
INTRODUCTION
Aldose reductase (EC1.1.1.21, AKR1B1, AR) is a member of the aldo-keto reductase (AKR) protein family, which plays important roles in nuclear receptor signaling, inflammatory responses, osmoregulation, endobiotic and xenobiotic detoxification, hormone synthesis and action, cellular metabolism and reproduction etc. [1]. For glucose metabolism, AR serves as the first and the rate-limiting enzyme for the polyol pathway (PP) to reduce glucose to sorbitol, while sorbitol is further oxidized by sorbitol dehydrogenase (SDH) to generate fructose [2].
In the liver, AR was found to be transiently expressed during embryogenesis [3]. In adult animals, hepatic AR expression or activity is barely detectable or absent [3, 4]. A number of recent studies, nonetheless, have shown that hepatic AR can be significantly induced and activated under a variety of stress conditions or in diseased livers. In humans or rodents, AR and aldo-keto reductase family 1B10 (AKR1B10, also known as AR-like-1), were often among the most significantly up-regulated genes in many types of cancers including cervical cancer, colon cancer, leukemia, pancreatic cancer and hepatocellular carcinoma (HCC) [4–11]. Consistent with the transcriptomic analyses, many proteomic studies also indicated very significant elevations in the protein expression of AR and AKR1B10 in human liver cancer tissues [5, 7, 8, 12]. More interestingly, some reports showed that AR/AKR1B10 mRNA expression levels is an independent predictor of prognosis in HCC patients [10, 11]. In spite of these studies, however, very little studies were focus on the mechanism of the aberrantly overexpressed AR/AKR1B10 contribute to the development or progression of various types of cancers.
Potential mechanisms are aberrant overexpression/activation of hepatic AR and/or Polyol Pathway (PP)-associated overt oxidative stress and inflammation, which are believed to contribute significantly to the development of cancers [4, 13, 14]. Studies also suggest that inhibition of oxidative stress or inflammation is helpful with cancer prevention or treatment. For instance, trans-aldolase deficiency-induced hepatocarcinogenesis was associated with activation of AR that can be prevented by treatment with N-acetylcysteine [15]. In rats, diethylnitrosamine (DEN)-induced hepatocarcinogenesis was also associated with activation of AR and treatment with a ROS scavenger dially sulfide significantly ameliorated DEN-induced HCC [16].
Aberrant overexpression/activation of hepatic AR/PP may also contribute to lactate over-production, as in the well-known “Warburg effect” or aerobic glycolysis, whereby cancer cells exhibit increased conversion of glucose to lactate, even in the presence of sufficient oxygen [17]. Aberrant AR/PP-mediated hepatic over-production of fructose were shown to reprogram cellular glucose-lipid metabolism to significantly affect the development of obesity, metabolic syndrome, nonalcoholic fatty liver disease, and nonalcoholic steatohepatitis [18–21], all of which are important risk factors for the development of HCC. Fructose by itself was suggested to be able to promote tumorigenesis, in part by inducing metabolic reprogramming and lactate over-production [22–25]. However, the relationship between AR and lactate-production/Warburg effect has been unclear.
In the present study, we investigated the potential roles of AR in the development of HCC. The effects of AR overexpression and AR knockdown/knockout on lactate formation, the expression of inflammatory cytokines, and the most important Warburg effect regulating pathway, the AKT/mTOR signaling pathway, were evaluated in vitro in cultured liver cancer cells and in vivo in the livers of DEN-induced transgenic HCC model mice.
RESULTS
Overexpression of AR enhanced whereas knockdown of AR suppressed cancer cell proliferation, colony formation, and migration, invasion and wound-healing
In humans, microarray analyses identified AR and AKR1B10 mRNA up-regulated in the development of hepatitis C virus (HCV)-associated HCC [26, 27]. AR mRNA ranked at the top 3% and 7% of the significantly altered genes in HCV-positive HCC and HCV-positive cirrhosis respectively, in comparison with the HCV-negative normal subjects (Figure 1A, p < 0.001). Meanwhile, AKR1B10 mRNA ranked at the top 2% and 9% of the significantly altered genes in HCC and cirrhosis respectively (p < 0.001).
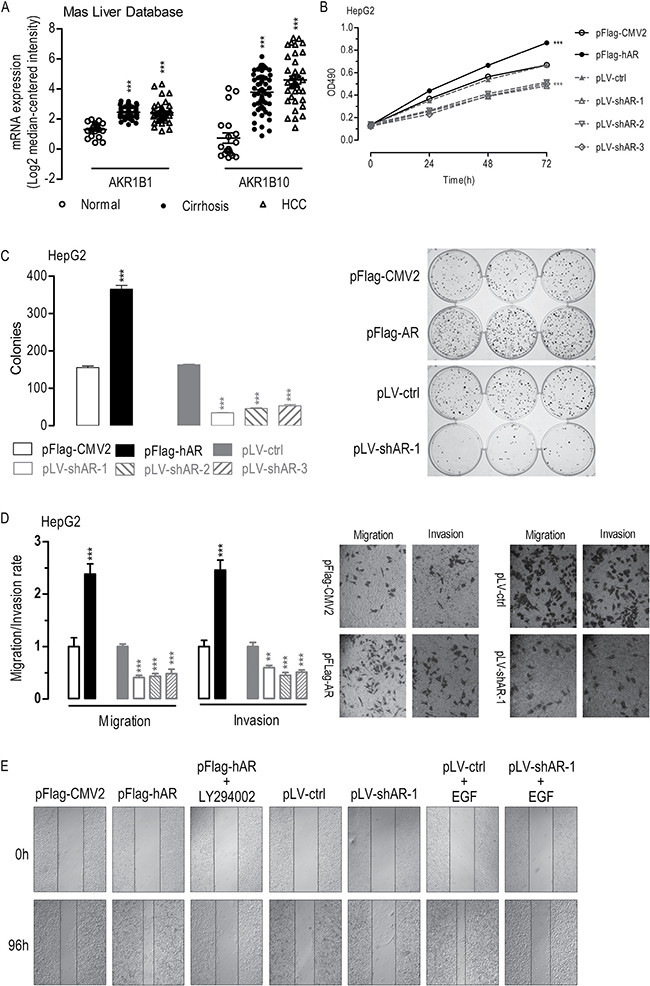
Figure 1: Effects of AR overexpression or knockdown on cell proliferation, migration, invasion, colony formation, and wound-healing in HepG2 cells. Dot plots showing AR and AKR1B10 mRNA expression in clinical liver samples as studied by Mas et al. (A) Overexpression of AR enhanced whereas knockdown of AR suppressed cell proliferation (B) (n = 6), colony formation (C) (n = 6), migration and invasion (D) (n = 6), and wound healing (E) (n = 3). Data were expressed as the mean ± SEM. **p < 0.01; ***p < 0.001, compared to pFlag-CMV2 or pLV-ctrl transfected cells.
To evaluate the effects of AR overexpression or knockdown on hepatocarcinogenesis, we performed transfection studies using HepG2 or SMMC-7721 liver cancer cells with a plasmid overexpressing AR or three plasmids overexpressing shRNAs against AR (Supplementary Table 1). In HepG2 cells, AR overexpression significantly enhanced HepG2 cell proliferation (Figure 1B), colony formation (Figure 1C), migration and invasion (Figure 1D), and wound-healing (Figure 1E). In contrast, shRNA-mediated knockdown of AR significantly suppressed HepG2 cell proliferation (Figure 1B), colony formation (Figure 1C), migration and invasion (Figure 1D), and wound-healing (Figure 1E). Remarkably, inhibition the phosphorylation of AKT1 by LY294002 significantly suppressed AR-induced wound-healing (Figure 1E) [28]. Furthermore, although small but not significant difference in wound-healing was found between the AR knockdown cells (pLV-shAR-1 transfected) and the control cells (pLV-ctrl-transfected), knockdown of AR suppressed wound-healing in EGF-stimulated cells (pLV-ctrl+EGF versus pLV-shAR-1+EGF) [29]. Similar effects of AR overexpression or knockdown on cell proliferation, colony formation, migration and invasion, and wound-healing observed in SMMC-7721 liver cancer cells (Supplementary Figure 1). Together these results suggested that aberrantly overexpressed AR promote oncogenic transformation or metastasis.
Overexpression of AR stabilized whereas knockdown of AR destabilized protein expression of AKT1 and AKT2
Since AR-induced wound-healing was largely suppressed by LY294002 treatment, we investigated how AR might affect the activity or expression of AKT1 and AKT2. Our qPCR analyses revealed that both AR overexpression and AR knockdown in HepG2 cells did not affect the mRNA expression of AKT1 and AKT2 (Supplementary Figure 2). Western blot analyses indicated that serine-473 phosphorylated AKT1 (pS473-AKT1), AKT1, AKT2, and several AKT1 and AKT2 down-stream signaling proteins, including serine-256 phosphoryalted-FOXO1 (pS256-FOXO1) and the key pathway of Warburg Effects [30] (including mTOR [31], HIF-1α [32], PKM2 [32], and another mTOR-regulated protein SREBP [33]), dose-dependently up-regulated in HepG2 cells overexpressing AR, with the exception of FOXO1 (Figure 2A). Consistent with this, these proteins dose-dependently down-regulated in AR-knockdown cells.
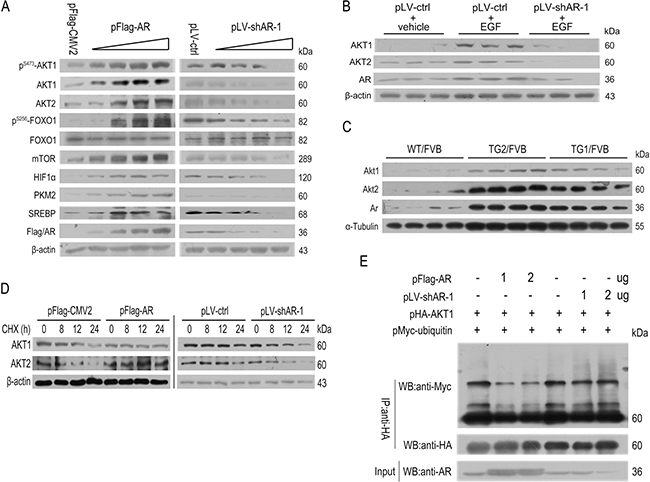
Figure 2: The effects of AR overexpression or knockdown on AKT/mTOR signaling. (A) Overexpression of AR enhanced whereas knockdown of AR suppressed the proteins expression of AKT/mTOR pathway. (B) Knockdown of AR prevented EGF-stimulated up-regulation of AKT1 and AKT2 (n = 3). (C) Stabilization of Akt1/Akt2 by AR overexpression in 34 wk old male TG1/FVB and TG2/FVB mice (n = 4). (D) Overexpression of AR stabilized AKT1/2 whereas knockdown of AR destabilized AKT1/2. (E) Overexpression of AR suppressed AKT1 ubiquitination whereas knockdown of AR promoted AKT1 ubiquitination.
Consistent with previous publication [29, 34, 35], knockdown of AR suppressed EGF-induced up-regulation of AKT1 and AKT2 in HepG2 cells (Figure 2B). In vivo, the Akt1 and Akt2 proteins significantly increased in liver tissues of liver-specific human AR-overexpressing transgenic FVB mice (TG1/FVB and TG2/FVB) (Figure 2C).
In the case of Akt1/2/3 isoforms have the similar structure (Supplementary Figure 3), following experiments performed using human Akt1 as a representation of Akt family. Also previous studies of mice showed that Akt1 were more important to cell growth, whereas Akt2 mediated glucose metabolism [36–38], this study focused on Akt1 only. Further co-immunoprecipitation analyses indicated that AR overexpression markedly suppressed the binding of MYC-ubiquitin to AKT1, whereas this effect was not significantly in AR knockdown cells (Figure 2D and 2E). AR overexpression thus might stabilized AKT1 in part through preventing proteosome-mediated degradation of ubiquitinated AKT1.
AR interacted with the kinase domain of AKT1
To further explore the molecular mechanisms of AR stabilized AKT1 and AKT2, immunoprecipitation (IP) assays were performed. In HEK293T cells, Flag-tagged AR co-precipitated with HA-tagged AKT1, using either anti-Flag or anti-HA antibody (Figure 3A). Furthermore, the E. coli expressing His-tagged AKT1 combined E. coli expressing GST-tagged AR but not the empty vector pGEX-4T1-GST (Figure 3B).
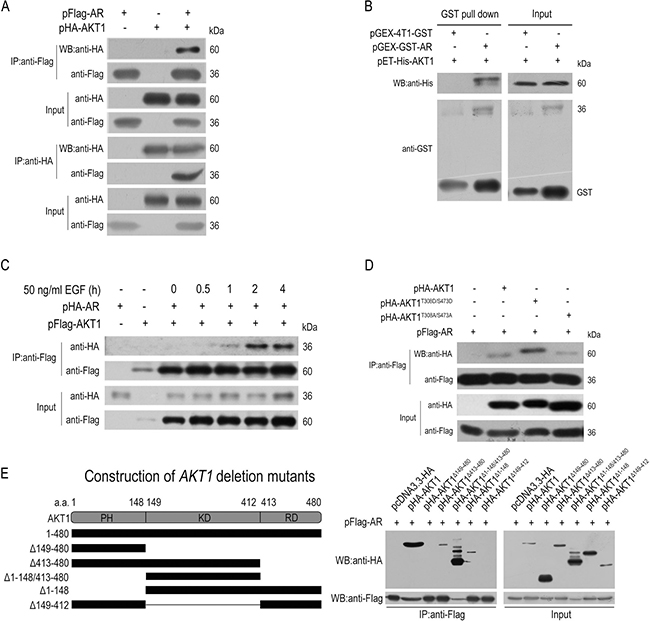
Figure 3: AKT1 interacts with AR physically. (A) Co-immunoprecipitation between the plasmid-encoded AKT1 and AR in HEK293T cells using anti-Flag or anti-HA antibodies. (B) In vitro GST pull-down of AR-AKT1 (both expressed in bacteria) complex. (C) EGF treatment time dependently enhanced the AR-AKT1 interaction. (D) Constitutively-active AKT1 (HA-AKT1T308D/S473D) had a higher affinity for AR than the WT AKT1 or constitutively inactive AKT1 (HA-AKT1T308A/S473A). (E) AR physically interacts with the kinase domain (KD) but not the PH/helix or the RD domain of AKT1.
Also by co-immunoprecipitation analyses, the AR-AKT1 interaction enhanced time-dependently by EGF treatment in HepG2 cells (Figure 3C). Moreover, AR had a higher affinity for a constitutively-active AKT1 (AKT1T308D/S473D) than either the WT AKT1 or a constitutively-inactive AKT1 (AKT1T308A/S473A) (Figure 3D).
Then, co-immunoprecipitation analyses were tested using 3 truncated mutants of AR and 5 truncated mutants of AKT1 (Supplementary Table 2) [39]. Probably due to the lack of distinctive structural domains [40, 41], three AR deletion mutants co-precipitated with the wildtype (WT) AKT1 protein, although the N-terminal deletion mutant (Flag-ARΔ1–100) had a much weaker affinity (Supplementary Figure 4). In contrast to this, HA-AKT1D413–480, HA-AKT1Δ1–148/413–480 and HA-AKT1Δ1–148 co-precipitated with the WT AR protein, but not HA-AKT1Δ149–480 and HA-AKT1Δ149–412 (Figure 3E), which indicated that the kinase domain of AKT1 alone was sufficient for the direct protein-protein interaction with AR.
AKT1 was essential for AR-induced significant alterations in AKT/mTOR signaling, lactate formation, and TNFα/IL-6 mRNA expression
To evaluate the effects of AKT1 in AR-induced hepatocarcinogenesis, transfection rescue studies were performed in HepG2 cells. In HepG2 cells, AR overexpression-induced AKT1, mTOR, HIF1a and PKM2 protein expression up-regulation (pFlag-CMV2+siCtrl transfected versus pFlag-AR+siCtrl transfected) was significantly diminished in cells co-treated with siAKT1 (pFlag-AR+siCtrl transfected versus pFlag-AR+siAKT1 co-transfected) (Figure 4A). Conversely, AR knockdown-induced AKT1, mTOR, and PKM2 down-regulation (pLV-ctrl+pcDNA3.3-HA transfected versus pLV-shAR-1+pcDNA3.3-HA transfected) was significantly restored in AKT1 co-overexpression cells (pLV-shAR-1+pcDNA3.3-HA transfected versus pLV-shAR-1+pHA-AKT1 co-transfected), although HIF1a were not altered significantly. Additionally, inactivation of AKT1 by LY294002 also reversed AR-induced mTOR, HIF1a and PKM2 up-regulation (Figure 4B).
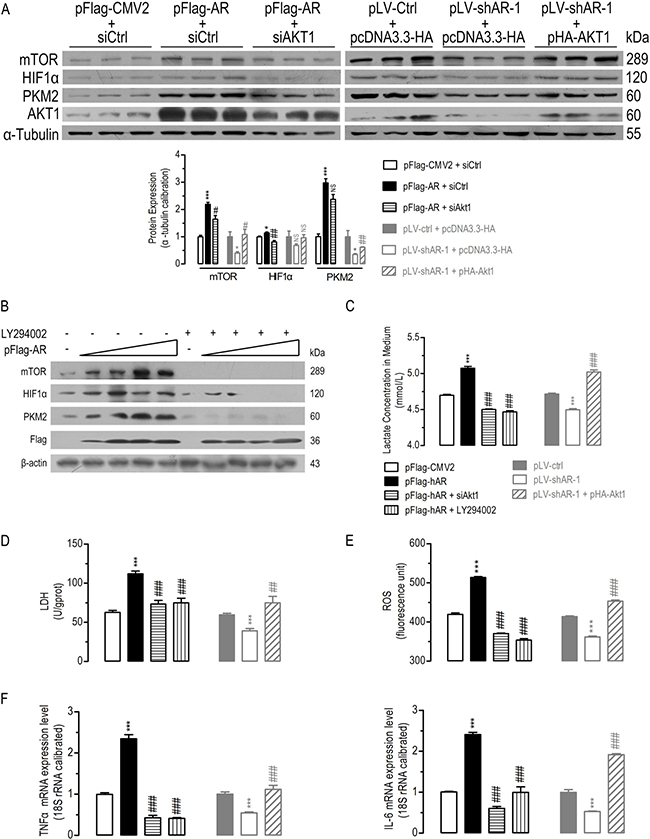
Figure 4: AKT1 was essential for AR-induced alterations in AKT/mTOR signaling, lactate formation and TNFα/IL-6 mRNA expression. AKT1 was essential for AR-induced significantly disturbed protein expression of mTOR, HIF1a, and PKM2 in HepG2 cells (A) (n = 3). Inhibition of AKT1 phosphorylation by LY294002 significantly diminished AR overexpression-induced mTOR, HIF1α, and PKM2 protein expression (B). AKT1 was essential for AR-induced significantly disturbed lactate formation (C) (n = 6), LDH activity (D) (n = 6), ROS (E) (n = 6), TNFa/IL-6 mRNA expression (F) (n = 6) in HepG2 cells. Data were expressed as the mean ± SEM. NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001 (compared to pFlag-CMV2+siCtrl or pLVctrl+pcDNA3.3-HA transfected cells); #p < 0.05; ##p < 0.01; ###p < 0.001 (compared to pFlag-AR+siCtrl or pLV-shAR-1+pcDNA3.3-HA transfected cells).
In comparison with the control cells, AR overexpression increased lactate formation (Figure 4C) and LDH activity (Figure 4D), AKT1 knockdown by siRNA (siAKT1) or inactivation by LY294002 treatment significantly diminished AR overexpression-induced lactate formation and LDH activity increasing. Meanwhile, AR knockdown suppressed lactate formation and LDH activity, but AKT1 overexpression significantly restored AR knockdown-induced lactate formation and LDH activity decreasing.
Since AR and AKT both regulated ROS and inflammatory signals [42–46], TNFα and IL-6 mRNA were analyzed by qPCR. Knockdown/inactivation AKT1 inhibited AR overexpression-induced ROS (Figure 4E) and TNFα/IL-6 mRNA expression increasing (Figure 4F). Whereas overexpression of AKT1 restored AR knockdown-induced ROS and TNFα/IL-6 mRNA expression decreasing. These in vitro experiments suggested that AKT1 was essential for AR-induced dys-regulations in AKT/mTOR signaling, metabolic reprogramming, antioxidant defense and inflammatory responses in HCC cells.
Liver-specific AR overexpression tended to promote whereas Ar deficiency tended to suppress DEN-induced HCC
To examine the effects of AR regulated hepatic Akt/mTor signaling, lactate formation, inflammatory response gene expression and liver cancer development in vivo, HCC was induced in 2-wk old WT/FVB, TG2/FVB, TG1/FVB, WT/B6 and KO/B6 [47, 48] male mice by a single injection of DEN at the dosage of 25 mg/kg body weight. In FVB mice, DEN-treated AR-overexpressing mice (TG2/FVB, TG1/FVB) had significantly higher tumor incidence (% mice with tumors > 1 μm), visible tumor number per mouse, maximal tumor size and accumulated tumor size (in diameter, mm) than the DEN-treated control mice (WT/FVB) (Figure 5), but not in body or liver weight (Supplementary Figure 5). Conversely, a significant amelioration in tumor incidence, visible tumor number per mouse, maximal tumor size and accumulated tumor size was observed in Ar deficient B6 mice (KO/B6) as compared to the control mice (WT/B6) (Figure 5), with a few minor exceptions. Together, these data suggested that liver-specific AR overexpression promote whereas Ar deficiency/knockdown suppress HCC tumorigenesis or progression.
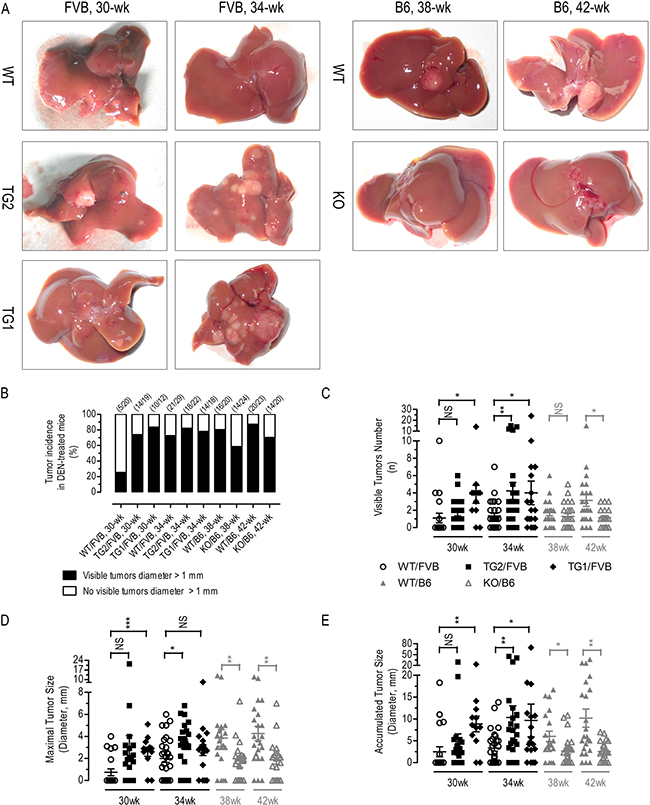
Figure 5: DEN-induced HCC in liver-specific AR overexpressing transgenics and Ar knockout mice. Typical liver photos (A) tumor incidence (B) (n = 12–29), visible tumor number (C) (n = 12–29), maximal tumor size (D) (n = 12–29), and accumulated tumor size (E) (n = 12–29) of different groups of DEN-treated mice. Numerical data were expressed as the mean ± SEM. NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001 (TG2/FVB or TG1/FVB versus WT/FVB, KO/B6 versus WT/B6).
Significant alterations in Akt/mTor signaling, lactate formation, and Tnfα/Il-6 mRNA expression in the liver tissues of AR-overexpressing transgenic and Ar knockout mice
As demonstrated by immunohistochemical (IHC) analyses, liver-specific AR overexpression significantly increased hepatic Akt1 expression, whereas Ar deficiency significantly reduced hepatic Akt1 expression in DEN-treated mice (Figure 6A). Moreover, AR overexpression increased cell proliferation marker Ki67, whereas Ki67 significantly suppressed in Ar deficient mice (Figure 6A). In AR overexpression mice exposed to DEN for 34 wk, hepatic protein levels of mouse pS473-Akt1, total Akt1/Akt2, pS256-FoxO1, mTor, Hif1α, Pkm2 and Srebp significantly increased (Figure 6B). Conversely, in Ar deficient mice exposed to DEN for 42 wk, the assayed proteins significantly reduced, except FoxO1.
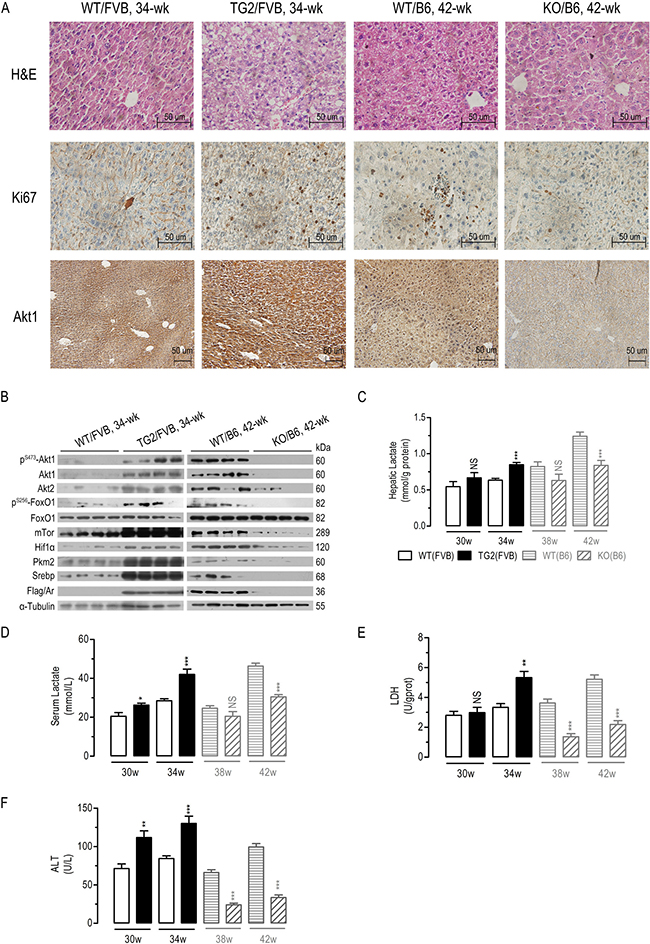
Figure 6: In vivo effects of liver-specific AR overexpression or Ar deficiency on mouse Akt/mTor signaling, LDH activity, and serum and hepatic lactate concentration. (A) Hepatic protein expression of Ki67/Akt1 in four groups of DEN-treated mice as analyzed by immunohistochemistry (A). Hepatic protein expression of Akt/mTOR signalingin four groups of DEN-treated mice (B) (n = 4). Hepatic lactate levels (C) (n = 6–8), serum lactate levels (D) (n = 6–8), hepatic LDH activity (E) (n = 6–8), serum ALT levels (F) (n = 6–8) in eight groups of DEN-treated mice. Numeric data were expressed as the mean ± SEM. NS, not significant; *p < 0.05; **p < 0.01; ***p < 0.001, compared to WT/FVB or WT/B6 respectively.
Also consistent with the in vitro studies, AR overexpression increased serum and hepatic lactate concentration, liver LDH and ALT activities, and hepatic Tnfα/Il-6 mRNA expression (Figure 6C–6F and Supplementary Figure 6), although the differences in hepatic lactate, LDH activity and Tnfa were not significantly of 30 wk. Conversely, Ar knockout decreased serum and hepatic lactate concentration, liver LDH and ALT activities, and hepatic Tnfα/Il-6 mRNA expression (Figure 6C–6F and Supplementary Figure 6), with the exception that the differences in Tnfα mRNA, serum and hepatic lactate were not significantly of 38 wk. In general, these data indicated that liver AR expression regulate Akt/mTor signaling, lactate formation, and Tnfα/Il-6 expression in vivo.
DISCUSSION
Dozens of studies have reported that abnormal AKT1 activates in diabetes, cardiovascular diseases and various cancers [49–51]. Novel protein interactions with AKT/mTOR pathway members have been commonly reported to efficiently regulate AKT1 kinase activity in cancers [39, 52]. In the present study we found that overexpressed AR interacted with AKT1 to increase AKT/mTOR signaling, which in turn promoted Warburg effects, lactate production, oxidative stress, and inflammation and thus contributed to hepatocarcinogenesis (Figures 1 and 4). A series of co-immunoprecipitation assays established a protein-protein interaction between AR and the kinase domain of AKT1 (Figure 3), leading to the stabilization AKT1 (Figure 2D and 2E) and eventually significant augmentation of AKT/mTOR signaling (Figure 2A). As a consequence of its interaction with AKT1, the overexpressed AR augmented AKT/mTOR signaling (Figures 2A, 2C and 6D) and tended to enhanced lactate formation and hepatic inflammation (Figures 4 and 6). Conversely, AR knockdown suppressed lactate formation and inflammation. In cultured HepG2 cells, we further demonstrated that AKT1 was essential for AR-induced dysregulation of AKT/mTOR signaling, metabolic reprogramming, antioxidant defense and inflammatory responses (Figure 4). We also clearly demonstrated that liver-specific AR overexpression leads to abnormal augmentation in hepatic AKT/mTOR signaling (Figure 6A) and enhanced HCC development (Figure 5). By contrast, oncogenic AKT/mTOR signaling and HCC development appears to be significantly ameliorated in mice deficient in Ar, with a few minor exceptions. Together, these data suggest that aberrantly overexpressed/over-activated hepatic AR promotes HCC development, at least in part by interacting with the oncogenic AKT1 to augment AKT/mTOR signaling.
Cancer cell metabolism is characterized by the so-called Warburg effects, the manifestations of which include enhanced glucose uptake and glycolysis, reduced oxidative phosphorylation and increased lactate secretion [53]. What might have long been overlooked, however, is the fact that the high intracellular glucose present in cancer cells very likely will trigger the overexpression of AR and/or over-activation of PP. It has been estimated that when glucose is abundant, more than 30% of glucose can be channeled into the AR/PP, which can lead to the over-production of fructose [2]. In mammalian cells, fructose also has a tendency to be converted into lipid, uric acid and lactate [54]. In this regard, it is very likely that overexpression of AR/over-activation of PP in cancer cells contribute significantly to cancer-associated metabolic reprogramming, in part by increasing synthesis of lipid, lactate and uric acid. In this investigation, we clearly demonstrated that AR overexpression promotes, and AR inhibition inhibits, lactate formation. The increase in lactate secretion from cancer cells, on the other hand, might be attributable to two distinct mechanisms: 1) high glucose directly activates AR/PP leading to the over-production of fructose and lactate; 2) overexpressed AR interacts with AKT1 to augment AKT/mTOR, HIF1a and PKM2 signaling, eventually leading to increased flux through the aerobic glycolysis to enhance lactate formation. Lactate secretion due to the over-activation of AR/PP or the augmented AKT/mTOR signaling due to AR-AKT1 interaction in cancer cells therefore might account for a significant portion of the total lactate formation, which was attributed mostly to the Warburg effects previously.
More interestingly, some reports showed that AR/AKR1B10 overexpression promotes the development of resistance against various chemotherapeutic drugs [4, 55, 56]. In addition, various studies were revealed that increased expression of AR and AKR1B10 is involved in carcinogenesis and drug resistance [10, 11, 55], and AR/AKR1B10 inhibitors could be potentially effective drugs for cancer therapeutics [55, 56]. Zopolrestat, an AR/AKR1B10 inhibitor, was found to provide additional therapeutic effects in liver cancer [57]. Epalrestat, an AKR1B1 inhibitor, significantly suppresses cancer stem cell properties, tumorigenicity, and metastasis of basal-like breast cancer cells through regulating the NF-κB pathway [58]. Consistent with previous observations, AR knockdown was found to increase susceptibility to chemotherapeutic agents, while AR expression led to tumor cell resistance to anticancer drugs (Supplementary Figure 7). Therefore, suppression of AR by inhibitors or siRNAs has the potential to serve as an adjuvant therapeutic strategy for cancers [59]. Until very recently, however, no AR inhibitor was evaluated in clinical human cancer therapy. Fidarestat, another AR inhibitor, has already been passed through the FDA’s Phase-III clinical trials and has proven safe for human use, without irreversible toxicity [4]. Thus, this drug could soon be used for various cancer therapies, though clinical studies of combination therapies using known chemotherapeutic drugs with AR inhibitors/siRNAs are needed to further assess clinical toxicity and risks.
In summary, we demonstrated in this investigation that when overexpressed in liver cells, AR may mediate over-activation of PP, causing over-production of fructose, lactate, and ROS and altered expression of inflammatory response genes. Overexpressed hepatic AR may also interact with AKT1 to augment AKT-mTOR signaling, further promoting metabolic reprogramming and dysregulation of antioxidant defense and inflammatory responses. Over-activation of the polyol pathway and AR-augmented abnormal AKT-mTOR signaling may act synergistically to promote tumorigenesis in the liver. More importantly, interfering with AR/PP expression/activation may be an effective adjuvant strategy for clinical cancer therapy.
MATERIALS AND METHODS
Clinical mRNA expression analysis
Clinical Akr1b1 or Akr1b10 mRNA expression were analysed in cirrhosis and HCC on www.oncomine.org. Searching “Akr1b1” or “Akr1b10” gene expression in Mas Liver Database (GSE14323) with the following parameters “clinical specimen” in sample type, “mRNA” in Data type, “Liver cancer” in cancer type.
Liver-specific human AR overexpressing transgenics and Ar deficient knockout mice
The liver-specific human AR-overexpressing transgenic FVB (TG1/FVB and TG2/FVB) mice were generated by Dan Song at Yun-qing Yang’s lab of Xiamen University. The liver-specific human AR-overexpressing transgenic FVB (TG1/FVB and TG2/FVB) mice, Ar knockout C57BL/6 mice (KO/B6) and their controls (WT/FVB or WT/B6) were also kind gifts from Prof. Yun-qing Yang and only used for this project.
Mice were bred and maintained under a standard 12–12 h light-dark cycle, and were fed standard rodent chow and water ad libitum, and housed in the barrier facility of the Laboratory Animal Center, Xiamen University. All animal experiments were performed according to the protocols approved by the Institutional Animal Use and Care Committee of Xiamen University, China.
Hepatocarcinoma (HCC) induction in mice
For the induction of HCC, the male mice of liver-specific human AR-overexpressing transgenic FVB (TG1/FVB and TG2/FVB), Ar knockout C57BL/6 (KO/B6) and their controls (WT/FVB or WT/B6) were injected intraperitoneally with diethylnitrosamine (DEN) at 25 mg/kg body weight at the age of 2 wk (Cat# 049k1613v, Sigma-Aldrich, St. Louis, MO, USA) [60]. DEN-treated TG/FVB and WT/FVB male mice were sacrificed either 30 or 34 wk of age, while DEN-treated KO/B6 and WT/B6 male mice were sacrificed either 38 or 42 wk of age. Liver lobes were photographed and tumors > 1 mm in diameter on liver surface were counted. The diameters were measured using a vernier caliper. Liver tissues were also dissected for further analyses. The phenotype data of two independent DEN-induced HCC mice models were analysed together in Figure 5. All experimental procedures involving animals were performed in accordance with the animal protocols approved by the Laboratory Animal Center of Xiamen University.
Other procedures
All of the other procedures are established standard techniques and are described in the Supplementary Files.
Statistical analyses
All statistical analyses were performed with the GraphPad Prism 5.0 software. Values are expressed as the means ± SEM. The Student’s t-test (two-tailed) for pair-wise comparisons. A probability value (p) < 0.05 was considered to be significant, those < 0.01 or < 0.001 more so.
Abbreviations
AKR1B10, aldo-keto reductase family 1B10, AR-like-1; AKT, protein kinase B, PKB; ALT, alanine aminotransferase; AMPK, AMP-activated kinase; AR, aldose reductase, aldo-keto reductase family 1B1; DEN, diethylnitrosamine; EGF, epidermal growth factor; FOXO1, forkhead box protein O1; HIF1α, hypoxia-inducible factor-1alpha; IL-6, interleukin 6; LDH, lactate dehydrogenase; mTOR, mechanistic target of rapamycin; PI3K, phosphatidylinositide 3-kinase; PKM2, pyruvate kinase isozymes M2; TNFα, tumor necrosis factor alpha.
ACKNOWLEDGMENTS
We would like to thank Prof. Sheng-Cai Lin for providing series plasmids of AKT1 and AKT2; Prof. Yun-Qing Yang for providing the liver-specific human AR-overexpressing and Ar knockout transgenic mice.
CONFLICTS OF INTEREST
The authors have no financial conflicts of interest.
FUNDING
This work was supported by grants from the National Natural Science Foundation of China [grant number 81602563, 81673661, 81670936, 81672871 and 81602148 to Guo-Dong Ye, Shu-Yu Yang, Cheng-Fu Cai, Qi-Cong Luo, Wang-Yu Cai], the project of Scientific and Technical department of Fujian Province [grant numbers 2016J01632 to Cheng-Fu Cai]. The project was also supported through a grant from the Open Research Fund of State Key Laboratory of Cellular Stress Biology, Xiamen University.
REFERENCES
1. Penning TM, Drury JE. Human aldo-keto reductases: Function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys. 2007; 464:241–50.
2. Yabe-Nishimura C. Aldose reductase in glucose toxicity: a potential target for the prevention of diabetic complications. Pharmacol Rev. 1998; 50:21–33.
3. Samanta BK, Chandra NC, Ghosh S, Mukherjee KL. Aldose metabolism in developing human fetal brain and liver. Experientia. 1984; 40:1420–22.
4. Tammali R, Srivastava SK, Ramana KV. Targeting aldose reductase for the treatment of cancer. Curr Cancer Drug Targets. 2011; 11:560–71.
5. Cao D, Fan ST, Chung SS. Identification and characterization of a novel human aldose reductase-like gene. J Biol Chem. 1998; 273:11429–35.
6. Fukumoto S, Yamauchi N, Moriguchi H, Hippo Y, Watanabe A, Shibahara J, Taniguchi H, Ishikawa S, Ito H, Yamamoto S, Iwanari H, Hironaka M, Ishikawa Y, et al. Overexpression of the aldo-keto reductase family protein AKR1B10 is highly correlated with smokers’ non-small cell lung carcinomas. Clin Cancer Res. 2005; 11:1776–85.
7. Saraswat M, Mrudula T, Kumar PU, Suneetha A, Rao Rao TS, Srinivasulu M, Reddy B. Overexpression of aldose reductase in human cancer tissues. Med Sci Monit. 2006; 12:CR525–29.
8. Scuric Z, Stain SC, Anderson WF, Hwang JJ. New member of aldose reductase family proteins overexpressed in human hepatocellular carcinoma. Hepatology. 1998; 27:943–50.
9. Laffin B, Petrash JM. Expression of the Aldo-Ketoreductases AKR1B1 and AKR1B10 in Human Cancers. Front Pharmacol. 2012; 3:104.
10. Jin J, Liao W, Yao W, Zhu R, Li Y, He S. Aldo-keto Reductase Family 1 Member B 10 Mediates Liver Cancer Cell Proliferation through Sphingosine-1-Phosphate. Sci Rep. 2016; 6:22746.
11. Ha SY, Song DH, Lee JJ, Lee HW, Cho SY, Park CK. High expression of aldo-keto reductase 1B10 is an independent predictor of favorable prognosis in patients with hepatocellular carcinoma. Gut Liver. 2014; 8:648–54.
12. Zeindl-Eberhart E, Haraida S, Liebmann S, Jungblut PR, Lamer S, Mayer D, Jäger G, Chung S, Rabes HM. Detection and identification of tumor-associated protein variants in human hepatocellular carcinomas. Hepatology. 2004; 39:540–49.
13. Obrosova IG. Increased sorbitol pathway activity generates oxidative stress in tissue sites for diabetic complications. Antioxid Redox Signal. 2005; 7:1543–52.
14. Chatzopoulou M, Pegklidou K, Papastavrou N, Demopoulos VJ. Development of aldose reductase inhibitors for the treatment of inflammatory disorders. Expert Opin Drug Discov. 2013; 8:1365–80.
15. Hanczko R, Fernandez DR, Doherty E, Qian Y, Vas G, Niland B, Telarico T, Garba A, Banerjee S, Middleton FA, Barrett D, Barcza M, Banki K, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest. 2009; 119:1546–57.
16. Ibrahim SS, Nassar NN. Diallyl sulfide protects against N-nitrosodiethylamine-induced liver tumorigenesis: role of aldose reductase. World J Gastroenterol. 2008; 14:6145–53.
17. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33.
18. Qiu L, Wu X, Chau JF, Szeto IY, Tam WY, Guo Z, Chung SK, Oates PJ, Chung SS, Yang JY. Aldose reductase regulates hepatic peroxisome proliferator-activated receptor alpha phosphorylation and activity to impact lipid homeostasis. J Biol Chem. 2008; 283:17175–83.
19. Qiu L, Lin J, Xu F, Gao Y, Zhang C, Liu Y, Luo Y, Yang JY. Inhibition of aldose reductase activates hepatic peroxisome proliferator-activated receptor-alpha and ameliorates hepatosteatosis in diabetic db/db mice. Exp Diabetes Res. 2012; 2012:789730.
20. Qiu L, Lin J, Ying M, Chen W, Yang J, Deng T, Chen J, Shi D, Yang JY. Aldose reductase is involved in the development of murine diet-induced nonalcoholic steatohepatitis. PLoS One. 2013; 8:e73591.
21. Lanaspa MA, Ishimoto T, Li N, Cicerchi C, Orlicky DJ, Ruzycki P, Rivard C, Inaba S, Roncal-Jimenez CA, Bales ES, Diggle CP, Asipu A, Petrash JM, et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat Commun. 2013; 4:2434.
22. Enzmann H, Ohlhauser D, Dettler T, Bannasch P. Enhancement of hepatocarcinogenesis in rats by dietary fructose. Carcinogenesis. 1989; 10:1247–52.
23. Liu H, Heaney AP. Refined fructose and cancer. Expert Opin Ther Targets. 2011; 15:1049–59.
24. Monzavi-Karbassi B, Hine RJ, Stanley JS, Ramani VP, Carcel-Trullols J, Whitehead TL, Kelly T, Siegel ER, Artaud C, Shaaf S, Saha R, Jousheghany F, Henry-Tillman R, Kieber-Emmons T. Fructose as a carbon source induces an aggressive phenotype in MDA-MB-468 breast tumor cells. Int J Oncol. 2010; 37:615–22.
25. Port AM, Ruth MR, Istfan NW. Fructose consumption and cancer: is there a connection? Curr Opin Endocrinol Diabetes Obes. 2012; 19:367–74.
26. Mas VR, Maluf DG, Archer KJ, Yanek K, Kong X, Kulik L, Freise CE, Olthoff KM, Ghobrial RM, McIver P, Fisher R. Genes involved in viral carcinogenesis and tumor initiation in hepatitis C virus-induced hepatocellular carcinoma. Mol Med. 2009; 15:85–94.
27. Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, Fiel I, Thung S, Mazzaferro V, Bruix J, Bottinger E, Friedman S, Waxman S, Llovet JM. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology. 2007; 45:938–47.
28. Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ, Lee H, Kim K, Kim J, An IS, Kim J, Um HD, Park IC, Lee SJ, et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res. 2012; 22:873–85.
29. Ramana KV, Tammali R, Srivastava SK. Inhibition of aldose reductase prevents growth factor-induced G1-S phase transition through the AKT/phosphoinositide 3-kinase/E2F-1 pathway in human colon cancer cells. Mol Cancer Ther. 2010; 9:813–24.
30. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. 2011; 108:4129–34.
31. Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, Khuri FR, Sun SY. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components in the mTOR axis and induces autophagy. Cancer Res. 2009; 69:8967–76.
32. Huang D, Li T, Li X, Zhang L, Sun L, He X, Zhong X, Jia D, Song L, Semenza GL, Gao P, Zhang H. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Reports. 2014; 8:1930–42.
33. Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR, Kaestner KH, Sabatini DM, Birnbaum MJ. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell Metab. 2011; 14:516–27.
34. Grassi ML, Palma CS, Thomé CH, Lanfredi GP, Poersch A, Faça VM. Proteomic analysis of ovarian cancer cells during epithelial-mesenchymal transition (EMT) induced by epidermal growth factor (EGF) reveals mechanisms of cell cycle control. J Proteomics. 2017; 151:2–11.
35. Chen C, Liang F, Chen B, Sun Z, Xue T, Yang R, Luo D. Identification of demethylincisterol A3 as a selective inhibitor of protein tyrosine phosphatase Shp2. Eur J Pharmacol. 2017; 795:124–33.
36. Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans. 2007; 35:231–35.
37. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001; 276:38349–52.
38. Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB β). Science. 2001; 292:1728–31.
39. Wang YW, Lin KT, Chen SC, Gu DL, Chen CF, Tu PH, Jou YS. Overexpressed-eIF3I interacted and activated oncogenic Akt1 is a theranostic target in human hepatocellular carcinoma. Hepatology. 2013; 58:239–50.
40. Howard EI, Sanishvili R, Cachau RE, Mitschler A, Chevrier B, Barth P, Lamour V, Van Zandt M, Sibley E, Bon C, Moras D, Schneider TR, Joachimiak A, Podjarny A. Ultrahigh resolution drug design I: details of interactions in human aldose reductase-inhibitor complex at 0.66 A. Proteins. 2004; 55:792–804.
41. Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008; 40:553–624.
42. Tammali R, Ramana KV, Srivastava SK. Aldose reductase regulates TNF-alpha-induced PGE2 production in human colon cancer cells. Cancer Lett. 2007; 252:299–306.
43. Yadav UC, Ramana KV, Aguilera-Aguirre L, Boldogh I, Boulares HA, Srivastava SK. Inhibition of aldose reductase prevents experimental allergic airway inflammation in mice. PLoS One. 2009; 4:e6535.
44. Ramana KV, Fadl AA, Tammali R, Reddy AB, Chopra AK, Srivastava SK. Aldose reductase mediates the lipopolysaccharide-induced release of inflammatory mediators in RAW264.7 murine macrophages. J Biol Chem. 2006; 281:33019–29.
45. McNamara CR, Ahuja R, Osafo-Addo AD, Barrows D, Kettenbach A, Skidan I, Teng X, Cuny GD, Gerber S, Degterev A. Akt Regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PLoS One. 2013; 8:e56576.
46. Gu FM, Li QL, Gao Q, Jiang JH, Zhu K, Huang XY, Pan JF, Yan J, Hu JH, Wang Z, Dai Z, Fan J, Zhou J. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer. 2011; 10:150.
47. Liu H, Luo Y, Zhang T, Zhang Y, Wu Q, Yuan L, Chung SS, Oates PJ, Yang JY. Genetic deficiency of aldose reductase counteracts the development of diabetic nephropathy in C57BL/6 mice. Diabetologia. 2011; 54:1242–51.
48. Yang JY, Tam WY, Tam S, Guo H, Wu X, Li G, Chau JF, Klein JD, Chung SK, Sands JM, Chung SS. Genetic restoration of aldose reductase to the collecting tubules restores maturation of the urine concentrating mechanism. Am J Physiol Renal Physiol. 2006; 291:F186–95.
49. Hers I, Vincent EE, Tavaré JM. Akt signalling in health and disease. Cell Signal. 2011; 23:1515–27.
50. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005; 4:988–1004.
51. Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, Delogu S, Zimmermann A, Ericsson J, Brozzetti S, Staniscia T, Chen X, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011; 140:1071–83.
52. Franke TF. Intracellular signaling by Akt: bound to be specific. Sci Signal. 2008; 1:pe29.
53. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011; 11:325–37.
54. Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr. 2002; 76:911–22.
55. Matsunaga T, Suzuki A, Kezuka C, Okumura N, Iguchi K, Inoue I, Soda M, Endo S, El-Kabbani O, Hara A, Ikari A. Aldo-keto reductase 1B10 promotes development of cisplatin resistance in gastrointestinal cancer cells through down-regulating peroxisome proliferator-activated receptor-γ-dependent mechanism. Chem Biol Interact. 2016; 256:142–53.
56. Zemanova L, Hofman J, Novotna E, Musilek K, Lundova T, Havrankova J, Hostalkova A, Chlebek J, Cahlikova L, Wsol V. Flavones Inhibit the Activity of AKR1B10, a Promising Therapeutic Target for Cancer Treatment. J Nat Prod. 2015; 78:2666–74.
57. Moriguchi H, Zhang Y, Mihara M, Sato C. A therapeutic method for the direct reprogramming of human liver cancer cells with only chemicals. Sci Rep. 2012; 2:280.
58. Wu X, Li X, Fu Q, Cao Q, Chen X, Wang M, Yu J, Long J, Yao J, Liu H, Wang D, Liao R, Dong C. AKR1B1 promotes basal-like breast cancer progression by a positive feedback loop that activates the EMT program. J Exp Med. 2017; 214:1065–79.
59. Lee EK, Regenold WT, Shapiro P. Inhibition of aldose reductase enhances HeLa cell sensitivity to chemotherapeutic drugs and involves activation of extracellular signal-regulated kinases. Anticancer Drugs. 2002; 13:859–68.
60. Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, Karin M. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007; 317:121–24.