
INTRODUCTION
Bladder cancer is the most common malignancy of the urinary tract and the second most common malignancy of the urogenital tract following prostate cancer in the United States [1]. The American Cancer Society estimates 76,960 new cases and 16,390 deaths of bladder cancer in the US with male: female ratio of 3:1. These figures indicate that bladder cancer is now the fourth most common among men and the ninth most common in women (http://www.cancer.org). The primary risk factors for bladder cancer include environmental and occupational exposures to chemical carcinogens [2] as tobacco smoke/metabolites, aromatic hydrocarbons, house paints, fungicides, plastics, and heavy metals [2–4]. Also, N-nitroso compounds (NOCs) used in chemically preserved food products are associated with risk for bladder cancer [5]. A family history of bladder cancer is linked to two-fold higher risk, but bladder cancer affected families are not common and no high-penetrance genes have been identified [6–8].
Bladder cancer typically arises from the urothelium, the well-differentiated transitional epithelium that lines the urinary bladder. The majority of bladder cancers diagnosed in developed countries is of transitional cell histology and is known as urothelial carcinoma (UC). Bladder cancer develops via two clinically and pathologically distinct routes: papillary and non-papillary forms of disease [9, 10] (Figure 1). About 75–80% of urinary bladder tumors are superficial papillary lesions known as non-muscle invasive urothelial cancer (NMIUC) and are referred to as low-grade intra-urothelial neoplasia. These tumors can be multifocal and may recur after local excision, but typically they do not invade the bladder wall or metastasize. Development of low grade lesions is associated with molecular aberrations in the oncogene RAS, FGFR3, and deletions of 9q [11]. Twenty to twenty-five percent of patients are present with solid, non-papillary tumors that invade the detrusor muscle which is called muscle invasive urothelial cancer (MIUC). These originate from carcinoma in situ (CIS) or severe dysplasia. These tumors have a high tendency to give rise to distant metastases. Hence, they are referred to as high-grade intra-urothelial neoplasia and are associated with alterations in p53, retinoblastoma (Rb), and PTEN [12]. Treatment of bladder cancer and the efficacy of such treatment vary profoundly depending on the clinical stage and associated risk factors [13–15]. The primary treatment of NMIUC is transurethral resection of bladder tumors (TURBT) [16] followed by intravesical therapy either by instillation of mitomycin C or with Bacillus Calmette-Guerin (BCG) [17, 18]. These treatments result in longer disease-free survival in patients with low-grade disease, but recurrence is common in patients with high-grade disease with progression to locally advanced muscle invasive and metastatic disease [17, 18]. High risk patients or those with recurrence after TURBT are treated with several cycles of BCG, which promotes immunoreaction against cancer cells [19]. BCG treatment requires frequent surveillance over long periods of time with adverse effects including severe urocystitis or systemic tuberculosis [20, 21]. Clinical management of MIUC primarily focuses on cystectomy (surgical removal of bladder) with neoadjuvant or adjuvant chemotherapy and/or radiation therapy for selected individuals [22, 23]. Metastatic bladder cancer generally treated by a multidrug chemotherapy regimen consisting of methotrexate, vinblastine, adriamycin, and cisplatin (MVAC) or, alternatively, gemcitabine plus cisplatin (GC) [16]. In addition to the side effects of chemotherapy, bladder cancer is highly resistant and upon relapse, effective treatment options are limited [24]. There has been no new approved treatment for bladder cancer in the past 30 years [25, 26]. Therefore, there is a dire unmet need to better understand the pathobiology of bladder cancer, identify diagnostic and prognostic biomarkers of disease progression and develop new therapeutics. To achieve these goals, several experimental models of bladder cancers are developed and undoubtedly, they present useful tools for molecular and functional analyses, and preclinical pharmacological assessment.
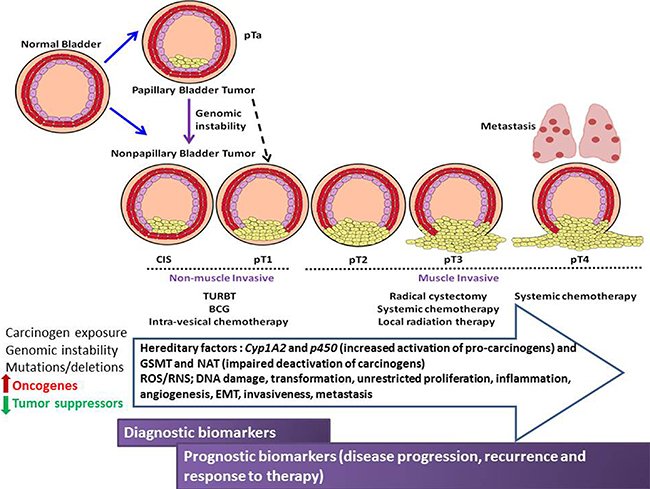
Figure 1: Schematic illustration of the pathological stages of urothelial cancer of the bladder, therapeutic intervention, environmental, genetic and molecular influences of urothelial carcinogenesis, progression and metastasis. Opportunities for diagnostic/prognostic biomarker discovery are shown compared to disease stage.
Bladder cancer in vivo models
An ideal animal model of bladder cancer should recapitulate its human counterpart with similar histopathological features, natural course of tumor growth and progression. Most importantly, these models should possess a high reproducibility, predictive and translatability value to allow mechanistic, chemo-preventive and therapeutic studies that can be furthered into human clinical trials. The most commonly used species for animal research are small rodents, such as mice and rats. Rodents have a lower urinary tract that is homologous to that of humans. Also, bladder cancer is not very common in rodents unless they are induced by a chemical carcinogen [27] or oncogenes [28]. These small animals can be easily housed, maintained at a low cost and are useful in generating valuable information regarding bladder carcinogenesis. Mouse strains such as C57B6, BALBC, and ICR, and rat strains such as Wistar, Sprague-Dawley and Fisher are most commonly used for bladder cancer research [29]. Some rat strains such as Brown Norway and DA/Han show high incidence of spontaneous bladder tumors, and thus can be used as experimental models without treatment with chemical carcinogens [30]. Larger animals such as dogs, rabbits, guinea pigs and hamsters have all been used in the past as bladder cancer models [31]. However, their use is limited due to financial and ethical constraints.
Animal models of bladder cancer (Figure 2) can be categorized as autochthonous (spontaneous) and non-autochthonous (transplantable). The first are either chemically induced models or genetically engineered models. The transplantable models can be further sub-classified as syngeneic (murine bladder cancer cells implanted into immunocompetent or transgenic mice) and xenografts (human bladder cancer cells implanted into immune-deficient mice). These models can be further divided based on the site of tumor as orthotopic (tumor growth occurs within bladder) and heterotopic (tumor growth occurs outside of the bladder).

Figure 2: Summary of the available mouse models of urinary bladder cancer.
Autochthonous (spontaneous) models
Carcinogen induced model
Bladder cancer is caused by continuous exposure to chemical carcinogens such as tobacco, aromatic amines and chlorinated hydrocarbons [32]. Therefore, it is essential to examine the relationship between chemical carcinogenesis and the development of urothelial carcinoma of the bladder. The first urothelial carcinogenesis model was induced in rats [33–35], thereafter several carcinogens were developed in various species, including mice, rats, and dogs [36]. Urothelial carcinoma can be induced predominantly in rodents with the use of several chemical carcinogens. The major chemical urothelial carcinogens are N-Butyl-N-(4-hydroxybutyl)nitrosamine (BBN), N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide (FANFT), and N-methyl-N-nitrosourea (MNU). Most of these carcinogenic agents have aromatic amine components. The carcinogen models develop over a prolonged period of time recapitulating the prolonged human exposure.
BBN was first identified as a bladder carcinogen in rodents [34, 37] and is detected in tobacco smoke and environmental and infectious metabolites [29]. BBN-induced cancer models can be established either orally by adding it to drinking-water and is degraded to N-butyl-N-(3-carboxypropyl)-nitrosamine, which has carcinogenic effects on the urothelium when cleared in the urine [29]. BBN-exposed mice develop various pathological features, including hyperplasia, dysplasia, CIS, and invasive tumors, as well as metastases that are histologically and genetically similar to human bladder tumors arising from extensive tobacco use [29]. In line with this, Williams et al. [38] compared gene expression profiles of urothelial carcinoma for three different species: mouse, rat, and human. Several human genes homologous to those differentially expressed in carcinogen-induced rodent tumors were also differentially expressed in human bladder cancer and were associated with progression to muscle-invasive disease. The overall gene expression profiles of rodent tumors tracked with those of invasive human tumors rather than those of non-muscle-invasive tumors. The cell cycle associated genes such as cell division cycle 20 (CDC20), cell division cycle 2 (CDC2), cyclins D1 and B2 (CCND1 and CCNB2), mitotic arrest–deficient 2, Saccharomyces cerevisiae, homolog-like 1 (MAD2L1), and cyclin A2 (CCNA2) were primarily differentially expressed between normal and cancerous urothelium in all three species [38]. Furthermore, Lu et al. [39] reported that changes in several protein/mRNA levels were consistent in BBN murine model versus human bladder cancer, thereby reinforcing the reliability of this model.
BBN-induced rodent tumors, predominantly mouse tumors, have p53 mutations, or mutations in genes related to the p53 pathway, especially in high-grade tumors [37, 40]. H-Ras mutations are observed occasionally in mouse and rat models, although BBN-induced carcinogenesis occurs more efficiently in H-Ras transgenic mice. BBN-induced tumors show high levels of Epidermal growth factor receptor (EGFR) [37, 41]. Nrf2–/– null mutation in both Nrf2 alleles and p27kip null mutation in both p27 alleles resulted in higher sensitivity to BBN than the wild type mouse [42–44].
BBN chemical carcinogenesis model in transgenic mice proved to be a powerful tool to identify the mechanism of action of tumor suppressor and oncogenes as well as the pre-neoplastic lesions that are not clinically encountered [45]. In this respect, the role of the matricellular glycoprotein, secreted protein acidic and rich in cysteine (SPARC) in the pathobiology of bladder cancer was reported [45]. This model showed that genetic ablation of SPARC augmented BBN-induced bladder carcinogenesis, progression and metastasis. The authors identified that SPARC restrains urothelial carcinogenesis, progression and metastasis through an effect on cancer cells inhibiting carcinogen-induced cell cycle deregulation and on stromal cell plasticity, inhibiting their acquisition of a pro-inflammatory cancer associated phenotype. In another study, utilizing Rgs6–/– mice, the role for RGS6 as a tumor suppressor was reported as they displayed accelerated pathological lesions than Rgs6+/+ mice [46]. The investigators identified RGS6 as a tumor suppressor modulating ATM/p53 and RASSF1A, the two critical signaling pathways that are often dysregulated in urothelial carcinoma. BBN-induced rodent models were analyzed by genome-wide analysis of copy number aberrations in bladder lesions [47]. The tumor promoting effect of phospholipase Cε (PLCε) was also studied using BBN in PLCε knockout mice [46]. The tumor promoting effect of PLCε was demonstrated by a decreased incidence of ensuing urothelial lesions in PLCε knockout mice compared to their wildtypes as well as a significant downregulation of PLCε downstream targets such as VEGF-A and COX-2 [46]. Array CGH revealed that several mouse chromosome regions, including the Cyp2a5 and Cyp2a22 loci on mouse chromosome 7qA3, were amplified in invasive bladder cancer. Interestingly, the human CYP2A6 gene, ortholog of the mouse Cyp2a5 gene was found to be upregulated in human cell lines and in resected human bladder cancer specimens [47]. BBN model can be also used to study the impact of conditional cell/tissue specific knockout/knockin of genes; for example, the effect of conditional knockout of estrogen receptor α and β [48] as well as androgen receptor [49]. Moreover, this model was used to evaluate the preventive and therapeutic efficacy of chemotherapeutics [46, 50–52].
MNU is another genotoxic carcinogen that acts directly on the urothelium by intra-vesical instillation and leads to constitutive DNA methylation [53]. The primary advantage of this model is that papilloma and carcinoma appear after 12 weeks and MNU is the only carcinogen to produce bladder cancer at a single dose [35]. However, MNU is intrinsically unstable and should be stored at a low temperature and protected from light. Therefore, it may result in experimental inconsistencies due to its decomposition and altered carcinogenic potency over time.
FANFT is an indirect chemical carcinogen that stimulates the bladder mucosa, and the bladder tumors mostly develop into transitional cell carcinoma (TCC) after it is fed to rodents for 5 to 8 months [54]. FANFT is not currently common as it is identified as an environmental pollutant and is hazardous for human health.
Genetically engineered models
The transgenic mouse, or genetically engineered mouse (GEM), are useful research systems that are engineered to carry cloned oncogenes or lack tumor-suppressing genes, and allows investigation of human disease associated genetic abnormalities in vivo. They allow studies of individual as well as multiple mutational events which eventually orchestrates bladder tumorigenesis. Several methods are employed to generate GEM models. Mice with germ line deletion may not allow investigation of the gene function if knockout leads to premature death or embryonic lethality and do not allow for the clear distinction of the tissue/cell-specific contribution of a given gene in the disease. An approach that avoids these limitations is a conditional gene knock-in or knockout involving the Cre-loxP system, which allows for studying gene function in specific cell and tissue types [55]. Urothelial-specific Cre system is available and is typically used to selectively achieve gene knockin/knockout in the bladder epithelium. The Cre-loxP system is based on the bacteriophage P1 wherein Cre recombinase acts on palindromic sequences called loxP sites that have been genetically engineered into the specific sites in the mouse genome. Cre recombinase can then excise the genomic sequence between two loxP sites. Therefore, mouse alleles containing loxP sites can be used for introducing mutations, or for a temporally or spatially controlled gene knockout. Majority of the bladder cancer GEM models have used the mouse Uroplakin II (UpkII) promoter which is the 3.6 kb 5′-upstream sequence of mouse UpkII gene [56]. Uroplakins are membrane integral proteins that constitute the major differentiation products of the urothelium. Zhang et al. demonstrated that SV40 (Simian virus 40) mice generated using UPII promoter developed CIS urothelial carcinoma with low copy number of SV40T transgene and those with high copies developed CIS along with invasive and metastatic transitional carcinoma [57]. Ayala de la Pena and colleagues [58] developed the promoter known as Upk II which was used to generate mice expressing the oncogene SV40 large T-antigen that can inactivate p53 and retinoblastoma (Rb) proteins [58]. GEM models are widely used for investigating specific gene functions including, H-RAS, p53, RB, PTEN, fibroblast growth factor receptor (FGFR), and epidermal growth factor receptor (EGFR), in the development of bladder cancer. H-RAS was amongst the initially identified oncogenes in human bladder cancer [59]. RAS activation can occur through overexpression of H-Ras in the urothelium, which occurs in more than 50% of bladder cancers [60]. A transgenic model was developed to allow targeted expression of a constitutively active H-Ras in the urothelium using the UpkII promoter [44]. This mutation resulted in early onset urothelial hyperproliferation, which progressed to low-grade, papillary, non-invasive tumors. This study revealed a strong relation between tumor latency and H-Ras copy number. Mice with low-copy (one or two copies) number of genes showed shorter tumor latency and developed non-invasive lesions compared to mice with high-copy numbers (above 30 copies). He et al. developed a conditional compound mice expressing oncogenic HRAS and lacking p53 gene expression. This model resulted in CIS and muscle-invasive urothelial carcinoma with focal squamous differentiation similar to human counterpart. This study suggested that the RTK/RAS pathway activation along with p53 deficiency can be used as a combinatorial therapeutic marker to indicate urothelial carcinoma progression [61]. FGFR3 is another gene that is often associated with low grade bladder cancer. Ahmad et al., [62] developed a model using UpkII promoter to induce expression of mutated Fgfr3 to the mouse urothelium. Mutated Fgfr3 alone was insufficient to induce urothelial tumorigenesis. When Fgfr3 mutations were introduced along with overexpression of K-Ras or β-catenin inactivating mutations, mice developed tumors indicating that Fgfr3 cooperates with other mutations to drive tumorigenesis [42, 62, 63]. In addition, mutations and/or deletions in the p53 gene are among the most common genetic alterations found in human bladder cancer [11, 25]. Consistently, He and colleagues [64] demonstrated that urothelial-specific deletion of both copies of Rb did not augment urothelial proliferation, but conversely, this deletion fostered activation of the p53 pathway and led to apoptosis. Double-mutant mice that were null for both p53 and Rb did not exhibit accelerated tumorigenesis, though they became extremely sensitive to BBN induced carcinogenesis and invasiveness [64]. This study suggested that genetic ablation of both p53 and Rb is essential for the progression of UCC, but insufficient to initiate invasive UCC by itself. Allelic loss of Tp53 alone was sufficient to drive tumorigenesis but not invasiveness of urothelial cancer [43, 65]. The PTEN-PI3K-AKT pathway has been implicated in bladder cancer and reports suggest that deletion of PTEN occurs frequently in invasive UCC [25, 42, 66]. Studies crossing Uroplakin-Cre mice with reported Pten floxed mice revealed the co-operation of activation of β-catenin and with Pten deletion to drive urothelial carcinogenesis [42]. Expression of the activated form of β-catenin led to the formation of localized hyper-proliferative urothelial lesions by 3 months, which did not progress to malignancy. Targeted deletion of Pten and expression of activated β-catenin caused papillary UCC that exhibited increased pAKT signaling and were dependent on mammalian target of rapamycin (mTOR). Importantly, in human UCC, there was a significant correlation between high levels of β-catenin and pAKT (and low levels of PTEN). Moreover, targeted deletion of both p53 and Pten [67] using an adenovirus expressing Cre recombinase delivered directly into the bladder of mice with floxed alleles of p53 and Pten (p53fl/flPtenfl/fl), developed bladder tumors with 100% penetrance after 24 weeks, with metastasis to local lymph nodes and distant sites, including the spleen, liver and diaphragm. This work strengthened the notion that urothelial bladder cancer is a complex disease with no single driver mutation, but requires multiple mutations to induce bladder tumorigenesis. Yoo et al, [68] developed a GEM model by deleting exons 4–5 of the Pten gene using the FabpCre system, in which the expression of Cre recombinase was placed under the control of transcriptional regulatory elements from a fatty acid-binding protein (Fabp) gene. Crossing Fabp-Cre mice with Ptenfl/fl mice resulted in loss of Pten in all of the cell layers of the urothelium as well as prostate, vagina and intestine. Of note that Fabp-Cre mice unexpectedly developed lesions in the terminal small intestine, cecum, colonic mucosa and other pelvic organs including urinary bladder [69]. Fabp-Cre-Ptenfl/fl mice developed urothelial hyperplasia and UCC by 13.5 months (~44 weeks) of age with decreased levels of p27, increased levels of p21, and a decrease in proliferation rates in the urothelium [68]. These observations were distinctive from concurrent prostate lesions. Thus investigators concluded that deletion of Pten induces distinct tissue-specific pathways that influence tumor and progression [68].
Another approach to conditionally manipulate genes in the urothelium is the direct injection of adenovirus-Cre in the bladder lumen. This method has been used to investigate the roles of the main driver mutations as K-ras, p53, Pten and Rb in the temporal and spatial development of urothelial cancer [67, 70, 71]. These studies further identified therapeutic targets downstream of the aforementioned mutations. Of note, Yang and colleagues [71] observed that the simultaneous inactivation of p53 and activation of K-ras induces quick formation of spindle-cell sarcoma in the soft tissues adjacent to the bladder but slow formation of urothelial hyperplasia inside the bladder. These results strongly suggest that the effect of oncogene regulation to produce either hyperplasia or carcinogenesis greatly depends on the tissue type. Most importantly, studies with the GEM models that recapitulate disease progression allowed the preclinical validation of the key pathways involved in urothelial cancer progression from NMI to MI disease [26, 67, 70, 72] and paved the way to clinical trials for intra-vesical delivery of single or combinatorial chemotherapeutics to prevent disease progression in high risk patients with early stage disease [26].
Non-autochthonous (transplantable) models
The transplantable models (xenografts and syngeneic) can be categorized as heterotopic and orthotopic models based on site of tumor cell implantation. It is essential to choose representative and reproducible cell lines for transplantable models. A large number of human and rodent urinary bladder cancer cell lines are available representing different origins, grades and stages of urothelial carcinoma, and mirror many of the genetic, morphologic, and gene expression alterations observed in human urothelial carcinoma [73]. Several cell lines were established from invasive and metastatic tumors, which are advantageous in the investigation of late tumor progression and metastatic lesions [74, 75] (Table 1). Important resources with detailed information regarding genetic alterations in cell lines are the Catalog of Somatic Mutations in Cancer (COSMIC) at the Sanger Institute (Cambridge, United Kingdom) [76], the Cancer Genomic Atlas (TCGA) for tumor samples [25], the Cancer Cell Line Encyclopedia (CCLE) [77], and the Genomics of Drug Sensitivity in Cancer [48]. It is imperative that cell lines are authenticated to ensure the use of correctly identified uncontaminated cell lines in research studies. Therefore, it is recommended to purchase cell lines from authenticated cell repositories. Identity verification with Short tandem repeat (STR) profiling establishes a DNA fingerprint for every human cell line and may be used as a record of the line. STR profiling uses multiplex PCR to simultaneously amplify several polymorphic markers in the human genome. Each cell line exhibits a pattern of repeats which constitutes the unique STR identity profile [78].
Table 1: Genetic characterization of major oncogenes and tumor suppressor genes in human bladder cancer cell lines
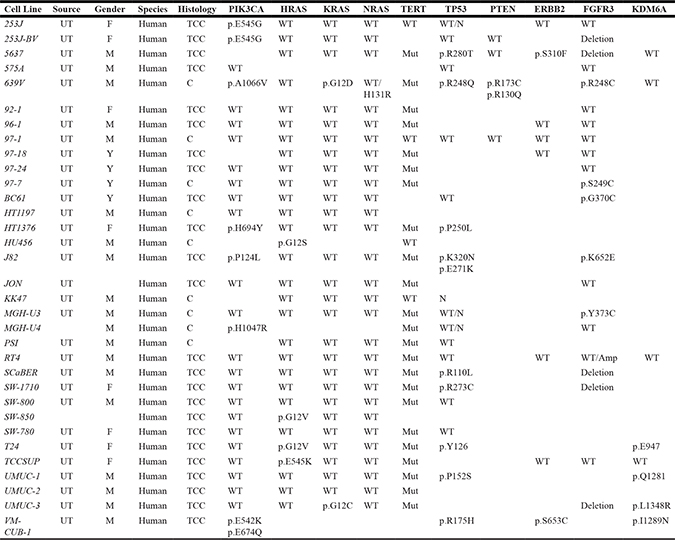
C, Carcinoma; TCC, transitional cell carcinoma; F, female; M, male; UT, urinary tract; WT, wild type; Mut, Mutant; N, copy number neutral;
Y, Y chromosome detected.
Orthotopic models
Orthotopic models allow evaluation of tumor behavior in an organ-specific microenvironment. These models can be established by injecting urothelial cancer cells into the bladder lumen of recipient hosts. Transitional cells preferentially establish on an altered urothelial surface. For this purpose, the natural protective epithelium lining the surface of the urinary bladder has to be damaged to facilitate tumor cell adhesion. The mechanical damage can be achieved by electrical cauterization and epithelial abrasion [79–85] as well as chemical denudation with HCl [86, 87], N-methyl-N-nitrosourea [88], silver nitrate [80] followed by tumor cell instillation.
Several factors determine the efficient tumor take rate, such as the bladder preconditioning, cell concentration, the instillation volume and the tumor cell dwell time in bladder [89]. An instillation volume of 50–100 μL is typically used with mice weighing < 30 g. Higher volumes can result in peri-urethral leakage and reflux to the upper urinary tract. Therefore, it is highly suggested to ensure complete emptying of the bladder before instillation as it prevents over-distension of the mouse bladder [89]. Also, it has been previously reported that an increase in the tumor cell dwell time augments duration for which the cancer cells are in contact with the bladder mucosa, resulting in an increase in the tumor inoculation rate [90, 91]. Other than technical limitations, orthotopic models only metastasize to local lymph nodes and do not allow studies of metastasis or recurrence.
Heterotopic transplantable models
These imply injecting/transplanting rodent or human bladder cancer cells or tissues in a rodent in an ectopic site other than tissue of origin. Thus, these models can be syngeneic (rodent cells/tissue in immunocompetent or transgenic rodent) or xenografts (human cells/tissue in immuno-deficient mouse). These models are adopted especially if the original site is not technically convenient for inoculation such as the urinary bladder. These models are easy to establish, easy to manage, cost-effective and are widely used in mechanistic studies as well as in evaluating the efficacy of novel therapeutic agents [92]. Several transplantable models are used in complementary way to study different aspects of tumor growth, and metastasis as well as the effect of tumor-stromal interactions [26].
Syngeneic models
Syngeneic models are generated by implanting rodent bladder cancer cells or tissues into syngeneic, immunocompetent or transgenic animals. Rodent bladder cancer cell lines such as AY27 and MBT2 were initially induced by administering C3H/He mouse and Fischer 344 rat strains with FANFT) [86, 93], whereas MB49 was induced by feeding 7,12-Dimethylbenzanthracene to the C57BL/6 mouse strain [94]. Implantation of urothelial cancer cells in syngeneic hosts can be orthotopic (intravesical or heterotopic (subcutaneous, renal capsule implantation or experimental metastasis model). For instance, the use of the immunocompetent host in syngeneic models allows for the study of intravesical BCG treatment or gene therapy [79, 81, 95–100]. The murine MB49 bladder tumor model is quite similar to human bladder cancer which makes it an interesting model to study novel gene- and immunotherapies. Loskog et al. generated a subcutaneous mouse model using MB49 cells in C57BL/6 female and male mice [97]. Loskog et al. generated a MB49 metastatic tumor model using stably transfected luciferase MB49 cells, which could be monitored by in vivo bioluminescence. Another spontaneous metastatic model using syngeneic MB49 cell line revealed the significance of vasoconstrictor protein endothelin-1 (ET-1) in the early-establishment of metastasis. This study revealed that circulating tumor cells generate a strong inflammatory response in the lungs mediated by the ET-1/endothelin-1 receptor A (ETAR) axis and this allows metastatic colonization. Also, the pharmacologic inhibition of ETAR by ZD4054 prior to injection of tumor cells significantly decreased the early inflammatory response as well as the development of lung metastases. Therefore, this spontaneous metastatic model enabled us to determine that tumor ET-1 expression and ETAR activity are essential for metastatic lung colonization but their functional role is less significant in established primary or metastatic tumors. These data provide a major evidence that preclinical evaluation of new therapeutics should be done in the adjuvant setting using models of metastatic colonization [101]. The significance of tumor expression of the proteoglycan versican and chemokine CCL2 (also known as MCP1) in promoting bladder cancer lung metastasis was investigated using syngeneic MB49 metastatic model [102]. The expression of isoforms V1 and V3 of the proteoglycan versican is reduced by RhoGTP dissociation inhibitor 2 (RhoGDI2), which subsequently suppresses lung metastasis post tail-vein injection in Ccl2–/– and Ccr2–/– mice. Our findings emphasized the significance of CCL2 and CCR2 in GDI2-versican–mediated lung metastasis [102]. Additionally, using a syngeneic model of spontaneous metastasis in which SPARC-proficient MB49 cells were injected SC in SP–/– and SP+/+ mice, host-SPARC not only inhibited the in vivo growth, invasiveness, angiogenesis and inflammation of primary tumors, but also mitigated spontaneous lung metastasis through inhibition of the pre-conditioning of the inflammatory pre-metastatic and metastatic lung niche [45]. Syngeneic models are both time and cost-effective, and reproducible. These model systems proved to be very useful in developing and validating new therapies at targeting the multistep cascade of tumor growth, progression, spontaneous metastasis and organotropism.
Xenograft models
Xenografts imply the implantation of human tumor cells or tissues in immunocompromised mice. Xenografts can be subdivided into: orthotopic (intravesical) or heterotopic (subcutaneous, renal capsule implantation, or intravenous or intracardiac for experimental metastasis).
Orthotopic xenografts
Chade et al. [80] described the use of intravesical 0.2% trypsin before treatment for 30 min, and mechanical bladder injury immediately before cancer cells are instilled for 3 h. Microscopic examination of the bladder 10 days after implantation revealed bladder tumors in 80–100% of mice, but the procedure-related death was high. Jager et al. [103] developed a novel high precision approach for orthotopic xenograft implantation. Bladder cancer cell lines such as UMUC1, UMUC3, and UMUC13 were inoculated into 10-week old athymic nude mice by percutaneous injection under ultrasound guidance. This model enabled monitoring of tumor volume, measuring in vivo tumor perfusion with microbubble contrast agents, and injecting therapeutic agents into the tumor under ultrasound guidance. The orthotopic tumors exhibit an increased microvessel density, high growth factors expression and proteolytic enzyme activity compared to those of the subcutaneous tumors. Moreover, rodents have a lower urinary tract which is comparable to humans, and neoplasms in the bladder are morphologically very similar, with a similar phenotype to human urinary carcinoma with respect to tumorigenesis and gene expression. However, orthotopic model establishment techniques are confounded by technical variability, with variable outcomes, complications and success.
Subcutaneous tumor xenografts
Typically, human cancer cells are implanted in the flank or hind leg of immune-deficient mice. The subcutaneous model is the most commonly adopted model. This model is easy to establish, easy to manage, cost-effective and is used in mechanistic studies, and in evaluating the efficacy of novel therapeutic agents [92]. Another advantage of subcutaneous tumors is that they can be used to study local recurrence after excision. However, since subcutaneous tumors are not established at the original site, the urinary bladder, they do not recapitulate the real tumor microenvironment. In addition, subcutaneous xenografts do not metastasize, thus, cannot be used for studying some aspects of tumor biology specifically metastasis [104–106].
Experimental metastasis models
They are used to study the mechanism of metastasis after tail vein or intracardiac injection of human cancer cells directly into the circulation. The inoculated cells require an initial adaptation phase in which the tumor cells acclimate to a new microenvironment. Cells that survive the turbulence in circulation grow and metastasize in the distant organs such as the lungs [107, 108]. The developed metastases can be isolated and cultured in vitro as metastatic derived isogenic cell clones that exhibit higher tumorigenic and metastatic potential compared with the parental cell line [107, 108].
Tissue recombination models
Tissue recombination involves the isolation of fetal bladder mesenchyme from rats and then recombination with genetically altered human cell lines [67, 109]. It is possible to isolate mesenchymal tissue (and urothelium) from transgenic mice, enabling investigators to study the role of specific stromal contributions in bladder cancer progression. Another study reported the use of RT4 urothelial carcinoma cell line for tissue recombination experiments to identify the implications of p53 alterations and PTEN loss in urothelial carcinoma [67]. DeGraff et al. determined the influence of decreased FOXA1 expression for urothelial tumorigenicity using the tissue recombination system [109]. They used the non-tumorigenic RT4 cell line engineered to exhibit a low expression of FOXA1 and conducted tissue recombination experiments to determine the influence of decreased FOXA1 expression on bladder cancer cell proliferation. The resultant recombinants demonstrated enhanced RT4 proliferation and tumor volume. Majumdar et al. [110] identified the important role for endophilin A1 or SH3 domain containing GRB2 Like 2 (Sh3gl2) in bladder cancer using this model with RT4 depleted of Sh3gl2. They demonstrated that Sh3gl2 expression significantly declined in early-stage as well as in advanced bladder cancer, and silencing Sh3gl2 increased tumor volume. Therefore, this model enabled to understand the role of Sh3gl2 in bladder cancer as a predictor of the therapeutic response to RTK inhibitors.
The primary advantages of tissue recombination model include its relative cost-effectiveness, engineering flexibility and its ability to allow investigators to determine the impact of genetic alterations in tissue differentiation milieu. Tissue recombination is an efficient technique that allows testing the in vivo functions of different oncoproteins or tumor suppressors. These models offer an advantage over in vitro cell culture systems as they allow the determination of the influence of tumor microenvironment and facilitate the identification of stromal targets in cancer therapy. However, this model cannot recapitulate all aspects of the genetics, tumor heterogeneity, and complex etiology of bladder cancer. Also, the chances of discrepancies introduced through physical dissociation, lentiviral transduction, and transplantation could be high. Tissue recombination is a multistep approach which can be technically demanding and calls for enormous attention at all steps to avoid variability.
Patient-derived xenograft (PDX) models
PDX models involve the direct transplantation of primary human tumor tissues from patients into immune-deficient mice. This model precludes the use of highly passaged cell lines [111] and allows studying relative contributions of the stroma and urothelium, as it realistically restores the original environment of the human tumor in a mouse. In 1979, the earliest attempts were made for establishment of bladder cancer xenograft models by implanting patient-derived tumor tissues [112–115], and the characterization was restricted to phenotypic analyses, such as Hematoxylin and Eosin (H&E) or immunocytochemical staining, without molecular or genomic analyses. Park et al. [116] developed a bladder cancer PDX model and were the first to use established genomic analyses techniques such as short tandem repeat genotyping, mutational analysis, and array-CGH along with conventional histologic analysis. This model retained the histopathological features and molecular profile of the original tumors and showed the clinico-pathological heterogeneity of bladder cancer as well. Also, the concept of co-targeting PI3K and MAPK signaling for certain types of bladder cancer were initiated in pre-clinical studies using PDX models [117]. PDX models derived from patient tumor tissues tend to reflect the biological characteristics of human tumors more precisely than tumor cell lines. Fichtner et al. [118] reported that tumor cell lines may lack similarity to the original tumor and may not mimic the exact clinical situation. Additionally, tumor cells in culture are devoid of stromal and endothelial elements [119]. Thus, to cope with the microenvironment, these tumor cells undergo genetic and epigenetic changes that occur gradually over serial passages resulting in a phenotype different from the original tumor [120]. Moreover, PDX models are representative of the original tumor heterogeneous phenotype compared with cell lines and these models. However, some of the limitations of the use of primary human tissue xenografts include (i) patient-derived tumors may not establish a xenograft [121], (ii) Interference of mouse stroma with human stroma [121], (iii) tumor take rate may be as low as 35% [89, 122], (iv) time taken for tumor establishment may be long (~4–5 months), and (v) heterogeneous tumor tissue transplants may require a high number of replicates for statistical validation. Nevertheless, once PDX models are fully validated, it may have several clinical applications, including screening of multiple therapeutic agents simultaneously and interpreting the mechanisms of resistance. This will contribute towards turning molecularly targeted therapy into personalized cancer therapy.
Hollow fiber (HF) model
This model utilizes semipermeable biocompatible fibers that can be loaded with cancer cells and implanted surgically in animals, which can be treated by chemotherapeutic agents [123–126]. Moon et al. [127] filled polyvinylidene fluoride (PVDF) hollow fibers with human bladder cancer cell lines (CRL2742, 253JP, SW1710, HTB9) and surgically implanted these fibers subcutaneously and intraperitoneally into athymic nude mice. Subsequently, these mice were treated with gemcitabine, cisplatin, paclitaxel and after 6 days these fibers were recovered to determine cell viability. Although tedious, it is a cost-effective screening method because multiple cell lines can be evaluated for drug cytotoxicity within a short duration (assay time < 2 weeks).
Monitoring and evaluation of animal models for bladder cancer
Methods for tumor growth evaluation and therapeutic effects in animal models depend on the model system used. In subcutaneous tumors, measurement of subcutaneous tumor mass and imaging of tumor cells labeled with a luminescent or a fluorescent tag are instrumental for monitoring tumor growth and evaluation of diagnostic and responses summarized in? Detection of orthotopic and spontaneous urethral tumors cannot rely only on palpable bladder mass, weight loss, and urinalysis. A transurethral mini cystoscope is used as a non-invasive method for detecting and monitoring superficial tumors [129]. However, similar to white light cystoscopy used in the clinic, it is unreliable for differentiation between low- vs high-grade cancer. It can neither assess level of invasion or CIS nor differentiate CIS from inflammation [130]. The combination of mini trans-urethral cystoscopy with optical imaging modalities and intravesical injection of fluorescent or photosensitive dyes as fluorescein or proto-porphyrin precursors significantly improved the diagnostic value of mini-cystoscopy [130]. Other conventional imaging methods in animal studies include abdominal ultrasound, positron emission tomography-computerized tomography (PET-CT) and magnetic resonance imaging (MRI) can be used for early tumor detection as early as 14 days after tumor inoculation in mice [131]. High frequency and high resolution intravesical ultrasound (HRUS) were recently reported to demonstrate high sensitivity in monitoring tumor growth in orthotopic bladder cancer mouse model as a rapid, efficient and comparatively inexpensive imaging modality [132, 133]. Chan et al. utilized targeted contrast enhanced micro US imaging that enabled detection of vascular changes during bladder cancer development and progression [89]. Photoacoustic imaging (PAI) is a hybrid imaging modality that couples optical and ultrasound imaging in real time [134]. In this technology, biological tissue is exposed to a short laser pulse (nano-second) that is absorbed by hemoglobin and other chromophores in tissue resulting in thermo-elastic expansion, which produces broadband pulses (MHz) of acoustic energy. These pulses propagate to the tissue surface and are detected by an array of ultrasound transducers [135]. As ultrasound scattering is two to three orders of magnitude weaker than optical scattering in biological tissues [136], PAI can provide a better resolution than optical imaging for depths greater than 1 mm. In a recent study, three non-invasive optical imaging modalities including, BLI, HRUS and PAI were combined to increase sensitivity and improve accurate quantification of tumor growth in vivo. This multi-modality evaluation technique provided significant anatomical, molecular, and functional information [137]. Intensity-focused HIFU uses high-intensity ultrasound waves to precisely treat only the solid malignant tumors with minimal damage to surrounding tissue. HIFU ablation leads to coagulative thermal necrosis and cavitation damage due to absorption of ultrasound energy during transmission in tissue [87]. Imaging and diagnostic approaches described above are valuable means of non-invasive tumor evaluation, specially monitoring response to therapeutics in the live animal. These methods are often coupled and validated with biomarker validation from biological samples in the live animals or with histology and immunohistochemistry from bladder tissues dissected after animal euthanasia [137–139].
Bioluminescence (BLI) is another commonly used optical imaging modality for both in vitro and in vivo non-invasive monitoring of molecular and cellular activities. BLI is considered to be a far superior imaging methodology compared with fluorescence, as BLI does not need excitation and avoids the auto-fluorescence background signal and can be combined with other modalities as theranostic approach [140]. Jäger and colleagues combined BLI with percutaneous ultrasound-guided injection of luciferase-tagged cells bladder cancer cells into the anterior bladder wall. They monitored xenograft growth and perfusion in vivo longitudinally during therapy and were able to inject therapeutic agents directly into the tumor under ultrasound guidance [103].
For preclinical molecular assessment, the availability of linked color markers in lentiviral systems proved to be valuable tools for phenotypic analyses and metastasis tracking. This facilitates monitoring the dynamic changes in fluorescent signals, and drug responsiveness can be noninvasively tracked in host animal [141]. Fluorescence Imaging (FLI) is one of the new imaging tools, commonly used to monitor in vivo processes with reporters such as green fluorescent protein (GFP), red fluorescent protein (RFP) and near-infrared proteins [141, 142]. More fluorescent probes in the infrared (IR) spectrum have been developed to either tag tumor cells, or generate reporter mice to combine non-invasive imaging, tumor size monitoring and the response to therapy, along with tracking the expression of genes or molecules in specific cell types [122, 143–147]. Recently, an optimized CRISPR-Cas9 based light-inducible gene expression system in bladder cancer cell was developed [148] to control Tp53 gene expression in a dose-dependent manner and inhibit proliferation of cancer cell by modulating expression of p53 gene [148].
CONCLUSIONS AND PERSPECTIVES
Mouse models of urothelial cancer of the bladder present useful tools for advancing the early diagnosis of the disease, implementing tests for high risk population for primary prevention and early diagnosis and stratification of patients for a given therapy. Mouse models that represent the natural history of the disease provide the advantage of studying the early premalignant stages of the disease and allow for capturing of key biological and molecular features of human cancers that are not clinically encountered. They can be exploited not only for identification of early diagnostic biomarkers, but also for identification of biomarkers related to specific genetic factors associated with the human disease. The use of transgenic and genetically-engineered mouse models can help recognize the challenges associated with modelling tumor heterogeneity, tumor-stromal interactions and the contribution of the stromal compartment and the immune system to disease progression. Furthermore, the use of transplantable models (syngeneic and xenografts) can be combined to carefully study the events in the multistep cascades of cancer progression, invasiveness and metastasis. Combining multiple models together with imaging technologies will enable the generation of robust models that can further our knowledge of the pathobiology of the disease and allow for the discovery and validation of novel diagnostic and prognostic biomarkers and personalized therapies.
ACKNOWLEDGMENTS AND FUNDING
This work is supported by R01 CA193437 to NS.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013; 63:11–30.
2. Letasiova S, Medve’ova A, Sovcikova A, Dusinska M, Volkovova K, Mosoiu C, Bartonova A. Bladder cancer, a review of the environmental risk factors. Environ Health. 2012; 11:S11.
3. Andrew AS, Schned AR, Heaney JA, Karagas MR. Bladder cancer risk and personal hair dye use. Int J Cancer. 2004; 109:581–586.
4. Jankovic S, Radosavljevic V. Risk factors for bladder cancer. Tumori. 2007; 93:4–12.
5. Catsburg CE, Gago-Dominguez M, Yuan JM, Castelao JE, Cortessis VK, Pike MC, Stern MC. Dietary sources of N-nitroso compounds and bladder cancer risk: findings from the Los Angeles bladder cancer study. Int J Cancer. 2014; 134:125–135.
6. Aben KK, Baglietto L, Baffoe-Bonnie A, Coebergh JW, Bailey-Wilson JE, Trink B, Verbeek AL, Schoenberg MP, Alfred Witjes J, Kiemeney LA. Segregation analysis of urothelial cell carcinoma. Eur J Cancer. 2006; 42:1428–1433.
7. Kantor AF, Hartge P, Hoover RN, Fraumeni JF Jr. Familial and environmental interactions in bladder cancer risk. Int J Cancer. 1985; 35:703–706.
8. Murta-Nascimento C, Silverman DT, Kogevinas M, Garcia-Closas M, Rothman N, Tardon A, Garcia-Closas R, Serra C, Carrato A, Villanueva C, Dosemeci M, Real FX, Malats N. Risk of bladder cancer associated with family history of cancer: do low-penetrance polymorphisms account for the increase in risk? Cancer Epidemiol Biomarkers Prev. 2007; 16:1595–1600.
9. Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, Kamat AM, Siefker-Radtke AO, Tuziak T, Sabichi AL, Grossman HB, Benedict WF, Czerniak B. Focus on bladder cancer. Cancer Cell. 2004; 6:111–116.
10. Spiess PE, Czerniak B. Dual-track pathway of bladder carcinogenesis: practical implications. Arch Pathol Lab Med. 2006; 130:844–852.
11. Wu XR. Urothelial tumorigenesis: a tale of divergent pathways. Nature reviews Cancer. 2005; 5:713–725.
12. McConkey DJ, Lee S, Choi W, Tran M, Majewski T, Lee S, Siefker-Radtke A, Dinney C, Czerniak B. Molecular genetics of bladder cancer: Emerging mechanisms of tumor initiation and progression. Urologic oncology. 2010; 28:429–440.
13. Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. 2009; 374:239–249.
14. Prasad SM, Decastro GJ, Steinberg GD. Urothelial carcinoma of the bladder: definition, treatment and future efforts. Nat Rev Urol. 2011; 8:631–642.
15. Resnick MJ, Bassett JC, Clark PE. Management of superficial and muscle-invasive urothelial cancers of the bladder. Curr Opin Oncol. 2013; 25:281–288.
16. Clark PE, Agarwal N, Biagioli MC, Eisenberger MA, Greenberg RE, Herr HW, Inman BA, Kuban DA, Kuzel TM, Lele SM, Michalski J, Pagliaro LC, Pal SK, et al. Bladder cancer. J Natl Compr Canc Netw. 2013; 11:446–475.
17. Gontero P, Bohle A, Malmstrom PU, O’Donnell MA, Oderda M, Sylvester R, Witjes F. The role of bacillus Calmette-Guerin in the treatment of non-muscle-invasive bladder cancer. European urology. 2010; 57:410–429.
18. Redelman-Sidi G, Glickman MS, Bochner BH. The mechanism of action of BCG therapy for bladder cancer—a current perspective. Nat Rev Urol. 2014; 11:153–162.
19. Herr HW, Morales A. History of bacillus Calmette-Guerin and bladder cancer: an immunotherapy success story. J Urol. 2008; 179:53–56.
20. Elmamoun MH, Christmas TJ, Woodhouse CR. Destruction of the bladder by single dose Mitomycin C for low-stage transitional cell carcinoma (TCC)—avoidance, recognition, management and consent. BJU Int. 2014; 113:E34–38.
21. Pommier JD, Ben Lasfar N, Van Grunderbeeck N, Burdet C, Laouenan C, Rioux C, Pierre-Audigier C, Meybeck A, Choudat L, Benchikh A, Nguyen S, Bouvet E, Yeni P, et al. Complications following intravesical bacillus Calmette-Guerin treatment for bladder cancer: a case series of 22 patients. Infect Dis (Lond). 2015; 47:725–731.
22. Dash A, Pettus JA 4th, Herr HW, Bochner BH, Dalbagni G, Donat SM, Russo P, Boyle MG, Milowsky MI, Bajorin DF. A role for neoadjuvant gemcitabine plus cisplatin in muscle-invasive urothelial carcinoma of the bladder: a retrospective experience. Cancer. 2008; 113:2471–2477.
23. Zargar H, Espiritu PN, Fairey AS, Mertens LS, Dinney CP, Mir MC, Krabbe LM, Cookson MS, Jacobsen NE, Gandhi NM, Griffin J, Montgomery JS, Vasdev N, et al. Multicenter assessment of neoadjuvant chemotherapy for muscle-invasive bladder cancer. European urology. 2015; 67:241–249.
24. Yafi FA, North S, Kassouf W. First- and second-line therapy for metastatic urothelial carcinoma of the bladder. Current oncology (Toronto, Ont). 2011; 18:e25–34.
25. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014; 507:315–322.
26. Kobayashi T, Owczarek TB, McKiernan JM, Abate-Shen C. Modeling bladder cancer in mice: opportunities and challenges. Nature reviews Cancer. 2015; 15:42–54.
27. Clayson DB, Fishbein L, Cohen SM. Effects of stones and other physical factors on the induction of rodent bladder cancer. Food Chem Toxicol. 1995; 33:771–784.
28. Oliveira PA, Colaco A, De la Cruz PL, Lopes C. Experimental bladder carcinogenesis-rodent models. Experimental oncology. 2006; 28:2–11.
29. Vasconcelos-Nobrega C, Colaco A, Lopes C, Oliveira PA. Review: BBN as an urothelial carcinogen. In Vivo. 2012; 26:727–739.
30. van Moorselaar RJ, Ichikawa T, Schaafsma HE, Jap PH, Isaacs JT, van Stratum P, Ramaekers FC, Debruyne FM, Schalken JA. The rat bladder tumor model system RBT resembles phenotypically and cytogenetically human superficial transitional cell carcinoma. Urol Res. 1993; 21:413–421.
31. Crallan RA, Georgopoulos NT, Southgate J. Experimental models of human bladder carcinogenesis. Carcinogenesis. 2006; 27:374–381.
32. Babjuk M, Burger M, Zigeuner R, Shariat SF, van Rhijn BW, Comperat E, Sylvester RJ, Kaasinen E, BohleA, Palou Redorta J, Roupret M. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2013. European urology. 2013; 64:639–653.
33. Druckrey H, Preussmann R, Ivankovic S, Schmidt CH, Mennel HD, Stahl KW. [Selective induction of bladder cancer in rats by dibutyl- and n-butyl-n-butanol-nitrosamine]. [Article in German]. Z Krebsforsch. 1964; 66:280–290.
34. Fukushima S, Hirose M, Tsuda H, Shirai T, Hirao K. Histological classification of urinary bladder cancers in rats induced by N-butyl-n-(4-hydroxybutyl)nitrosamine. Gan. 1976; 67:81–90.
35. Hicks RM, Wakefield JS. Rapid induction of bladder cancer in rats with N-methyl-N-nitrosourea. I. Histology. Chem Biol Interact. 1972; 5:139–152.
36. Cohen SM. Urinary bladder carcinogenesis. Toxicol Pathol. 1998; 26:121–127.
37. Masui T, Dong Y, Yamamoto S, Takada N, Nakanishi H, Inada K, Fukushima S, Tatematsu M. p53 mutations in transitional cell carcinomas of the urinary bladder in rats treated with N-butyl-N-(4-hydroxybutyl)-nitrosamine. Cancer Lett. 1996; 105:105–112.
38. Williams PD, Lee JK, Theodorescu D. Molecular credentialing of rodent bladder carcinogenesis models. Neoplasia. 2008; 10:838–846.
39. Lu Y, Liu P, Wen W, Grubbs CJ, Townsend RR, Malone JP, Lubet RA, You M. Cross-species comparison of orthologous gene expression in human bladder cancer and carcinogen-induced rodent models. Am J Transl Res. 2010; 3:8–27.
40. Cohen SM. Comparative pathology of proliferative lesions of the urinary bladder. Toxicol Pathol. 2002; 30:663–671.
41. Yao R, Lemon WJ, Wang Y, Grubbs CJ, Lubet RA, You M. Altered gene expression profile in mouse bladder cancers induced by hydroxybutyl(butyl)nitrosamine. Neoplasia. 2004; 6:569–577.
42. Ahmad I, Morton JP, Singh LB, Radulescu SM, Ridgway RA, Patel S, Woodgett J, Winton DJ, Taketo MM, Wu XR, Leung HY, Sansom OJ. beta-Catenin activation synergizes with PTEN loss to cause bladder cancer formation. Oncogene. 2011; 30:178–189.
43. Cheng J, Huang H, Pak J, Shapiro E, Sun TT, Cordon-Cardo C, Waldman FM, Wu XR. Allelic loss of p53 gene is associated with genesis and maintenance, but not invasion, of mouse carcinoma in situ of the bladder. Cancer research. 2003; 63:179–185.
44. Zhang ZT, Pak J, Huang HY, Shapiro E, Sun TT, Pellicer A, Wu XR. Role of Ha-ras activation in superficial papillary pathway of urothelial tumor formation. Oncogene. 2001; 20:1973–1980.
45. Said N, Frierson HF, Sanchez-Carbayo M, Brekken RA, Theodorescu D. Loss of SPARC in bladder cancer enhances carcinogenesis and progression. The Journal of clinical investigation. 2013; 123:751–766.
46. Jiang T, Liu T, Li L, Yang Z, Bai Y, Liu D, Kong C. Knockout of phospholipase Cepsilon attenuates N-butyl-N-(4-hydroxybutyl) nitrosamine-induced bladder tumorigenesis. Molecular medicine reports. 2016; 13:2039–2045.
47. Kanemoto K, Fukuta K, Kawai N, Tozawa K, Ochiai M, Okamoto K, Ohnami S, Sakamoto H, Yoshida T, Kanai Y, Katoh M, Yasui T, Kohri K, et al. Genomic Landscape of Experimental Bladder Cancer in Rodents and Its Application to Human Bladder Cancer: Gene Amplification and Potential Overexpression of Cyp2a5/CYP2A6 Are Associated with the Invasive Phenotype. PloS one. 2016; 11:e0167374.
48. Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, Iorio F, Surdez D, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012; 483:570–575.
49. Johnson DT, Hooker E, Luong R, Yu EJ, He Y, Gonzalgo ML, Sun Z. Conditional Expression of the Androgen Receptor Increases Susceptibility of Bladder Cancer in Mice. PloS one. 2016; 11:e0148851.
50. Matsuo T, Miyata Y, Asai A, Sagara Y, Furusato B, Fukuoka J, Sakai H. Green Tea Polyphenol Induces Changes in Cancer-Related Factors in an Animal Model of Bladder Cancer. PloS one. 2017; 12:e0171091.
51. Shang Z, Li Y, Zhang M, Tian J, Han R, Shyr CR, Messing E, Yeh S, Niu Y, Chang C. Antiandrogen Therapy with Hydroxyflutamide or Androgen Receptor Degradation Enhancer ASC-J9 Enhances BCG Efficacy to Better Suppress Bladder Cancer Progression. Mol Cancer Ther. 2015; 14:2586–2594.
52. Gao Y, Shi Q, Xu S, Du C, Liang L, Wu K, Wang K, Wang X, Chang LS, He D, Guo P. Curcumin promotes KLF5 proteasome degradation through downregulating YAP/TAZ in bladder cancer cells. Int J Mol Sci. 2014; 15:15173–15187.
53. Reis LO, Pereira TC, Favaro WJ, Cagnon VH, Lopes-Cendes I, Ferreira U. Experimental animal model and RNA interference: a promising association for bladder cancer research. World J Urol. 2009; 27:353–361.
54. Spry LA, Zenser TV, Cohen SM, Davis BB. Role of renal metabolism and excretion in 5-nitrofuran-induced uroepithelial cancer in the rat. The Journal of clinical investigation. 1985; 76:1025–1031.
55. Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000; 26:99–109.
56. Lin JH, Zhao H, Sun TT. A tissue-specific promoter that can drive a foreign gene to express in the suprabasal urothelial cells of transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92:679–683.
57. Zhang ZT, Pak J, Shapiro E, Sun TT, Wu XR. Urothelium-specific expression of an oncogene in transgenic mice induced the formation of carcinoma in situ and invasive transitional cell carcinoma. Cancer research. 1999; 59:3512–3517.
58. Ayala de la Pena F, Kanasaki K, Kanasaki M, Tangirala N, Maeda G, Kalluri R. Loss of p53 and acquisition of angiogenic microRNA profile are insufficient to facilitate progression of bladder urothelial carcinoma in situ to invasive carcinoma. The Journal of biological chemistry. 2011; 286:20778–20787.
59. Reddy EP, Reynolds RK, Santos E, Barbacid M. A point mutation is responsible for the acquisition of transforming properties by the T24 human bladder carcinoma oncogene. Nature. 1982; 300:149–152.
60. Apolo AB, Milowsky M, Bajorin DF. Clinical states model for biomarkers in bladder cancer. Future oncology (London, England). 2009; 5:977–992.
61. He F, Melamed J, Tang MS, Huang C, Wu XR. Oncogenic HRAS Activates Epithelial-to-Mesenchymal Transition and Confers Stemness to p53-Deficient Urothelial Cells to Drive Muscle Invasion of Basal Subtype Carcinomas. Cancer research. 2015; 75:2017–2028.
62. Ahmad I, Singh LB, Foth M, Morris CA, Taketo MM, Wu XR, Leung HY, Sansom OJ, Iwata T. K-Ras and beta-catenin mutations cooperate with Fgfr3 mutations in mice to promote tumorigenesis in the skin and lung, but not in the bladder. Dis Model Mech. 2011; 4:548–555.
63. Ahmad I, Patel R, Liu Y, Singh LB, Taketo MM, Wu XR, Leung HY, Sansom OJ. Ras mutation cooperates with beta-catenin activation to drive bladder tumourigenesis. Cell Death Dis. 2011; 2:e124.
64. He F, Mo L, Zheng XY, Hu C, Lepor H, Lee EY, Sun TT, Wu XR. Deficiency of pRb family proteins and p53 in invasive urothelial tumorigenesis. Cancer research. 2009; 69:9413–9421.
65. Gao J, Huang HY, Pak J, Cheng J, Zhang ZT, Shapiro E, Pellicer A, Sun TT, Wu XR. p53 deficiency provokes urothelial proliferation and synergizes with activated Ha-ras in promoting urothelial tumorigenesis. Oncogene. 2004; 23:687–696.
66. Tsuruta H, Kishimoto H, Sasaki T, Horie Y, Natsui M, Shibata Y, Hamada K, Yajima N, Kawahara K, Sasaki M, Tsuchiya N, Enomoto K, Mak TW, et al. Hyperplasia and carcinomas in Pten-deficient mice and reduced PTEN protein in human bladder cancer patients. Cancer research. 2006; 66:8389–8396.
67. Puzio-Kuter AM, Castillo-Martin M, Kinkade CW, Wang X, Shen TH, Matos T, Shen MM, Cordon-Cardo C, Abate-Shen C. Inactivation of p53 and Pten promotes invasive bladder cancer. Genes & development. 2009; 23:675–680.
68. Yoo LI, Liu DW, Le Vu S, Bronson RT, Wu H, Yuan J. Pten deficiency activates distinct downstream signaling pathways in a tissue-specific manner. Cancer research. 2006; 66:1929–1939.
69. Saam JR, Gordon JI. Inducible gene knockouts in the small intestinal and colonic epithelium. J Biol Chem. 1999; 274:38071–38082.
70. Seager CM, Puzio-Kuter AM, Patel T, Jain S, Cordon-Cardo C, Mc Kiernan J, Abate-Shen C. Intravesical delivery of rapamycin suppresses tumorigenesis in a mouse model of progressive bladder cancer. Cancer Prev Res (Phila). 2009; 2:1008–1014.
71. Yang X, La Rosa FG, Genova EE, Huber K, Schaack J, Degregori J, Serkova NJ, Li Y, Su LJ, Kessler E, Flaig TW. Simultaneous activation of Kras and inactivation of p53 induces soft tissue sarcoma and bladder urothelial hyperplasia. PloS one. 2013; 8:e74809.
72. Delto JC, Kobayashi T, Benson M, McKiernan J, Abate-Shen C. Preclinical analyses of intravesical chemotherapy for prevention of bladder cancer progression. Oncotarget. 2013; 4:269–276. doi: 10.18632/oncotarget.852.
73. Gabriel U, Bolenz C, Michel MS. Experimental models for therapeutic studies of transitional cell carcinoma. Anticancer research. 2007; 27:3163–3171.
74. Truschel ST, Ruiz WG, Shulman T, Pilewski J, Sun TT, Zeidel ML, Apodaca G. Primary uroepithelial cultures. A model system to analyze umbrella cell barrier function. The Journal of biological chemistry. 1999; 274:15020–15029.
75. Celis A, Rasmussen HH, Celis P, Basse B, Lauridsen JB, Ratz G, Hein B, Ostergaard M, Wolf H, Orntoft T, Celis JE. Short-term culturing of low-grade superficial bladder transitional cell carcinomas leads to changes in the expression levels of several proteins involved in key cellular activities. Electrophoresis. 1999; 20:355–361.
76. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010; 38:D652–657.
77. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–607.
78. Almeida JL, Cole KD, Plant AL. Standards for Cell Line Authentication and Beyond. PLoS Biol. 2016; 14:e1002476.
79. Bisson JF, Parache RM, Droulle P, Notter D, Vigneron C, Guillemin F. A new method of implanting orthotopic rat bladder tumor for experimental therapies. Int J Cancer. 2002; 102:280–285.
80. Chade DC, Andrade PM, Borra RC, Leite KR, Andrade E, Villanova FE, Srougi M. Histopathological characterization of a syngeneic orthotopic murine bladder cancer model. International braz j urol. 2008; 34:220–226; discussion 226–229.
81. Gunther JH, Frambach M, Deinert I, Brandau S, Jocham D, Bohle A. Effects of acetylic salicylic acid and pentoxifylline on the efficacy of intravesical BCG therapy in orthotopic murine bladder cancer (MB49). J Urol. 1999; 161:1702–1706.
82. Werthman PE, Drazan KE, Rosenthal JT, Khalili R, Shaked A. Adenoviral-p53 gene transfer to orthotopic and peritoneal murine bladder cancer. J Urol. 1996; 155:753–756.
83. Dobek GL, Godbey WT. An orthotopic model of murine bladder cancer. Journal of visualized experiments. 2011.
84. Soloway MS, Masters S. Urothelial susceptibility to tumor cell implantation: influence of cauterization. Cancer. 1980; 46:1158–1163.
85. Yang Xh, Ren Ls, Wang Gp, Zhao Ll, Zhang H, Mi Zg, Bai X. A New Method of Establishing Orthotopic Bladder Transplantable Tumor in Mice. Cancer Biology & Medicine. 2012; 9:261–265.
86. Soloway MS. Intravesical and systemic chemotherapy of murine bladder cancer. Cancer research. 1977; 37:2918–2929.
87. Zhang Z, Xu X, Zhang X, Chen X, Chen Q, Dong L, Hu Z, Li J, Gao J. The therapeutic potential of SA-sCD40L in the orthotopic model of superficial bladder cancer. Acta Oncol. 2011; 50:1111–1118.
88. Weldon TE, Soloway MS. Susceptibility of urothelium to neoplastic cellular implantation. Urology. 1975; 5:824–827.
89. Chan E, Patel A, Heston W, Larchian W. Mouse orthotopic models for bladder cancer research. BJU Int. 2009; 104:1286–1291.
90. Cheon J, Moon DG, Cho HY, Park HS, Kim JJ, Gardner TA, Kao C. Adenovirus-mediated suicide-gene therapy in an orthotopic murine bladder tumor model. Int J Urol. 2002; 9:261–267.
91. Smith EB, Schwartz M, Kawamoto H, You X, Hwang D, Liu H, Scherr DS. Antitumor effects of imidazoquinolines in urothelial cell carcinoma of the bladder. J Urol. 2007; 177:2347–2351.
92. Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol. 2007; 170:793–804.
93. Soloway MS, Martino C, Hyatt C, Marrone JC. Immunogenicity of N-[-4-(5-nitro-2-furyl)-2-thiazolyl]formamide-induced bladder cancer. Natl Cancer Inst Monogr. 1978; 293–300.
94. Summerhayes IC, Franks LM. Effects of donor age on neoplastic transformation of adult mouse bladder epithelium in vitro. J Natl Cancer Inst. 1979; 62:1017–1023.
95. Fodor I, Timiryasova T, Denes B, Yoshida J, Ruckle H, Lilly M. Vaccinia virus mediated p53 gene therapy for bladder cancer in an orthotopic murine model. J Urol. 2005; 173:604–609.
96. Hanel EG, Xiao Z, Wong KK, Lee PW, Britten RA, Moore RB. A novel intravesical therapy for superficial bladder cancer in an orthotopic model: oncolytic reovirus therapy. J Urol. 2004; 172:2018–2022.
97. Loskog AS, Fransson ME, Totterman TT. AdCD40L gene therapy counteracts T regulatory cells and cures aggressive tumors in an orthotopic bladder cancer model. Clin Cancer Res. 2005; 11:8816–8821.
98. Miyazaki J, Nishiyama H, Yano I, Nakaya A, Kohama H, Kawai K, Joraku A, Nakamura T, Harashima H, Akaza H. The therapeutic effects of R8-liposome-BCG-CWS on BBN-induced rat urinary bladder carcinoma. Anticancer research. 2011; 31:2065–2071.
99. Reis LO, Ferreira U, Billis A, Cagnon VH, Favaro WJ. Anti-angiogenic effects of the superantigen staphylococcal enterotoxin B and bacillus Calmette-Guerin immunotherapy for nonmuscle invasive bladder cancer. J Urol. 2012; 187:438–445.
100. Shen ZJ, Wang Y, Ding GQ, Pan CW, Zheng RM. Study on enhancement of fibronectin-mediated bacillus Calmette-Guerin attachment to urinary bladder wall in rabbits. World J Urol. 2007; 25:525–529.
101. Said N, Smith S, Sanchez-Carbayo M, Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. The Journal of clinical investigation. 2011; 121:132–147.
102. Said N, Sanchez-Carbayo M, Smith SC, Theodorescu D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. The Journal of clinical investigation. 2012; 122:1503–1518.
103. Jager W, Moskalev I, Janssen C, Hayashi T, Awrey S, Gust KM, So AI, Zhang K, Fazli L, Li E, Thuroff JW, Lange D, Black PC. Ultrasound-guided intramural inoculation of orthotopic bladder cancer xenografts: a novel high-precision approach. PloS one. 2013; 8:e59536.
104. Black PC, Dinney CP. Bladder cancer angiogenesis and metastasis—translation from murine model to clinical trial. Cancer metastasis reviews. 2007; 26:623–634.
105. Lodillinsky C, Rodriguez V, Vauthay L, Sandes E, Casabe A, Eijan AM. Novel invasive orthotopic bladder cancer model with high cathepsin B activity resembling human bladder cancer. J Urol. 2009; 182:749–755.
106. Wilmanns C, Fan D, Obrian C, Radinsky R, Bucana C, Tsan R, Fidler I. Modulation of Doxorubicin sensitivity and level of p-glycoprotein expression in human colon-carcinoma cells by ectopic and orthotopic environments in nude-mice. Int J Oncol. 1993; 3:413–422.
107. Overdevest JB, Thomas S, Kristiansen G, Hansel DE, Smith SC, Theodorescu D. CD24 offers a therapeutic target for control of bladder cancer metastasis based on a requirement for lung colonization. Cancer research. 2011; 71:3802–3811.
108. Smith SC, Nicholson B, Nitz M, Frierson HF Jr, Smolkin M, Hampton G, El-Rifai W, Theodorescu D. Profiling bladder cancer organ site-specific metastasis identifies LAMC2 as a novel biomarker of hematogenous dissemination. Am J Pathol. 2009; 174:371–379.
109. DeGraff DJ, Clark PE, Cates JM, Yamashita H, Robinson VL, Yu X, Smolkin ME, Chang SS, Cookson MS, Herrick MK, Shariat SF, Steinberg GD, Frierson HF, et al. Loss of the urothelial differentiation marker FOXA1 is associated with high grade, late stage bladder cancer and increased tumor proliferation. PloS one. 2012; 7:e36669.
110. Majumdar S, Gong EM, Di Vizio D, Dreyfuss J, Degraff DJ, Hager MH, Park PJ, Bellmunt J, Matusik RJ, Rosenberg JE, Adam RM. Loss of Sh3gl2/endophilin A1 is a common event in urothelial carcinoma that promotes malignant behavior. Neoplasia. 2013; 15:749–760.
111. DeGraff DJ, Robinson VL, Shah JB, Brandt WD, Sonpavde G, Kang Y, Liebert M, Wu XR, Taylor JA 3rd. Current preclinical models for the advancement of translational bladder cancer research. Mol Cancer Ther. 2013; 12:121–130.
112. Kovnat A, Armitage M, Tannock I. Xenografts of human bladder cancer in immune-deprived mice. Cancer research. 1982; 42:3696–3703.
113. Kovnat A, Buick RN, Connolly JG, Jewett MA, Keresteci AG, Tannock IF. Comparison of growth of human bladder cancer in tissue culture or as xenografts with clinical and pathological characteristics. Cancer research. 1984; 44:2530–2533.
114. Russell PJ, Raghavan D, Gregory P, Philips J, Wills EJ, Jelbart M, Wass J, Zbroja RA, Vincent PC. Bladder cancer xenografts: a model of tumor cell heterogeneity. Cancer research. 1986; 46:2035–2040.
115. Sufrin G, McGarry MP, Sandberg AA, Murphy GP. Heterotransplantation of human transitional cell carcinoma in athymic mice. J Urol. 1979; 121:159–161.
116. Park B, Jeong BC, Choi YL, Kwon GY, Lim JE, Seo SI, Jeon SS, Lee HM, Choi HY, Lee KS. Development and characterization of a bladder cancer xenograft model using patient-derived tumor tissue. Cancer science. 2013; 104:631–638.
117. Cirone P, Andresen CJ, Eswaraka JR, Lappin PB, Bagi CM. Patient-derived xenografts reveal limits to PI3K/mTOR- and MEK-mediated inhibition of bladder cancer. Cancer Chemother Pharmacol. 2014; 73:525–538.
118. Fichtner I, Slisow W, Gill J, Becker M, Elbe B, Hillebrand T, Bibby M. Anticancer drug response and expression of molecular markers in early-passage xenotransplanted colon carcinomas. Eur J Cancer. 2004; 40:298–307.
119. Li H, Fan X, Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. Journal of cellular biochemistry. 2007; 101:805–815.
120. Karam JA, Zhang XY, Tamboli P, Margulis V, Wang H, Abel EJ, Culp SH, Wood CG. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. European urology. 2011; 59:619–628.
121. Pan CX, Zhang H, Tepper CG, Lin TY, Davis RR, Keck J, Ghosh PM, Gill P, Airhart S, Bult C, Gandara DR, Liu E and de Vere White RW. Development and Characterization of Bladder Cancer Patient-Derived Xenografts for Molecularly Guided Targeted Therapy. PloS one. 2015; 10:e0134346.
122. Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, Gill H, Presti J Jr, Chang HY, van de Rijn M, Shortliffe L, Weissman IL. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:14016–14021.
123. Casciari JJ, Hollingshead MG, Alley MC, Mayo JG, Malspeis L, Miyauchi S, Grever MR, Weinstein JN. Growth and chemotherapeutic response of cells in a hollow-fiber in vitro solid tumor model. J Natl Cancer Inst. 1994; 86:1846–1852.
124. Hall LA, Krauthauser CM, Wexler RS, Hollingshead MG, Slee AM, Kerr JS. The hollow fiber assay: continued characterization with novel approaches. Anticancer Res. 2000; 20:903–911.
125. Mi Q, Lantvit D, Reyes-Lim E, Chai H, Zhao W, Lee IS, Peraza-Sanchez S, Ngassapa O, Kardono LB, Riswan S, Hollingshead MG, Mayo JG, Farnsworth NR, et al. Evaluation of the potential cancer chemotherapeutic efficacy of natural product isolates employing in vivo hollow fiber tests. J Nat Prod. 2002; 65:842–850.
126. Morrell A, Jayaraman M, Nagarajan M, Fox BM, Meckley MR, Ioanoviciu A, Pommier Y, Antony S, Hollingshead M, Cushman M. Evaluation of indenoisoquinoline topoisomerase I inhibitors using a hollow fiber assay. Bioorg Med Chem Lett. 2006; 16:4395–4399.
127. Moon KH, Han BK, Jeong SJ, Hong SK, Byun SS, Lee SE. In vivo Hollow Fiber Assay for Anticancer Drugs’ Responsiveness in a Bladder Cancer Model. Korean J Urol. 2008; 49:392–397.
128. Cao J, Campbell J, Liu L, Mason RP, Lippert AR. In Vivo Chemiluminescent Imaging Agents for Nitroreductase and Tissue Oxygenation. Anal Chem. 2016; 88:4995–5002.
129. Bolenz C, Wenzel M, Cao Y, Trojan L, Hacker A, Arancibia MF, Alken P, Michel MS. Newly developed mini-endoscope for diagnosis and follow-up of orthotopic bladder transitional-cell carcinoma in vivo. J Endourol. 2007; 21:789–794.
130. Zlatev DV, Altobelli E, Liao JC. Advances in imaging technologies in the evaluation of high-grade bladder cancer. Urol Clin North Am. 2015; 42:147–157, vii.
131. Kikuchi E, Xu S, Ohori M, Matei C, Lupu M, Menendez S, Koutcher JA, Bochner BH. Detection and quantitative analysis of early stage orthotopic murine bladder tumor using in vivo magnetic resonance imaging. J Urol. 2003; 170:1375–1378.
132. Satoh H, Morimoto Y, Arai T, Asanuma H, Kawauchi S, Seguchi K, Kikuchi M, Murai M. Intravesical ultrasonography for tumor staging in an orthotopically implanted rat model of bladder cancer. J Urol. 2007; 177:1169–1173.
133. Foster WK, Ford NL. Investigating the effect of longitudinal micro-CT imaging on tumour growth in mice. Phys Med Biol. 2011; 56:315–326.
134. Kolkman RG, Brands PJ, Steenbergen W, van Leeuwen TG. Real-time in vivo photoacoustic and ultrasound imaging. Journal of biomedical optics. 2008; 13:050510.
135. Luke GP, Yeager D, Emelianov SY. Biomedical applications of photoacoustic imaging with exogenous contrast agents. Annals of biomedical engineering. 2012; 40:422–437.
136. Jose J, Willemink RG, Steenbergen W, Slump CH, van Leeuwen TG, Manohar S. Speed-of-sound compensated photoacoustic tomography for accurate imaging. Medical physics. 2012; 39:7262–7271.
137. Scheepbouwer C, Meyer S, Burggraaf MJ, Jose J, Molthoff CF. A Multimodal Imaging Approach for Longitudinal Evaluation of Bladder Tumor Development in an Orthotopic Murine Model. PloS one. 2016; 11:e0161284.
138. Cutz JC, Guan J, Bayani J, Yoshimoto M, Xue H, Sutcliffe M, English J, Flint J, LeRiche J, Yee J, Squire JA, Gout PW, Lam S, et al. Establishment in severe combined immunodeficiency mice of subrenal capsule xenografts and transplantable tumor lines from a variety of primary human lung cancers: potential models for studying tumor progression-related changes. Clin Cancer Res. 2006; 12:4043–4054.
139. Wang Y, Revelo MP, Sudilovsky D, Cao M, Chen WG, Goetz L, Xue H, Sadar M, Shappell SB, Cunha GR, Hayward SW. Development and characterization of efficient xenograft models for benign and malignant human prostate tissue. Prostate. 2005; 64:149–159.
140. Keyaerts M, Caveliers V, Lahoutte T. Bioluminescence imaging: looking beyond the light. Trends in molecular medicine. 2012; 18:164–172.
141. Bouvet M, Hoffman RM. Tumor imaging technologies in mouse models. Methods in molecular biology. 2015; 1267:321–348.
142. Hoffman RM. Advantages of multi-color fluorescent proteins for whole-body and in vivo cellular imaging. J Biomed Opt. 2005; 10:41202.
143. Pan HF, Ding JX, Liang RR, Tao ZD, Liu MW, Zhang SJ, Xu JH. [Quenched fluorescein: a reference dye for instrument response function of TCSPC]. [Article in Chinese]. Guang Pu Xue Yu Guang Pu Fen Xi. 2014; 34:2284–2288.
144. Redelman-Sidi G, Iyer G, Solit DB, Glickman MS. Oncogenic activation of Pak1-dependent pathway of macropinocytosis determines BCG entry into bladder cancer cells. Cancer Res. 2013; 73:1156–1167.
145. Volkmer JP, Sahoo D, Chin RK, Ho PL, Tang C, Kurtova AV, Willingham SB, Pazhanisamy SK, Contreras-Trujillo H, Storm TA, Lotan Y, Beck AH, Chung BI, et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:2078–2083.
146. Shin K, Lim A, Odegaard JI, Honeycutt JD, Kawano S, Hsieh MH, Beachy PA. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat Cell Biol. 2014; 16:469–478.
147. Shin K, Lim A, Zhao C, Sahoo D, Pan Y, Spiekerkoetter E, Liao JC, Beachy PA. Hedgehog signaling restrains bladder cancer progression by eliciting stromal production of urothelial differentiation factors. Cancer Cell. 2014; 26:521–533.
148. Lin F, Dong L, Wang W, Liu Y, Huang W, Cai Z. An Efficient Light-Inducible P53 Expression System for Inhibiting Proliferation of Bladder Cancer Cell. Int J Biol Sci. 2016; 12:1273–1278.