
INTRODUCTION
DNA mismatch repair (MMR) is pivotal in maintaining genomic stability in both prokaryotes and eukaryotes. There are nine mammalian MMR genes, including E. coli MutS homologues (MSH6), MutL homologues (MLH1, MLH3) and yeast post-meiotic segregation homologues (PMS1 and PMS2) [1–5]. In humans, MLH1, MSH2, MSH6 and PMS2 mutations increase susceptibility to multiple malignancies, most notably colorectal and endometrial cancers [1]. Mechanistically, mammalian MSH2-MSH6 and MSH2-MSH3 complexes form ‘sliding clamps’ that scan genomic DNA for sequence mismatches [6–9]. MSH2-MSH6 complex binds to single base-base mismatches and small insertions/deletion loops (IDLs), whereas MSH2-MSH3 complex is involved in the repair of larger IDLs [1] (IDL repair deficiency is commonly referred to as Microsatellite Instability, MSI). The MSH proteins interact with multiple proteins including the mammalian E.coli MutL homologues (MLH) and yeast post-meiotic segregation (PMS) homologue proteins, which have significant amino acid identity and structural similarity to the MLH proteins, among others (reviewed in [1]).
The MLH1-PMS2 complex is the primary MutL heterodimer that interacts with both MSH2-MSH6 and MSH2-MSH3 complexes, and is the only heterodimer that participates in repair of single base substitutions. There is partial redundancy in the functions of MLH1-PMS2 and MLH1-MLH3 complexes in IDL repair and DNA damage response [10, 11]. In meiosis, both MLH1-PMS2 and MLH1-MLH3 complexes promote recombination-mediated cross-over events [12–14], and MLH1-PMS2 also plays roles in antibody class switch recombination [15]. A third MutL complex, MLH1-PMS1, has also been reported, but its role in mammalian MMR is yet to be clearly defined.
HR is a critical pathway for repair of DNA double-strand breaks (DSBs). Briefly, in mammals recA-like protein (Rad51) forms complexes with single-stranded DNA (ssDNA) strands. Rad51-ssDNA filaments interact with double stranded DNA (dsDNA) and pair when homologous sequence contacts are made. Subsequently, strand exchange occurs and a hybrid dsDNA/ssDNA complex called a D-loop is formed that is processed by multiple HR sub-pathways, including double-strand break repair (DSBR), synthesis dependent strand annealing (SDSA) and break-induced replication (BIR) [16]. DSBR, SDSA and BIR all produce copy number-neutral HR repair. In contrast, when repetitive sequences flank a DSB, the mechanism of single strand annealing (SSA) can also be used for DSB repair [17]. SSA involves direct annealing between misaligned homologous repeat sequences and ssDNA recession. Consequently, SSA at repetitive homologous motifs causes deletion of one repeat sequence and the intervening sequences between repeats, resulting in deletion copy number alterations (CNAs). When flanked by direct repeats (DRs) or inverted repeats, DSBs stimulate HR and SSA several hundred fold [18].
Relatively small degrees of sequence heterogeneity can alter the frequency of DSB-induced HR and SSA in mammalian cells. For example, HR is decreased by >80% for DRs that are 1.2% divergent [19]. When the donor and recipient sequences are highly similar but not identical (homeologous), the repair process is referred as homeologous recombination (HeR) [16]. MMR proteins play important roles in preventing HeR and SSA. Specifically, MMR recognizes and repairs mismatches in DNA heteroduplex regions (regions that are formed by annealing single strands from different sources), removes non-homologous tails during HeR repair and disrupts HeR repair via heteroduplex rejection or anti-recombination (recently reviewed in [16]).
MMR suppression of HeR was first observed in bacteria [20, 21] where it prevents rotation of mismatched DNA strands [22], and was subsequently found to occur in eukaryotes [23]. In the yeast Saccharomyces cerevisiae, functional mutations of Msh2p, Msh6p, Mlh1p or Pms1p (orthologues of mammalian PMS2) increase HeR between integrated reporters containing DRs with mismatch heterology [23–26], yet roles for Mlh2p/Mlh3p are poorly characterized. Additionally, individual functional mutations of Msh2p, Mlh1p or compound mutations of the three Mlh1p heterodimeric partners, Pms1p/Mlh2p/Mlh3p, increases SSA [27]. In mammalian cells, MutS homologues MSH2 and MSH6 can suppress HeR [24, 28–31], but the role of mammalian MutL homologues in mechanisms inhibiting HeR is less well characterized. Furthermore, whether or not the overall increased HeR and SSA rates from loss of MMR contribute to tumorigenesis is poorly understood.
In tumors, DSBs also occur frequently at chromosomal fragile sites (CFS). CFS are AT-rich repetitive sequences that are difficult-to-replicate during mitosis and manifest as gaps flanked by DSBs in different tumors [32–35]. Fragile sites FRA16D and FRA3B have increased DSB rates and are the most common CFS reported in tumors [36–39]. FRA3B and FRA16D are evolutionarily conserved across eukaryotes [36] and encode the tumor suppressors
FHIT and WWOX, respectively. Fragile histidine triad protein (FHIT) (also called bis(5′-adenosyl)-triphosphatase) spans 10 exons over a 1Mb region at fragile site FRA3B. FHIT is a member of histidine triad subfamily of nucleotide binding proteins. FHIT is a DNA hydrolase that metabolizes substrate AP3A (diadenosine 5-prime,5-triple prime-P(1),P(3)-triphosphate), which promotes DNA replication and inhibits stress response signaling [40]. CRCs and gastric carcinomas with MMR deficiency frequently also carry large deletions in FHIT [41]. FHIT deletions also frequently occur in lung cancer [36, 42] Fhit knockout mice develop gastric carcinomas and skin cancers [43, 44].
WWOX is a WW domain-containing oxidoreductase at FRA16D. WWOX contains 9 exons and spans 1.2 Mb [43]. Genomic deletions and other structural variants inactivating WWOX occur in almost one-third of solid tumors [45, 46]. WWOX suppresses TGFβ/SMAD signaling [46], and WWOX mouse mutants promote mammary tumor growth [36]. While loss of MMR in CRCs has been associated with increased frequency of FHIT [47] and WWOX [48] deletions, inactivation of FHIT and WWOX at FRA3B and FRA16D CFS has not been linked to HeR. Here, we report that mammalian MutL homologs MLH1, PMS2 and MLH3 participate in suppression of HeR and SSA. In addition to GFP reporter studies, we computationally identified 6,848 paired DRs with 97%–99.9% sequence identity mapping to unique locations in the mouse genome as substrates for endogenous genomic HeR and SSA. Confirming reporter gene studies in yeast, we demonstrate that mutation of Mlh1, Pms2, Mlh3 or Mlh3/Pms2 increases HeR in MEF DR sequences. Furthermore, for the 1–12 heterologous bases in each paired DR, we show that >94% of base substitution mutations are consistent with gene conversion (where a mismatched base in one direct repeat motif is replaced with its respective paired direct repeat heterologous base). Next, to assess SSA, we used gamma-irradiation to promote endogenous DSBs and show that Mlh1, Pms2 and Mlh3 mutations suppress CNAs, including recurrent CNA hotspots that we previously identified in mouse dMMR intestinal tumors. Finally, we analyze data from dMMR and MMR proficient (pMMR) CRCs with matched normal tissues from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov). These studies confirm high rates of human endogenous paired DR HeR and reveal that the intervening sequences between paired DRs are deleted more frequently in dMMR vs pMMR CRCs. Specifically, these dMMR recurrent CNA deletions include hotspots at chromosomal fragile sites FRA3B and FRA16D, which have flanking DR and IR sequences and high rates of spontaneous DSBs in CRCs. Overall, these data provide insights into mammalian MutL homolog inhibition of HeR and SSA and are consistent with a potential role for dMMR inhibition of homeologous recombination in tumor suppression.
RESULTS
Mlh1, Pms2 and Mlh3 suppress direct repeat motif reporter gene homeologous recombination
To understand the role of mammalian MutL homologues in HR and HeR, we modified the pDR-GFP homologous recombination reporter by introducing eight closely spaced mutations in GFP to monitor homeologous recombination, similar to neomycin-based reporters previously used to study the role of mammalian MSH2 in HeR [28] (Supplementary Figure 1). These reporters have 5′ and 3′ overlapping fragments of GFP in the same orientation with intervening puromycin selection cassette and a double stranded nuclease (I-SceI) binding site between the direct repeats. DR-GFP has 100% identity between overlapping sequences and DR-GFP8mu has 8 point mutations (1.2% heterology). Upon transient expression of I-SceI, a double strand break is induced in cells. DR-GFP and DR-GFP8mu (or pneo-Wt and pneo-8 mu) can be repaired by HR/HeR or SSA and scored as GFP+ or GFP- expressing cells by flow cytometry or neomycin positive cells in colony selection assay. We used these reporters for exploratory experiments in wild-type (Wt), Mlh1-/-, Pms2-/-, Mlh3-/-, and Pms2-/-;Mlh3-/- mouse embryonic fibroblasts (MEFs) matched for passage number.
In Wt MEFs transfected with I-SceI, comparisons of DR-GFP and DR-GFP8mu stable transfectants showed that 1.2% heterology suppressed HeR vs HR by 74 ± 3%, whereas MLH1-/- MEFs showed only 3 ± 4% suppression (Supplementary Figure 2) (P = 0.005, ANOVA followed by Dunnet’s test). Further experiments in Mlh3-/-, Pms2-/- and Pms2-/-;Mlh3-/- MEFs showed 77.5 ± 0.5%, 60 ± 5% and 13.5 ± 0.5% HeR suppression relative to HR (all p < 0.001, ANOVA followed by Dunnet’s test). These studies are consistent with a potential role for mouse Mlh1 in HeR suppression. Similar to previous findings in IDL repair and DNA damage response, these data also are consistent with partially redundant roles for PMS2 and MLH3 in HeR suppression, as results in Mlh1-/- MEFs were not significantly different from those in Mlh3-/-;Pms2-/- MEFs (P = 0.74, ANOVA followed by Dunnet’s test).
Identification of paired direct repeats in the mouse genome
To understand the roles of MutL homologues in HeR suppression of endogenous direct repeat sequences, we computationally identified paired DR motifs in the mouse genome with uniquely mapping locations and characteristics similar to DR-GFP8mu and DR-Neomut8 reporters. Using the Vmatch program [49], we identified 7,863 mouse DR pairs with the following parameters: 5′ and 3′ DR motifs each > 480 bp in the same orientation with 1–12 mismatched pairs (97%–99.9% identity) and intervening sequences ranging from 500bp to 50 kb (Supplementary Figure 3 and Supplementary Table 1). The average length of paired DRs is 720bp, ranging from 500 bp to 22,359 bp (chr5:105039588-105061946 and 105092514-105114872). The average intervening sequence between paired DRs is 2,157 bp, with the largest 49,972 bp (chr15:74830812-74880784). The set of DRs with 97%–99.9% identity covers 5,035,586bp in the mouse genome. There are 1,031 DRs that overlap with protein coding exons. Chromosome 7 has the highest density of DRs (734/Mbp), while chromosome 16 has the lowest (79/Mbp).
Mouse genome paired direct repeats have high rates of endogenous homeologous recombination and SSA
DSBs that occur between DRs stimulate both HR and SSA [50]. To understand the role of individual MutL homologues in suppression of HeR and SSA, we treated MEFs with γ-irradiation and cultured for two additional passages. Subsequently, we performed targeted capture of DNA from control and irradiated MEFs for the DR sequences from Mlh1-/-, Pms2-/-, Mlh3-/-, Mlh3-/-;Pms2-/- and Wt MEFs that are all derived from C57B/L6 genetic background and matched for passage number (n = 25). Targeted DR sequences were then analyzed on an Illumina HiSeq 2500. After mapping sequence reads to mouse genome (mm9), and filtering DR reads for mapping quality score >23 and sequence read depth >10 reads, we successfully obtained sequence data for 5,119 paired DR for further analysis.
We compared log-transformed and normalized mutation rates for each MEF genotype, both control and irradiated using multiple linear regression. The analysis revealed no systematic difference in mutation rates with irradiation induced DSBs (P > 0.15; ANOVA followed by Dunnet’s test). Compared with Wt MEF DRs, Mlh3-/-, Pms2-/-, Mlh3-/-;Pms2-/- and Mlh1-/- MEF DRs all had significantly higher mutation rates (all P < 0.001; ANOVA followed by Dunnet’s test) (Table 1).
Table 1: DR mutation rates and homologous recombination in dMMR MEFs
MEF Phenotype |
Overall DR mutations |
Mutations at specific DR sites |
Mutations outside specific DR sites |
GC consistent mutations at specific DR sites |
GC inconsistent mutation at specific DR sites |
% of GC-consistent mutations at specific DR sites |
GC consistent mutations at two contiguous DR sites |
|---|---|---|---|---|---|---|---|
Wt |
5.9 |
118.7 |
4.1 |
115.8 |
2.9 |
97.55686605 |
8.7 |
Irradiated Wt |
7.1 |
99.9 |
5.6 |
96.9 |
2.9 |
96.996997 |
8.8 |
Mlh3−/− |
41.6 |
1000.4 |
25.9 |
961.3 |
39.1 |
96.09156337 |
58.7 |
Irradiated Mlh3−/− |
42.5 |
1015.1 |
26.7 |
976.2 |
38.9 |
96.16786523 |
55.1 |
Pms2 −/− |
43.1 |
959.5 |
28 |
907.2 |
52.3 |
94.5492444 |
64.6 |
Irradiated Pms2 −/− |
42.4 |
953.4 |
27.5 |
904.1 |
49.4 |
94.82903293 |
58.6 |
Mlh3 −/− ; Pms2 −/− |
44.1 |
1021.7 |
28.1 |
983 |
38.7 |
96.21219536 |
67.7 |
Irradiated Mlh3 −/− ; Pms2 −/− |
45.1 |
1040 |
28.9 |
997.9 |
42.1 |
95.95192308 |
77.8 |
Mlh1 −/− |
61.2 |
1370.3 |
40 |
1306.8 |
63.5 |
95.36597825 |
118 |
Irradiated Mlh1 −/− |
67.1 |
1574.1 |
42.5 |
1523.5 |
50.7 |
96.78546471 |
136.2 |
Note: For all above, mutation rates were calculated per 10000 bp.
Next, we separately compared mutations at heterologous bases that differed between 5′ and 3′ regions of paired DRs (HeR-BP) and surrounding bases (non-HeR-BP) in DRs with no CNAs. HeR-BPs in DR sequences had significantly higher mutation rates than surrounding bases (Table 1) ranging from 18 to 40 fold higher respectively (all P < 0.001; ANOVA followed by Dunnet’s Test). In both Wt and dMMR MEFs, for mutations occurring at HeR-BPs, more than 94% of changes were at the corresponding base in the paired DR (e.g. if paired 5′ and 3′ DR HeR-BP sequences are gggCtaa and gggTtaa respectively, the corresponding heterologous mutation in the 3′DR consistent with gene conversion would be gggCtaa) (Figure 1).

Figure 1: Overview of mouse genome direct repeat sequences. (A) Schematic illustration of part of a 5′ and 3′ paired direct repeat (DR) sequence with 3 non-identical basepairs that can be monitored for HeR. Non-identical basepairs are shown in red and green. (B) Distribution of 5,119 paired DRs in the mouse genome. Red indicates location of a paired DR. Also see Supplementary Figure 3 for the distribution of DRs for each chromosome for mouse and human genomes. Figure was generated using PhenoGram1 [73].
The percentage of DR HeR-BP mutations consistent with gene conversion is significantly higher than chance (Table 1; Supplementary Table 1) for all MEF genotypes analyzed. This observation was consistent in both 5′ to 3′ and 3′ to 5′ direction paired DR sequence gene conversions (all P < 0.001, Chi-square). Similarly, for longer gene conversion tracts that include two consecutive heterologous bases with mutations consistent with gene conversion, we observed that Mlh3, Pms2, Mlh1 or combined Mlh3/Pms2 mutations also significantly increased the number of tracts vs Wt MEFs (Table 1). Overall, among the universe of mouse genome DRs, mutation of Mlh1, Pms2, Mlh3 or Mlh3/Pms2 significantly increased HeR-BP mutation rates consistent with gene conversion for mouse genome paired DRs vs Wt MEFs (all P<0.001, ANOVA followed by Dunnet’s Test) (Table 1). In conclusion, these data are consistent with high rates of endogenous gene conversion in mouse genome paired DR sequences in both Wt and dMMR MEFs. Individual functional mutations of Mlh3, Pms2, Mlh3 and Pms2 or Mlh1 all further increased rates of mutations significantly, consistent with gene conversion in DRs at HeR-BP, as well as overall mutation rates in non-HeR-BP. Mutation rates at HeR-BP were significantly higher than non-HeR-BP bases in DR sequences. Mlh1 mutation caused the greatest increase in HeR-BP and non-HeR-BP, but individual Mlh3, Pms2 or combined Mlh3 and Pms2 mutations resulted in similar increases (Table 1).
Increased DR copy number alterations caused by γ-irradiation induced DSBs in Mlh1, Pms2 and Mlh3 mutant MEFs
Previously, we identified dMMR enriched recurrent CNAs in mouse Mlh3-/-;Pms2-/- intestinal tumors on chromosomes 7 and 12, which were not present in paired normal mucosa [10]. To understand the role of different mammalian MutL homologues in suppression of mouse genomic DR SSA and CNA generation, we compared paired DR sequence read depth of control to γ-irradiated MEFs. Mlh3-/- and Pms2-/- MEFs had significantly increased DR CNA rates, predominantly deletions, vs Wt MEFs (P = 1.08 × 10-12 and P = 0.0203; Chi-square), as did Mlh3-/-Pms2-/- and Mlh1-/- MEFs (P = 5.91 × 10-13 and 2.22 × 10-115) (Figure 2; Supplementary Table 3). Importantly, Mlh1-/- MEFs had overall increased CNA rates vs all other MutL deficient MEFs tested, including Mlh3-/-;Pms2-/- (all p < 1.09 × 10-6; ANOVA followed by Dunnet’s Test). Strikingly, shared DR deletion hotspots were observed on mouse chromosomes 6 and 7 (chr6: 89127070-114500739 and chr7: 3957350-3958737; Figure 2). The chromosome 6 CNA deletion hotspot was observed in Mlh3-/-, Mlh3-/-;Pms2-/- and Mlh1-/- MEFs, but not Pms2 mutants, whereas the chromosome 7 CNA deletion cluster hotspot was observed in all MutL mutant MEFs tested (Figure 2). Additionally, in Mlh1-/- MEFs, a DR CNA amplification hotspot was observed on chromosome 12 (chr12: 114392088-117316196) (Figure 2). Cross-comparison of these data with previous genome-wide array comparative genomic hybridization (aCGH) studies of dMMR Mlh3-/-;Pms2-/- intestinal tumors vs paired normal mucosa revealed that the observed irradiated MEF CNA chromosome 7 and chromosome 12 hotspots overlapped with previously observed hotspot regions in Mlh3-/-;Pms2-/- intestinal tumors [10].
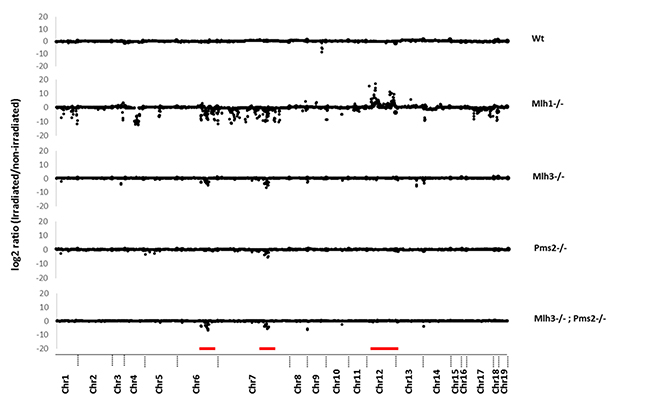
Figure 2: Copy number alterations in direct repeat motifs after irradiation induced double strand breaks. Y-axis shows log2 ratio of read depth comparing pre- and post-irradiated DRs. X-axis shows chromosomal location of each DR. MEF genotypes are indicated on the right. Red bars indicate CNAs in chromosome 6, 7 (deletions) and 12 (duplications).
In summary, mutation of Mlh3, Pms2 or Mlh1 each significantly increased DR CNA rates in γ-irradiated MEFs. Each MutL mutation caused a bias towards genomic deletions, likely due to increased SSA between homologous DR sequences. Mlh1-/- MEFs overall had increased CNA rates vs Mlh3, Pms2 or combined Mlh3;Pms2 mutations. On chromosome 6, an Mlh3 mutation dependent CNA deletion hotspot was observed that was also seen in Mlh1-/- but not Pms2-/- irradiated MEFs. A chromosome 7 CNA deletion hotspot shared by Mlh3, Pms2 and Mlh1 mutant MEFs was also seen that had been previously identified in Mlh3-/-;Pms2-/- deficient intestinal tumors. Furthermore, an Mlh1 (but not other MutL homologue) mutation dependent chromosome 12 CNA amplification hotspot, which had also been previously identified in Mlh3-/-;Pms2-/- intestinal tumors, was observed.
Human dMMR CRC paired DRs have increased rates of homeologous recombination
Using the same methodology and parameters for analysis as in the mouse genome, we identified 12,379 uniquely mappable DRs in the human genome, each carrying 1-12 heterologous bases per paired DR. We then evaluated whether MMR mutations increased paired DR HeR rates in matched normal mucosa-dMMR patient CRC by analyzing genome sequence data from The Cancer Genome Atlas (TCGA-COAD) [51]. We identified 10 dMMR CRCs with matched normal-tumor whole genome sequence with >30× average sequence coverage, and compared them to 10 consecutive pMMR CRCs in TCGA-COAD for somatic mutations in DRs.
As expected, dMMR CRCs have significantly higher mutation rates in DRs (1.36 × 10-4 vs 3.7 × 10-4 p < 0.001, chi-square test) (Table 2). Similar to our findings in DRs in mouse genome, in the human CRCs HeR-BP in DRs have significantly higher mutation rates than surrounding bases (mean 9.75 fold increase, p < 0.001, chi-square test). Secondly, consistent with gene conversion and similar to our findings in mouse MEFs, HeR-BP in TCGA dMMR and pMMR CRCs were found to be significantly enriched for the corresponding base substitution in their matched 5′ or 3′DRs (92.86% in dMMR CRCs and 95.44% in pMMR CRCs respectively, all p < 0.001, Chi Square test) (Table 2). HeR-BP mutations consistent with gene conversion were also significantly higher in dMMR vs pMMR CRCs (1.02 × 10-3 vs 3.2 × 10-4 respectively, Chi Square test). Additionally, dMMR CRCs also had higher rates of tracts consisting of two consecutive heterologous bases both with gene conversion consistent mutations (Table 2).
Table 2: DR mutation rates and homologous recombination in dMMR vs pMMR CRCs
hCRC TCGA sample ID |
Overall DR mutations |
Mutations at specific DR sites (HeR-BP) |
Mutations outside specific DR sites (non-HeR-BP) |
GC consistent mutations at specific DR sites (HeR-BP) |
GC inconsistent mutation at specific DR sites (HeR-BP) |
% of GC-consistent mutations at specific DR sites(HeR-BP) |
GC consistent mutations at two contiguous DR sites (HeR-BP) |
|
|---|---|---|---|---|---|---|---|---|
dMMR |
aa-3516 |
0.112 |
0.783 |
0.096 |
0.7 |
0.083 |
89.39974457 |
0.1056 |
aa-3518 |
0.101 |
0.524 |
0.09 |
0.5 |
0.024 |
95.41984733 |
0 |
|
d5-6540 |
0.093 |
0.805 |
0.076 |
0.748 |
0.058 |
92.91925466 |
0.1149 |
|
aa-a00r |
0.102 |
0.561 |
0.09 |
0.463 |
0.098 |
82.5311943 |
0.119 |
|
aa-a01r |
0.101 |
0.99 |
0.079 |
0.99 |
0 |
100 |
0.0359 |
|
a6-6781 |
0.097 |
1.082 |
0.074 |
1.022 |
0.059 |
94.45471349 |
0.0911 |
|
ad-6964 |
0.095 |
0.64 |
0.082 |
0.598 |
0.043 |
93.4375 |
0.0289 |
|
az-6601 |
0.093 |
0.868 |
0.075 |
0.821 |
0.046 |
94.58525346 |
0.1931 |
|
ad-a5ej |
0.081 |
0.664 |
0.067 |
0.649 |
0.015 |
97.74096386 |
0 |
|
qg-a5z2 |
0.078 |
0.649 |
0.064 |
0.62 |
0.029 |
95.53158706 |
0 |
|
pMMR |
aa-3514 |
0.027 |
0.236 |
0.022 |
0.236 |
0 |
100 |
0 |
aa-3534 |
0.038 |
0.387 |
0.029 |
0.364 |
0.023 |
94.05684755 |
0 |
|
aa-a01x |
0.021 |
0.249 |
0.015 |
0.228 |
0.021 |
91.56626506 |
0 |
|
ag-3593 |
0.032 |
0.357 |
0.023 |
0.357 |
0 |
100 |
0.0538 |
|
ag-3890 |
0.028 |
0.268 |
0.022 |
0.251 |
0.017 |
93.65671642 |
0 |
|
aa-3685 |
0.037 |
0.218 |
0.032 |
0.2 |
0.018 |
91.74311927 |
0 |
|
aa-a02y |
0.028 |
0.229 |
0.023 |
0.229 |
0 |
100 |
0 |
|
aa-a01v |
0.037 |
0.339 |
0.029 |
0.313 |
0.026 |
92.33038348 |
0 |
|
ad-a5ek |
0.027 |
0.301 |
0.021 |
0.286 |
0.015 |
95.0166113 |
0 |
|
qg-a5z1 |
0.035 |
0.404 |
0.026 |
0.389 |
0.015 |
96.28712871 |
0 |
Note: For all above, mutation rates were calculated per 10000 bp.
Overall, these human CRC data are consistent with studies in mouse MEFs and DRs in the mouse genome. In human DRs, both HeR-BPs and surrounding bases have overall elevated mutation rates when dMMR is compared to pMMR CRCs. However, HeR-BPs have higher mutation rates than surrounding DR bases, almost all mutations are consistent with a mechanism of gene conversion in both pMMR and dMMR CRCs, including tracts of gene conversion consistent mutations. Moreover, dMMR CRCs have higher HeR rates than pMMR CRCs.
Human dMMR CRC deletion hotspots are at chromosomal fragile sites encoding FHIT and WWOX
To understand whether dMMR CRCs have CNA hotspots similar to what we observed in MEFs, we investigated the distribution of genomic regions preferentially deleted only in dMMR CRCs as potential sites for SSA mediated genomic deletions. By comparing CNA deletion rates in 32 dMMR and 156 pMMR tumors, we found that the ratio of CNA deletions in DRs and intervening sequences compared to non-DR sequences is significantly higher in dMMR vs. pMMR tumors (Wilcoxon rank sum test P = 0.00947). Of note, this analysis also revealed two dMMR somatic deletion hotspots that mapped to FRA3B and FRA16D chromosomal fragile sites (CFS) (Figure 3). CFS are long AT-rich repeat genomic sequences that are sites of DNA polymerase stalling during genome replication and have very high rates of spontaneous DSBs in tumors. When DSBs occur at CFS, homologous recombination is known to play an important role in CFS repair. Loci FRA3B and FRA16D, which encode the tumor suppressor genes FHIT and WWOX respectively, are the two most frequently deleted chromosomal loci in human CRCs. Furthermore, higher rates of FHIT and WWOX deletions have previously been reported in dMMR and pMMR CRC cell lines and tumors [52–58].
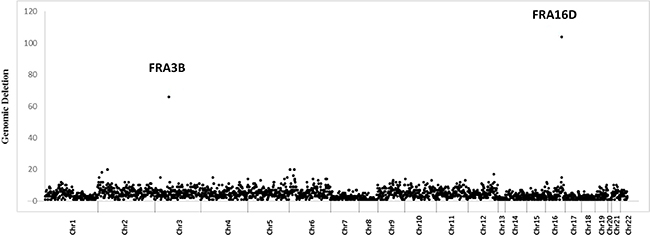
Figure 3: Increased somatic deletions at chromosomal fragile sites FRA3B and FRA16D in dMMR human CRCs. Density distribution (number/MB) of deleted probes across all human autosomes in dMMR tumors. FRA3B and FRA16D outliers are indicated.
Since FRA3B and FRA16D common chromosomal fragile sites have high rates of spontaneous DSBs, to understand whether increased SSA recombination between homologous repeat sequences could be a mechanism contributing to increased FRA3B and FRA16D CNA deletions in dMMR CRCs, we mapped each somatic deletion’s flanking breakpoints in genome sequence from 13 TCGA-CoAD dMMR and 77 pMMR CRCs with matched normal tissues and mean sequence coverage >10×. Using Vmatch parameters requiring >80% identity for > 80 bp in length within 1kb of deletion breakpoints, we identified 4 and 0 paired DRs, and 1 and 0 inverted paired DRs respectively in dMMR vs. pMMR CRCs (38.4% vs 0%, P = 0.0001 chi-square test). These dMMR CRC genomic deletions were flanked by DR and IR motifs ranging in size from 46 kb to 338 kb (Figure 4; Supplementary Table 4). Notably, these findings are consistent with high rates of spontaneous DSBs at the FRA3B and FRA16D chromosomal fragile sites in human dMMR CRCs that promote SSA and deletions at the FHIT and WWOX tumor suppressor loci.
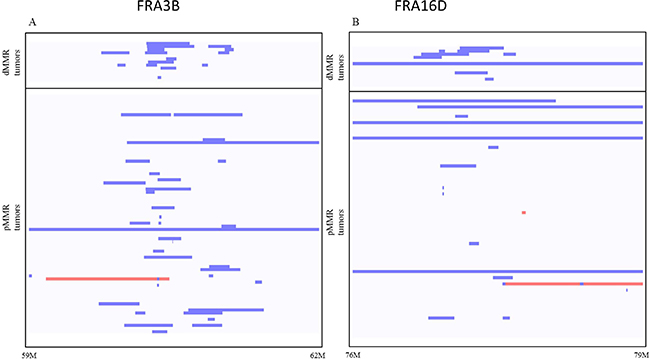
Figure 4: Somatic FRA3B (A) and FRA16D (B) genomic deletions (blue) and duplications (red) in dMMR and pMMR tumors. Analysis of whole genome sequencing (WGS) data of dMMR and pMMR human CRC tumors from TCGA-COAD. The rates of genomic deletions at FRA3B and FRA16D are significantly higher in dMMR than in pMMR human CRCs (p = 0.026 and p = 0.017, t-test). Blue lines indicate deletion, red lines indicate amplification.
DISCUSSION
DNA mismatch repair plays critical roles in maintaining genome integrity and inhibiting tumorigenesis. For repetitive sequences, suppression of HeR and SSA are important mechanisms to maintain genomic integrity. However, the role of individual mammalian MutL homologues in preserving genomic integrity by suppressing HeR and SSA between similar DNA sequences is poorly characterized. Furthermore, the roles of HeR and SSA in tumor suppression are unclear.
Building upon previous studies with HeR reporter genes [28], we demonstrate that genomic DRs can serve as in vivo endogenous reporters for measuring the levels of suppression or induction of HeR and SSA. In Wt MEFs, endogenous mutations in DRs are dominated by gene conversion, with HeR-BP mutation rates 33–40 fold higher than surrounding bases and >94% consistent with gene conversion from the corresponding DR paired sequence. Loss of function mutations in Mlh1, Pms2, Mlh3 and combined loss of function mutations in Mlh3;Pms2 further increase rates of mutations significantly, which is consistent with gene conversion in DRs at HeR-BP, as well as overall mutation rates in non-HeR-BP. Using both GFP reporter and genomic DRs, we extend previous reporter gene based studies of Saccharomyces cerevisiae Mlh1p and Pms1p (ortholog of Pms2) [23–26] to mammalian cells and show that mouse MutL homologs Mlh1 and Pms2 are involved in HeR suppression. Furthermore, we extend these findings and provide evidence for mammalian Mlh3 involvement in HeR suppression as well (summarized in Supplementary Figure 4A).
We evaluated DR mutations in human dMMR and pMMR CRCs by using available whole genome sequencing data from the TCGA database (http://cancergenome.nih.gov). In human genome DRs, HeR-BPs have higher mutation rates than surrounding DR bases. Almost all mutations are consistent with a mechanism of gene conversion in both pMMR and dMMR CRCs, including tracts of two consecutive gene conversion consistent mutations. Overall, these findings are largely consistent with MEF studies, supporting DRs as in vivo endogenous HeR reporters and demonstrating that dMMR CRCs have higher HeR rates than pMMR CRCs.
Next, using γ-irradiation to induce DSBs, we show that DR paired sequences can also be used to monitor SSA. Consistent with Saccharomyces cerevisiae Mlh1p reporter gene studies [27], loss of function mutations in Mlh1, Pms2 or Mlh3 each significantly increased DR CNA rates (summarized in Supplementary Figure 4B).
For all mutant MEFs, there was a bias towards DR genomic deletions, which is most likely due to increased SSA between homologous DR sequences. Similarly, analysis of TCGA data also support increased rates of SSA in dMMR compared to pMMR in human CRCs. MMR deficiency increased the rate of CNA deletions for both DRs and the intervening sequences between matching DR pairs.
Interestingly, analysis of MEF showed that Mlh1-/- cells have significantly higher overall CNA rates compared to Pms2-/-, Mlh3-/- or even Mlh3-/-;Pms2-/- MEFs. While we did not have Pms2-/-;Mlh3-/-;Pms1 triple mutant MEFs to study, given that Mlh1-Pms2, Mlh1-Mlh3 and Mlh1-Pms1 constitute all known MutL complexes, our data suggest a potential previously unexplored role for the Mlh1-Pms1 MutL complex in suppression of SSA.
Unexpectedly, in both mouse MEF and human TCGA studies, we identified evidence of specific CNA hotspots. For MEF with DSBs induced by gamma irradiation, hotspots included (a) a mouse chromosome 6 deletion hotspot shared by Mlh1-/-, Pms2-/-, Mlh3-/- and Mlh3-/-;Pms2-/- MEFs, (b) mouse chromosome 7 deletion hotspot shared by Mlh1-/-, Pms2-/-, Mlh3-/- and Mlh3-/-;Pms2-/- MEFs that we had been previously identified in Mlh3-/-;Pms2-/- deficient intestinal adenocarcinomas but not matched normal mucosa or Mlh3-/- adenomas [10] and (c) a mouse chromosome 12 amplification hotspot in Mlh1-/- MEFs, also previously seen in Mlh3-/-;Pms2-/- deficient intestinal adenocarcinomas [10]. Each of these regions is rich in highly repetitive elements. The mouse chromosome 6 deletion-hotspot occurs in a gene desert with no obvious unique feature, while the chromosome 7 hotspot contains a cluster of highly homologous paired immunoglobulin receptor (PiR) genes and the chromosome 12 hotspot contains many Immunglobulin Heavy chain (IgH) genes with a high degree of homology. We propose that mouse PiR and IgH gene clusters act as endogenous substrates for homeologous DR and inverted repeat (IR) formation. For chromosome 6 and 7 loci, the predominance of DR deletions is consistent with MutL suppression of SSA. However, for the chromosome 12 hotspot, there is a strong bias of CNAs to cause copy number gains. The precise mechanism of recombination at this latter locus and the precise features of each loci that specifically cause great dependence on MMR to suppress recombination are unclear at this time.
In human CRCs, analysis of DRs and IRs revealed two somatic deletion dMMR hotspots at chromosomal fragile sites (CFS) FRA3B (chr3:58,600,000–63,700,000) and FRA16D (chr16:79,200,000–81,700,000). CFS are long AT-rich repeat genomic sequences that are sites of DNA polymerase stalling during genome replication and have very high rates of spontaneous DSBs in tumors [59–61]. When DSBs occur at CFS, homologous recombination is known to play an important role in CFS repair [62–65]. FRA3B and FRA16D, which encode the tumor suppressor genes FHIT and WWOX respectively, are the two most frequently deleted chromosomal loci in human CRCs. Higher rates of FHIT and WWOX deletions have previously been reported in dMMR vs pMMR CRC cell lines and tumors [66, 67]. Mapping revealed in dMMR CRCs higher rates of DR and IR motifs in breakpoints flanking FRA3B and FRA16D. Since CFS have high endogenous rates of DSBs, our data are consistent with increased rates of SSA promoting deletions of FHIT and WWOX tumor suppressors in dMMR tumor.
In summary, known DNA mismatch repair tumor suppression mechanisms include base-substitution and small in/del mutation repairs as well as DNA damage response initiation. Our data provide the first evidence that homeologous recombination and SSA suppression are also important MMR tumor suppression mechanisms. Poly ADP (Adenosine Diphosphate)-Ribose Polymerase (PARP) inhibitors are an example of a successful mechanistically based therapy that exploits synthetic lethality with HR deficiency in ovarian and breast tumors. Our findings suggest that screens to identify drugs promoting synthetic lethality with increased HeR rates may similarly be effective for targeted treatment of dMMR tumors.
MATERIALS AND METHODS
Mouse embryonic fibroblast cell culture
Mouse embryonic fibroblasts were established from day 12.5 embryos isolated from the uteri of pregnant mice and matched for passage number as previously described [11]. In brief embryos were digested in Trypsin-EDTA 10 minutes in an incubator (37°C; 5% CO2) and washed with PBS. MEFS were then grown in 15% FBS and 100U/ml penicillin, 100 ug/ml streptomycin at 37C, 5% CO2.
Mouse embryonic fibroblast transfection
We constructed pDR-GFP8mu containing 8 silent mutations in the iGFP of pDR-GFP [28]. Primary MEFs were transfected with either pDR-GFP or pDR-GFP8mu and pCβASc by electroporation using a Nucleofactor device. Briefly, 2 × 107 cells in 1 ml of phosphate-buffered saline were electroporated with 20 to 25 μg of each uncut plasmid DNA in a 0.4-cm electrode-gap cuvette (250 V, 960 μF). Electroporated cells were aliquoted into four or five 10-cm-diameter dishes and kit for primary cells. As I-SceI is expressed and recombinant cells expressing GFP were analyzed using flow cytometry (BD-Biosciences) or colonies were selected in media 24 h after electroporation and were grown in selection G418 (200 μg/ml) media for 14 days before colony counts.
MEF gamma irradiation
Primary MEFS from all cell lines were cultured and irradiated with 5 Grays @ 1.27 Gy/Min. Subsequently the cells were fed fresh media and allowed to grow and passaged twice. The cells were then harvested and genomic DNA was extracted using DNeasy kit (Qiagen) per manufacturer’s protocol. All experiments were performed by irradiating exponentially growing cell cultures with 137Cs irradiator (Mark 1 irradiator, JL Shepherd & Associates).
Direct repeat region identification
To search for direct repeat (DR) regions in mouse genome (mm9) and human genome (hg19) we used the program Vmatch [68] using the following parameters: minimal length of repeat region 480 bp, minimal gap between DR regions 500 bp, and maximal gap 50Kb; minimal sequence identity between paired DRs >97%, and maximum number of non-identical base pairs is 12/DR.
Sequence analysis
Paired-end reads sequenced at targeted direct repeat regions were processed at Weill Cornell Genomic Core and then mapped to mouse reference genome (mm9) with BWA method [69]. After removing PCR duplicates, only uniquely mapped and properly paired reads were considered for depth calculation. Variants in the direct repeat regions were called with SAMtools software [70]. These variants were then filtered with standard thresholds for quality (q30), depth (50), mapping quality (30), and genotype quality (9\0). Mutation variant “Alt-allele” calls had median 41% mean allele frequency.
TCGA SNP array analysis
Level 2 and level 3 SNP array data were downloaded from TCGA data portal (http://cancergenome.nih.gov). For each probe, the ratio of normalized probe signal between tumor and the matched normal tissue was used to determine if there was a copy number alteration at the probe site. We considered a corresponding CNA at a site if tumor to normal ratio was >1.5 or <0.5. At each probe site, differential CNA between dMMR and pMMR tumors was calculated with Chi-squared analysis based on the 2 × 2 contingency table. If the expected values in the contingency table were less than 1, we used Fisher exact test following recommendations of Campbell et al. [71].
TCGA COAD whole genome sequence analysis
For 10 dMMR CRC patients with available whole genome sequencing tumor and matching normal germline DNA data, pre-processed BAM files were downloaded from TCGA database portal (http://cancergenome.nih.gov). In addition, for 10 pMMR CRC patients, tumor and matching normal whole genome sequence data were also downloaded from TCGA and READ portals. The selection of these patients was based on matching sequence depth in addition to same sequencing center, to reduce potential batch effects. Somatic point mutations for each CRC patient were identified using Mutect [72] method by joint calling the TCGA pre-processed tumor and normal BAM files.
Abbreviations
BP: Base pair; BIR: Break induced replication; CFS: Chromosomal fragile site; CNA: Copy number alteration; CO: Crossover; CRC: Colorectal cancers; dMMR: Deficient DNA mismatch repair; DR: Direct sequences; DSBs: Double-strand breaks; DSBR: Double-strand break repair; HR: Homologous recombination; HeR: Homeologous recombination; IDL: Insertion/deletion loops; pMMR: Proficient DNA mismatch repair; IR: Inverted repeats; ssDNA: Single-stranded DNA; SDSA: Synthesis dependent strand annealing; SSA: Single strand annealing
ACKNOWLEDGMENTS
We thank members of the Lipkin laboratory for helpful comments on this manuscript.
CONFLICTS OF INTEREST
No author has a conflicts of interest related to this manuscript to disclose.
FUNDING
SL acknowledges funding from R01 CA098626. ZHG and JS acknowledge funds from Weill Cornell Center for Advanced Digestive Care (CADC). ZHG also acknowledges start-up funds from Icahn School of Medicine at Mount Sinai.
REFERENCES
1. Kolodner RD. A personal historical view of DNA mismatch repair with an emphasis on eukaryotic DNA mismatch repair. DNA Repair (Amst). 2015; 7864:30095–30091.
2. Modrich P. Strand-specific Mismatch Repair in Mammalian Cells. The Journal of Biological Chemistry. 1997; 272:24727–24730.
3. Buermeyer AB, Deschenes SM, Baker SM, Liskay RM. Mammalian DNA Mismatch Repair. Annual Review of Genetics. 1999; 33:533–564.
4. Bellacosa A. Functional interactions and signaling properties of mammalian DNA mismatch repair proteins. Cell Death & Differentiation. 2001; 8:1076–1092.
5. Pena-Diaz J, Jiricny J. Mammalian mismatch repair: error-free or error-prone? Trends in Biochemical Sciences. 2012; 37:206–214.
6. Blackwell LJ, Martik D, Bjornson KP, Bjornson ES, Modrich P. Nucleotide-promoted release of hMutSalpha from heteroduplex DNA is consistent with an ATP-dependent translocation mechanism. J Biol Chem. 1998; 273:32055–32062.
7. Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997; 91:995–1005.
8. Acharya S, Wilson T, Gradia S, Kane MF, Guerrette S, Marsischky GT, Kolodner R, Fishel R. hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc Natl Acad Sci U S A. 1996; 93:13629–13634.
9. Jeong C, Cho WK, Song KM, Cook C, Yoon TY, Ban C, Fishel R, Lee JB. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat Struct Mol Biol. 2011; 18:379–385.
10. Chen PC, Kuraguchi M, Velasquez J, Wang Y, Yang K, Edwards R, Gillen D, Edelmann W, Kucherlapati R, Lipkin SM. Novel roles for MLH3 deficiency and TLE6-like amplification in DNA mismatch repair-deficient gastrointestinal tumorigenesis and progression. PLoS Genet. 2008; 4:e1000092.
11. Chen PC, Dudley S, Hagen W, Dizon D, Paxton L, Reichow D, Yoon SR, Yang K, Arnheim N, Liskay RM, Lipkin SM. Contributions by MutL homologues Mlh3 and Pms2 to DNA mismatch repair and tumor suppression in the mouse. Cancer Res. 2005; 65:8662–8670.
12. Kolas NK, Svetlanov A, Lenzi ML, Macaluso FP, Lipkin SM, Liskay RM, Greally J, Edelmann W, Cohen PE. Localization of MMR proteins on meiotic chromosomes in mice indicates distinct functions during prophase I. J Cell Biol. 2005; 171:447–458.
13. Lipkin SM, Moens PB, Wang V, Lenzi M, Shanmugarajah D, Gilgeous A, Thomas J, Cheng J, Touchman JW, Green ED, Schwartzberg P, Collins FS, Cohen PE. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat Genet. 2002; 31:385–390.
14. Manhart CM, Alani E. Roles for mismatch repair family proteins in promoting meioticcrossing over. DNA Repair (Amst). 2015.
15. Zanotti KJ, Gearhart PJ. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair (Amst). 2015; 7864:30076–30078.
16. Tham KC, Kanaar R, Lebbink JH. Mismatch repair and homeologous recombination. DNA Repair (Amst). 2016; 38:75–83.
17. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010; 11:196–207.
18. Finn KJ, Li JJ. Single-stranded annealing induced by re-initiation of replication origins provides a novel and efficient mechanism for generating copy number expansion via non-allelic homologous recombination. PLoS Genet. 2013; 9:1003192.
19. Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998; 18:93–101.
20. Putnam CD. Evolution of the methyl directed mismatch repair system in Escherichia coli. DNA Repair (Amst). 2015; 7864:30030–30036.
21. Roberts MS, Cohan FM. The effect of DNA sequence divergence on sexual isolation in Bacillus. Genetics. 1993; 134:401–408.
22. Tham KC, Hermans N, Winterwerp HH, Cox MM, Wyman C, Kanaar R, Lebbink JH. Mismatch repair inhibits homeologous recombination via coordinated directional unwinding of trapped DNA structures. Mol Cell. 2013; 51:326–337.
23. Datta A, Hendrix M, Lipsitch M, Jinks-Robertson S. Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc Natl Acad Sci U S A. 1997; 94:9757–9762.
24. Chen W, Jinks-Robertson S. The role of the mismatch repair machinery in regulating mitotic and meiotic recombination between diverged sequences in yeast. Genetics. 1999; 151:1299–1313.
25. Datta A, Adjiri A, New L, Crouse GF, Jinks Robertson S. Mitotic crossovers between diverged sequences are regulated by mismatch repair proteins in Saccaromyces cerevisiae. Mol Cell Biol. 1996; 16:1085–1093.
26. Selva EM, New L, Crouse GF, Lahue RS. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics. 1995; 139:1175–1188.
27. Sugawara N, Goldfarb T, Studamire B, Alani E, Haber JE. Heteroduplex rejection during single-strand annealing requires Sgs1 helicase and mismatch repair proteins Msh2 and Msh6 but not Pms1. Proc Natl Acad Sci U S A. 2004; 101:9315–9320.
28. Elliott B, Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol Cell Biol. 2001; 21:2671–2682.
29. Villemure JF, Abaji C, Cousineau I, Belmaaza A. MSH2-deficient human cells exhibit a defect in the accurate termination of homology-directed repair of DNA double-strand breaks. Cancer Res. 2003; 63:3334–3339.
30. Abuin A, Zhang H, Bradlet A. Genetic analysis of mouse embryonic stem cells bearing Msh3 and Msh2 single and compound mutations. Mol Cell Biol. 2000; 20:149–157.
31. De Wind N, Dekkar M, Berns A, Radman M, Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation toler-ance, hyperrecombination, and predisposition to cancer. Cell. 1995; 82:321–330.
32. Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Hum Mol Genet. 2005; 14 :197–205.
33. Gao G, Smith DI. WWOX, large common fragile site genes, and cancer. Exp Biol Med (Maywood). 2015; 240:285–295.
34. Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, Hickson ID. Replication stress activates DNA repair synthesis in mitosis. Nature. 2015; 528:286–290.
35. Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, Hickson ID. MUS81 promotes common fragile site expression. Nat Cell Biol. 2013; 15:1001–1007.
36. Huebner K, Croce CM. FRA3B and other common fragile sites: the weakest links. Nat Rev Cancer. 2001; 1:214–221.
37. Krummel KA, Denison SR, Calhoun E, Phillips LA, Smith DI. The common fragile site FRA16D and its associated gene WWOX are highly conserved in the mouse at Fra8E1. Genes Chromosomes Cancer. 2002; 34:154–167.
38. Dillon LW, Burrow AA, Wang YH. DNA Instability at Chromosomal Fragile Sites in Cancer. Curr Genomics. 2010; 11:326–337.
39. Ma K, Qiu L, Mrasek K, Zhang J, Liehr T, Quintana LG, Li Z. Common Fragile Sites: Genomic Hotspots of DNA Damage and Carcinogenesis. Int J Mol Sci. 2012; 13:11974–11999.
40. Barnes LD, Garrison PN, Siprashvili Z, Guranowski A, Robinson AK, Ingram SW, Croce CM, Ohta M, Huebner K. Fhit, a putative tumor suppressor in humans, is a dinucleoside 5',5"'-P1,P3-triphosphate hydrolase. Biochemistry. 1996; 35:11529–11535.
41. Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associatedt(3;8) breakpoint, is abnormal in digestive tract cancers. Cell. 1996; 84:587–597.
42. Chizhikov V, Chikina S, Gasparian A, Zborovskaya I, Steshina E, Ungiadze G, Samsonova M, Chernyaev A, Chuchalin A, Tatosyan A. Molecular follow-up of preneoplastic lesions in bronchial epithelium of former Chernobyl clean-up workers. Oncogene. 2002; 21:2398–2405.
43. Kuroki T, Tajima Y, Furui J, Kanematsu T. Common fragile genes and digestive tract cancers. Surg Today 2006; 36:1–5.
44. Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K. Muir-Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci U S A 2000; 97:4742–4747.
45. Iliopoulos D, Guler G, Han SY, Druck T, Ottey M, McCorkell KA, Huebner K. Roles of FHIT, WWOX fragile genes in cancer. Cancer Lett. 2006; 232:27–36.
46. Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochim Biophys Acta 2014; 1846:188–200.
47. Andachi H, Yashima K, Koda M, Kawaguchi K, Kitamura A, Hosoda A, Kishimoto Y, Shiota G, Ito H, Makino M, Kaibara N, Kawasaki H, Murawaki Y. Reduced Fhit expression is associated with mismatch repair deficiency in human advanced colorectal carcinoma. Br J Cancer. 2002; 87:441–445.
48. Vernole P, Muzi A, Volpi A, Terrinoni A, Dorio AS, Tentori L, Shah GM, Graziani G. Common fragile sites in colon cancer cell lines: role of mismatch repair, RAD51 and poly(ADP-ribose) polymerase-1. Mutat Res. 2011; 712:40–48.
49. Abouelhoda MI, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. Journal of Discrete Algorithms 2004; 2:53–86.
50. Chen W, Jinks-Robertson S. Mismatch repair proteins regulate heteroduplex formation during mitotic recombination in yeast. Mol Cell Biol. 1998; 18:6525–6537.
51. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
52. Briffa R, Um I, Faratian D, Zhou Y, Turnbull AK, Langdon SP, Harrison DJ. Multi-Scale Genomic, Transcriptomic and Proteomic Analysis of Colorectal Cancer Cell Lines to Identify Novel Biomarkers. PLoS ONE. 2015; 10:1–29.
53. Aldaz CM, Ferguson BW, Abba MC. WWOX at the crossroads of cancer, metabolic syndrome related traits and CNS pathologies. Biochimica et Biophysica Acta. 2014; 1846:188–200.
54. Alsop AE, Taylor K, Zhang J, Gabra H, Paige AJW, Edwards PAW. Homozygous deletions may be markers of nearby heterozygous mutations: The complex deletion at FRA16D in the HCT116 colon cancer cell line removes exons of WWOX. Genes Chromosomes & Cancer. 2008; 47:437–447.
55. Finnis M, Dayan S, Hobson L, Chenevix-Trench G, Friend K, Ried K, Venter D, Woollatt E, Baker E, Richards RI. Common chromosomal fragile site FRA16D mutation in cancer cells. Hum Mol Genet. 2005; 14:1341–1349.
56. Arlt MF, Miller DE, Beer DG, Glover TW. Molecular characterization of FRAXB and comparative common fragile site instability in cancer cells. Genes Chromosomes & Cancer. 2002; 33:82–92.
57. Mangelsdorf M, Ried K, Woollatt E, Dayan S, Eyre H, Finnis M, Hobson L, Nancarrow J, Venter D, Baker E, Richards RI. Chromosomal Fragile Site FRA16D, DNA Instability in Cancer. Cancer Research. 2000; 60:1683–1689.
58. Paige AJW, Taylor KJ, Stewart A, Sgouros JG, Gabra H, Sellar GC, Symth JF, Porteous DJ, Watson JEV. A 700-kb Physical Map of a Region of 16q23.2 Homozygously Deleted in Multiple Cancers and Spanning the Common Fragile Site FRA16D. Cancer Research. 2000; 60:1690–1697.
59. Thys RG, Lehman CE, Pierce LC, Wang YH. DNA Secondary Structure at Chromosomal Fragile Sites in Human Disease. Curr Genomics. 2015; 16:60–70.
60. Franchitto A. Genome Instability at Common Fragile Sites: Searching for the Cause of Their Instability. BioMed Research International. 2013; 2013:1–9.
61. Burrow AA, Williams LE, Pierce LCT, Wang YH. Over half of breakpoints in gene pairs involved in cancer-specific recurrent translocations are mapped to human chromosomal fragile sites. BMC Genomics. 2009; 10:1–11.
62. Schwartz M, Zlotorynski E, Goldberg M, Ozeri E, Rahat A, Sage C, Chen BPC, Chen DJ, Agami R, Kerem B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005; 19:2715–2726.
63. Lundin C, Erixon K, Arnaudeau C, Schultz N, Jenssen D, Meuth M, Helleday T. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002; 22:5869–5878.
64. Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol. 2001; 307:1235–1245.
65. Richardson C, Jasin M. Coupled homologous and nonhomologous repair of a double-strand break preserves genomic integrity in mammalian cells. Mol Cell Biol. 2000; 20:9068–9075.
66. Turner BC, Ottey M, Zimonjic DB, Potoczek M, Hauck WW, Pequignot E, Keck-Waggoner CL, Sevignani C, Aldaz CM, McCue PA, Palazzo J, Huebner K, Popescu NC. The Fragile Histidine Triad/Common Chromosome Fragile Site 3B Locus and Repair-deficient Cancers. Cancer Research. 2002; 62:4054–4060.
67. Tallec BL, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M. Common Fragile Site Profiling in Epithelial and Erythroid Cells Reveals that Most Recurrent Cancer Deletions Lie in Fragile Sites Hosting Large Genes. Cell Reports. 2013; 4:420–428.
68. Abouelhoda M, Kurtz S, Ohlebusch E. Replacing suffix trees with enhanced suffix arrays. Journal of Discrete Algorithms. 2004; 2.
69. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 14:1754–1760.
70. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 16:2078–2079.
71. Campbell I. Chi-squared and Fisher-Irwin tests of two-by-two tables with small sample recommendations. Stat Med. 2007; 26:3661–3675.
72. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Landers ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature Biotechnology. 2013; 31:213–219.
73. Wolfe D, Dudek S, Ritchie M, Pendergrass S. Visualizing genomic information across chromosomes with PhenoGram. BioData Mining 2013; 6.