doi: 10.18632/oncotarget.4602.
69. Livi CB, Hardman RL, Christy BA, Dodds SG, Jones D, Williams C, Strong R, Bokov A, Javors MA, Ikeno Y, Hubbard G, Hasty P, Sharp ZD. Rapamycin extends life span of Rb1+/- mice by inhibiting neuroendocrine tumors. Aging (Albany NY). 2013; 5: 100-110. doi: 10.18632/aging.100533.
70. Popovich IG, Anisimov VN, Zabezhinski MA, Semenchenko AV, Tyndyk ML, Yurova MN, Blagosklonny MV. Lifespan extension and cancer prevention in HER-2/neu transgenic mice treated with low intermittent doses of rapamycin. Cancer Biol Ther. 2014; 15: 586-592.
71. Leontieva O, Gudkov A, Blagosklonny M. Weak p53 permits senescence during cell cycle arrest. Cell Cycle. 2010; 9: 4323-4327.