
INTRODUCTION
Myelodysplastic syndromes (MDS) are a group of heterogeneous hematopoietic stem cell (HSC) disease, characterized by ineffective hematopoiesis, a variable degree of peripheral cytopenia, hypercellular bone marrow with morphologically defined dysplasia of cell lineages, and an increased propensity of evolving to acute myeloid leukemia (AML) [1, 2]. MDS often affects the elderly male patients with ages over 70 years. The incidence of MDS is reckoned about 3-5/100000 persons [3]. This incidence is predicted to escalate remarkably with an increase of ages [4, 5]. MDS comprises several different subtypes, including refractory anemia (RA), refractory anemia with ringed sideroblasts (RARS), refractory cytopenia with multilineage dysplasia (RCMD), refractory cytopenia with multilineage dysplasia and ringed sideroblasts (RCMD-RS), refractory anemia with excess blasts (RAEB), myelodysplastic syndrome unclassified (MDS-U), MDS associated with isolated del(5q) [6, 7].
It has been reported that approximately 70% of MDS patients have clonal chromosomal aberrations at initial diagnosis [8]. These chromosomal abnormalities have a great influence on the behavior of malignant cells, disease evolution, response to therapeutic drugs, and overall survival of MDS patients [9, 10]. A large variety of different chromosomal abnormalities have been depicted in MDS, such as loss or gain of chromosomal fragments, acquired uniparental disomy (UPD), and complex karyotypes [11]. Chromosomal loss may engender deletion of tumor suppressor genes (TSGs). Alternatively, chromosomal gain may activate oncogenes [12]. UPD defined as two copies of a chromosomal pair originate from one single parent during meiosis, may also increase genomic instability by activating oncogenes or inactivating TSGs in MDS [13]. Moreover, complex karyotype which is defined as the existence of ≥ 3 chromosomal aberrations usually contains both numerical and structural alterations [14]. Complex karyotype often implies an increased risk of progressing to AML and unfavorable outcomes in MDS patients [15]. These chromosomal aberrations appear to be mechanisms to interpret disease progression. Additionally, environmental risk factors may also engender the pathogenesis of MDS. For instance, iron overload-induced oxidative stress may inhibit hematopoiesis by altering the supportive bone marrow stroma environment [5]. Epigenetic alterations such as TET2 on chromosome 4q24 and IDH2 mutation on 15q26.1 for mitochondrial dysfunction also contribute to the pathogenesis of MDS [2]. Thus, detection of chromosomal abnormalities may afford valuable information for accurate diagnosis of MDS, and may also optimize current therapeutic strategies for MDS patients.
During the past several decades, a series of techniques have been developed for detecting chromosomal aberrations in MDS, including metaphase cytogenetics (MC), fluorescence in situ hybridization (FISH), spectral karyotyping (SKY), single nucleotide polymorphism arrays (SNP-A) genotyping, array-based comparative genomic hybridization (a-CGH), and targeted DNA sequencing. The advent of these techniques has contributed to the investigation of chromosomal changes in MDS, including unbalanced chromosomal deletions and gains as well as balanced translocations [16]. The chromosomal findings will enhance our understanding of the pathogenesis of MDS.
In this review, we will not only recapitulate the current knowledge of common chromosomal aberrations in MDS, but also summarize the techniques for detecting chromosomal aberrations in MDS. Specifically, we will also introduce the application, advantages and limitations of each technique.
COMMON CHROMOSOMAL ABERRATIONS IN MDS
Del(5q), trisomy 8, del(20q), del(7q), monosomy 7, and complex karyotypes are the commonest chromosomal aberrations in MDS [17]. Loss, gain, and UPD of genomic materials in these chromosomes are associated with the initiation and progression of MDS. So we focus on reviewing these common chromosomal aberrations in MDS (Table 1).
Table 1: Chromosomal aberrations in MDS
Aberration type |
Position |
Significance |
Reference |
|---|---|---|---|
del(5q) |
5q31 |
AML evolution |
21 |
trisomy 8 |
cT8M |
intermediate-risk |
38,39 |
del(20q) |
20q11.2-q13.1 |
exacerbate malignancy |
46,47 |
del (7q) |
7q22,7q34 |
contribute to hematopoietic aberration |
56 |
monosomy 7 |
-7 |
higher-risk, poor prognosis |
66–68 |
complex karyotype |
multiple |
unfavorable outcome |
76 |
Deletion 5q
Heterozygous, interstitial deletions of chromosome 5q (del(5q)) are the commonest cytogenetic aberration in MDS [18], which accounts for approximately 30% of MDS subtypes [19]. MDS patients with isolated del(5q) often have a good prognosis, however, when accompanied with additional chromosomal aberrations, their prognosis becomes unfavorable [20]. The chromosome band 5q31 is the most frequently deleted region, including 2 different commonly deleted regions (CDRs). The proximal 5q31.1-q31.2 region is putatively related to an increased risk of evolving to AML [21]. Another distal CDR located in the 5q32-q33 bands is considered to involve the pathogenesis of 5q− syndrome and often prefigures a favorable prognosis [22]. Haploinsufficiency of many candidate genes may potentially alter hematopoiesis, resulting in the phenotype of MDS patients with del(5q) and malignant transformation [23]. For instance, RPS14 gene encodes a ribosomal protein small subunit 14 which influences the maturation of erythroid progenitor cells [24, 25]. Haploinsufficiency of RPS14 gene may affect the p53 pathway, and the subsequent loss of p53 rescues erythropoiesis and contributes to clonal progression [26]. Pathogenetic mechanisms in del(5q) MDS seem to involve hemizygous mutations in addition to haploinsufficiency, and may be modified by other somatic alterations influencing genes on other chromosomes [27]. Moreover, selection of particular treatment may rely on the presence of specific chromosomal aberrations. Low-risk, transfusion-dependent MDS patients with del(5q) are reported to respond well to lenalidomide [28–30]. So accurate detection of del(5q) is not only important for precise diagnosis of MDS, but also vital for individualized treatment of MDS patients.
Trisomy 8
Trisomy 8 (+8) is one of the most frequent chromosomal gains in adult MDS patients [31], which accounts for 5% of all MDS patients in Western countries [32] and roughly 30-35% in Chinese MDS patients [33, 34]. According to the new revised IPSS (IPSS-R), isolated trisomy 8 in MDS is classified as intermediate cytogenetic risk group and should be considered with adequate evidence to diagnose MDS in patients with hypercellular or normal bone marrow [35]. Despite the association between particular chromosomal lesions and somatic mutations has not been clarified, several studies have reported that trisomy 8 was related to an IDH or ASXL1 mutation in MDS harboring trisomy 8 [36, 37].
Trisomy 8 (+8) can also be identified as a constitutional mosaicism (cT8M). A study have analyzed the existence of +8 in CD3+ lymphocytes and granulocytes from peripheral blood, as well as in oral mucosa cells from MDS patients with +8, in order to elucidate the incidence of cT8M in MDS and provide an accurate diagnostic and prognostic value for isolated +8. Cytogenetic analysis of peripheral blood found trisomy 8 in 5% to 65% of cells. FISH analysis also revealed trisomy 8 in 3% to 74% of granulocytes from all patients studied [38]. Complexity of chromosomal aberrations have a great impact on the overall survival (OS) of MDS patients. Those patients with isolated trisomy 8 have a median OS from 11 to 25 months, while patients with bone marrow blasts ≥ 5% combining trisomy 8 have relatively shorter OS and increased AML transformation [39].
Clonal heterogeneity has been regarded as a specific cytogenetic characteristic of MDS. Trisomy 8 may “come and go” as an independent clone or a single cell aberration. Usually, clonal evolution is a predictor for disease progression [3]. MDS patients with trisomy 8 and del(5q) as independent clone had a remarkably longer time to progress to AML than those with clonal evolution [40]. Analysis of whole gene expression revealed that most genes on chromosome 8 are overexpressed in AML trisomy 8. Hence the gene-dose effect may lead to leukemic progression of MDS with trisomy 8 [41]. Furthermore, MDS patients with trisomy 8 are more likely to respond to immunosuppressive agents than other subtypes of MDS [42].
Deletion 20q
An interstitial deletion of chromosome 20q (del(20q)), is also prevalent in MDS, accounting for 3–7% of all MDS patients [43, 44]. Isolated del(20q) has been found both in primary and therapy-related MDS patients. Those patients often manifest anemia and thrombocytopenia, which involve bone marrow dysplasia [45]. Del(20q) is considered to derive from a pluripotent stem cell and may exacerbate malignancy due to the deletion of tumor suppressor genes [46]. In the past several years, many studies have been initiated to detect the CDR on chromosome 20. The CDR can be narrowed on chromosomal bands from 20q11.2 to 20q13.1, with variable sizes from 2.6 to 10.4 Mb [47]. These CDRs often subsume several key genes that may affect the pathogenesis and course of MDS. For example, the E2F1 gene on band 20q11.2 encodes a transcription factor, which involves in cell cycle control, proliferation modulation and p53-mediated apoptosis. Increased levels and activity of E2F1 transcription factor have been observed in myelodysplastic bone marrow [48, 49]. Isolated del(20q) in MDS is a favorable recurrent chromosomal aberration, with higher reticulocyte counts, fewer bone marrow blasts, and an indolent clinical course [50, 51]. The survival of patients with a del(20q) was considered to be significantly longer than other MDS patients [52]. Thus, MDS patients with isolated del(20q) usually have a relatively favorable prognosis.
However, as the size of chromosome 20 is too small, the traditional cytogenetic analysis is difficult to pinpoint chromosomal regions for its deletion [53]. So MDS with del(20q) may be further stratified by additional cytogenetic and molecular techniques.
Deletion 7q
Deletion of chromosome 7q (del (7q)) is also frequently found in MDS and are associated with a poor prognosis [54]. The percentage of del(7q) cells is significantly higher in HSC and progenitor compartments than in lymphocytes of MDS patients [55]. Multiple investigations of MDS samples with interstitial del (7q) have identified 3 potential CDRs at chromosome bands 7q22, 7q34, and 7q35-36 [55]. Specifically, deletion of 7q22 in bone marrow cells could contribute to hematopoietic abnormalities, such as vandalized lymphoid repopulating potential, myeloid output discrepancy, and a remarkable proliferation of HSC [56]. Del (7q) may engender the haploinsufficiency of several critical genes implicated in hematological malignancies, subsuming MLL3, CUX1, and EZH2 [57–59], which are responsible for the leukemic progression of MDS [60]. Furthermore, UPD 7q and homozygous EZH2 mutation have been found in 10% of MDS patients. These chromosomal abnormalities often portend clonal evolution and highlight the vital role of del (7q) in the pathogenesis of MDS [61].
Monosomy 7
Monosomal karyotype (MK) is defined as the existence of a single autosomal monosomy related with at least one additional structural alteration in the same clone, or at least 2 autosomal monosomies [62]. Monosomy 7 is the most prevalent chromosomal abnormality of MDS in childhood, and often exists as the sole cytogenetically visible chromosomal aberration [63, 64]. Immunophenotypic analysis of immature stem and progenitor cell compartments from patients with monosomy 7, showed expansion and dominance of the malignant –7 clone in granulocyte, macrophage progenitors, and other CD45RA+ progenitor compartments [65]. The monosomy 7 clone had a relative disadvantage in erythroid differentiation [65].
Monosomy 7 has been regarded as an independent predictor of survival in patients with higher-risk MDS. The addition of MK as a binary variable could improve the predictive accuracy of current models to estimate the survival of patients with MDS [66]. For example, a recent study has retrospectively analyzed 2080 primary patients, in order to elucidate the prognostic significance of MK in Chinese MDS patients. They have found that MK was significantly related to elderly patients, higher bone marrow blasts and relatively unfavorable cytogenetics. Monosomies of chromosome 5/7 were significantly associated with shorter OS by multivariate analysis [67]. Another study has investigated if an MK is related to OS independent of the number of cytogenetic aberrations in a population-based MDS cohort. They have found that monosomy 7 was responsible for worse OS in the entire cohort (median 6 vs 39 months), including those with a coexisting complex karyotypes (6 vs 17 months) [68]. Thus, MK predicts inferior survival of complex karyotypes in MDS patients.
Isolated monosomy 7 or monosomy 7 plus one additional aberration is associated with a median survival of 14.0 months and thus with an intermediate risk [19]. Consequently, early stem cell transplantation is recommended as soon as a monosomy 7 clone was detected [69].
Monosomy 7 is also the commonest chromosome abnormality in the course of evolution from MDS to AML in patients with different bone marrow failure syndromes and DNA repair deficiencies [70]. Thus, there are underlying aberrations leading to these constitutional disorders that also predispose to MDS and AML.
Complex karyotype
Complex karyotype (CK) was defined as the existence of at least three chromosomal alterations and was especially prevalent in secondary MDS [71, 72]. Complex karyotypes and large number of chromosomal abnormalities may reflect an inherent chromosomal instability that contributes to disease progression. A higher incidence of complex karyotypes represents more aggressive disease [73]. The pathogenic mechanisms leading to complex karyotypes in MDS still remain vague. UPD may contribute to genomic instability by activating oncogenes and inactivating tumor suppressor genes, facilitating the development and progression of complex chromosomal aberrations [74]. Moreover, complex karyotypes in MDS may arise from gradual acquisition of genetic changes in individual cells during clonal evolution or by extensive chromosome fragmentation and reorganization at a single event known as chromothripsis [75].
Patients with complex karyotypes often imply an unfavorable outcome, a shorter median OS, only 3 months, and propensity toward malignant progression. Multiple chromosomal aberrations often portend an adverse prognosis and difficult treatment [76].
Analysis of complex karyotypes facilitates the identification of latent unbalanced chromosomal alterations and candidate regions of genes responsible for the progression of MDS. These regions can then be investigated further at the molecular level, which may render more accurate diagnosis of MDS and help to find potential targets for therapeutic interventions in the future.
TECHNIQUES FOR DETECTING CHROMOSOMAL ABERRATIONS IN MDS
Cytogenetic findings are important for the diagnosis, prognosis evaluation and treatment selection of MDS patients [77]. Many techniques have been developed to detect chromosomal aberrations in MDS, such as metaphase cytogenetic (MC) analysis, FISH, Array-CGH, SNP-Array, SKY, and NGS (Table 2). Although these techniques are varying in depth, scope and cost, they are important for detecting diverse chromosomal abnormalities in MDS.
Table 2: Different techniques for detecting chromosomal aberrations in MDS
Technique |
Application |
Advantage |
Shortcoming |
Price |
|---|---|---|---|---|
MC |
visible chromosomal aberrations |
Simple, whole chromosomal view |
Low resolution, Can’t detect UPD |
800 rmb |
FISH |
small and hidden chromosomal aberrations |
Not rely on proliferating cells, High sensitivity |
only detect particular chromosomal aberrations |
2000 rmb |
SKY |
Unknown and complex chromosomal aberrations |
display better pictures of karyotypes |
Can’t detect structural aberration, low resolution |
3500 rmb |
SNP-A |
Cryptic and complex chromosomal aberrations |
high-resolution, can detect UPD |
Can’t detect balanced translocation and inversion |
5000 rmb |
Array-CGH |
Detect CNV and UPD |
genome-wide analysis high-resolution |
Can’t detect balanced rearrangements, low-level mosaicism and polyploidy |
4500 rmb |
Sequencing |
CNV and structural variants, unknown mutation or aberrations |
genome-wide analysis improved sensitivity monitor clonal mutations |
Expensive, time-consuming, complicated bioinformatic analysis |
6000 rmb |
Metaphase cytogenetics
Metaphase Cytogenetics (MC) still remains the gold standard for detection of chromosomal aberrations in MDS [78]. This method can’t only identify unbalanced chromosomal lesions, including loss, gain, and trisomy (Figure 1), but also detect balanced chromosomal defects, such as translocation and inversion [79]. It can provide a whole chromosomal view of visible aberrations in chromosome number and structure simultaneously [80]. Furthermore, the simplicity of MC assay allows for the feasibility of discerning single cellular clones [81]. The chromosomal aberrations detected by MC often have strong prognostic value. These are the major advantages of MC.
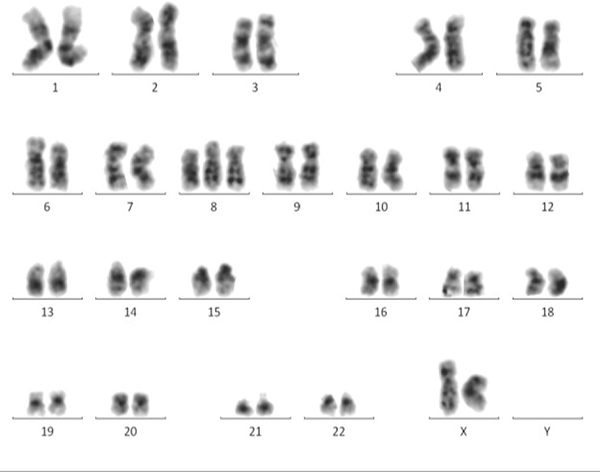
Figure 1: MC displays a whole chromosomal view of visible aberrations in a MDS patient. Trisomy 8 is distinctly revealed by MC.
However, about 40–50% of MDS patients don’t exhibit karyotype aberrations assessed by standard MC [82, 83]. The resolution of conventional MC is quite low. This method also requires proliferating cells, and to a large extent, relies on specialist experience for discriminating meaningful data [84]. As a result, traditional MC should be initiated by cytogenetic labs with rich experience in MDS.
Furthermore, even if 50% of MDS patients with abnormal karyotypes are identified by MC, it still can’t completely eliminate the existence of some cryptic chromosomal defects which often evades the detection by MC with relatively low resolution [85]. Most importantly, MC is unable to identify UPD because the chromosome banding patterns remain unaltered [86]. In some patients, MC may even fail to come up with informative results due to low resolution and non-dividing cells. Consequently, the technical limitations of MC may lead to underestimate of the extent of chromosomal abnormalities.
FISH
FISH is another important technique for molecular investigation of chromosomal alterations in MDS. FISH analysis can come up with valuable information involving the existence of small or hidden chromosomal abnormalities in patients with minor clones (Figure 2). FISH is also able to assess large numbers of interphase nuclei, so it can overcome some limitations of standard MC [87]. The diagnostic information from FISH is important for stratification of MDS into the appropriate subtypes and cytogenetic risk groups [88].
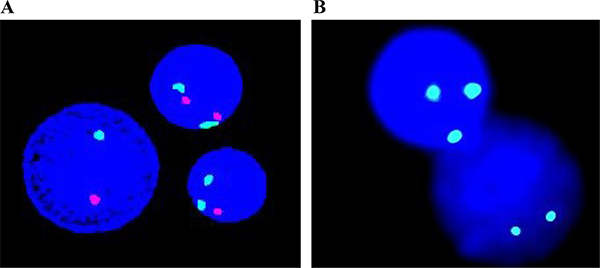
Figure 2: Based on individually designed probes, FISH helps to detect specific chromosomal aberrations in MDS. (A) D7Z1/D7S486 probe indicates deletion on 7p11.1-q11.1/7q31. (B) D8Z2 probe reveals trisomy 8.
Compared with conventional chromosome banding analysis, FISH has some remarkable advantages. First, FISH can be utilized for non-proliferating cells, a large amount of cells can be assessed with relatively less lab expenditure. Second, the sensitivity of FISH is comparatively higher than traditional MC analysis, so some submicroscopic chromosomal aberrations can also be identified by FISH [89]. Third, FISH with a panel of probes can also be used to monitor disease progression and response to therapy, especially if it could be performed on peripheral blood samples [90]. Moreover, FISH can also putatively be applied to monitor lower-risk patients receiving supportive care only. The detection of cytogenetic aberrations can potentially facilitate early therapeutic interventions [91].
However, FISH should only complement MC, it will not radically substitute classical chromosomal banding analysis for an initial diagnosis of MDS. The major limitation of FISH is that it only detect particular structural or numerical chromosomal aberrations at specific locus, so those chromosomal abnormalities regarding other regions may be neglected [92, 93]. In addition, the cutoffs for positive assessment of an overwhelming majority of FISH probes are limited to approximately 5%. When metaphase cells are fewer than 20, FISH is recommended to promote the accuracy for probing recurrent MDS-related chromosome aberrations [94]. So FISH is not normally recommended for an initial screening of cytogenetic aberrations in MDS.
SKY
Spectral karyotyping (SKY) is a novel technique for detecting chromosomal aberrations in myeloid malignancies. Based on the advancement of FISH, this technique has combined chromosome painting and multi-color fluorescence, enabling each of 23 chromosome pairs to be stained with a different color [95]. So SKY has supplemented the images of chromosomes by MC and FISH for exhibiting specific chromosomes.
The advantage of SKY is that it can unravel chromosomes of unknown origin, and clarify if one of the parents is a carrier of a balanced structural abnormality. SKY can also detect chromosomal rearrangements and minimal aberrations in MDS patients with complex karyotypes, displaying better pictures of karyotypes [96]. So SKY can overcome some defaults of the traditional banding methods, and reveal previously unrecognized chromosomal translocations. Furthermore, SKY is still crucial in detecting complex chromosomal abnormalities, so it also contributes to finding new MDS subgroups. In combination with other cytogenetic and molecular techniques, SKY may become a very powerful tool for the diagnosis, treatment and prognosis of MDS patients [97].
However, SKY can’t be used to detect structural aberrations, such as deletion, insertion, inversion, and duplication in the same clone, because these chromosomes are displayed with the same color. The resolution limit of SKY is roughly 1–2 Mb, similar to traditional chromosome banding techniques, so minor structural aberrations of less than one band cannot be visualized [98]. Therefore, SKY should combine with additional high-resolution techniques to pinpoint the site of chromosomal breakage in MDS.
Genome-wide SNP array
The rapid progress of high-resolution genome-wide single nucleotide polymorphism-array (SNP-A) technology is characterized by hybridization of sample DNA to probes specific for allelic variants in microarrays which can detect both CNV and UPD [99]. SNP-A can precisely pinpoint the location and size of submicroscopic chromosomal aberrations. Moreover, high-resolution SNP-A has become one of the most powerful techniques to detect complex chromosomal lesions in myeloid malignancies. For instance, a recent study has utilized Affymetrix CytoScan 750K microarray to detect chromosomal loss, gain, UPD, and complex karyotypes in 162 MDS patients. Approximately 34.57% of MDS patients with complex chromosomal abnormalities were identified by CytoScan 750K microarray [100]. So SNP-A has the potential to become a very useful diagnostic technique and may complement MC and FISH in clinical cytogenetic settings.
SNP-A has many advantages over conventional techniques. First, the resolution of SNP-A is much higher than MC or FISH. Those small cryptic chromosomal loss and gain can be identified by SNP-A. Second, SNP-A doesn’t require live proliferating cells, hence it can still yield diagnostic information when routine cytogenetic methods are not feasible. Third, cryptic UPD with preserved chromosomal bandings can also be detected by SNP-A [101].
However, there are still some limitations of SNP-A. It can’t identify balanced translocations and inversions. The sensitivity of SNP-A still remains a relatively low level. The median proportion of aberrant cell clones identifiable by SNP-A is 20–30% [102].
Two factors should be considered when applying SNP-A as a clinical cytogenetic tool. First, whether SNP-A could provide additional information to routine MC and FISH. Second, whether the chromosomal aberrations detected by SNP-A have any potential clinical significance [102]. Hence combined application of SNP-A with traditional cytogenetic techniques may maximize the detection rate of chromosomal abnormalities in MDS.
Microarray-based comparative genome hybridization (Array-CGH)
Array-CGH is an important technique for detecting CNV and UPD together in a single experiment. This method utilized competitive hybridization of differentially labeled fragmented sample DNA and control DNA to the genome at the microarray platform to detect chromosomal aberrations [103]. The fluorescence ratio of sample vs. control DNA hybridization signals is detected at different positions at the genome and yields information regarding the relative DNA copy number in the assayed genome in comparison with the normal diploid genome. Copy number alterations (CNA) of subtle chromosomal regions including potential candidate genes can be revealed [104]. The genomic resolution of the Array-CGH platform depends on the size of inserts, the space and length of DNA probes spotted on the array. So Array-CGH provides a genome-wide analysis of CNV at very high resolution. It has been reported that commercially available Array-CGH platforms have roughly 50-fold higher resolution than traditional cytogenetic methods, and can reveal chromosomal aberrations in 15% to 20% of samples [105].
The major advantage of Array-CGH over traditional cytogenetic methods is that it can detect DNA copy number alterations simultaneously at multiple loci in the genome, and can analyze a large number of genes on microarray in a single experiment [106]. Moreover, Array-CGH does not need a live, mitotically proliferating cells and can be initiated using DNA extracted from archived specimens. Analysis of Array-CGH is also objective, and feasible to automation, and can be implemented without special training or equipment [107]. However, balanced rearrangements, low-level mosaicism and polyploidy can’t be detected by Array-CGH [108].
In general, some additional cryptic chromosomal abnormalities detected by Array-CGH may improve the current diagnosis of MDS and help the assignment of appropriate phenotypes.
Sequencing-based technologies
More recently, the progress of targeted sequencing technology has also provided valuable information for detecting chromosomal aberrations. Next-generation sequencing (NGS) technology has been utilized to detect CNV and structural variants in myeloid malignant genomes [109, 110]. These sequencing-based technologies have several advantages over conventional cytogenetic methods: (1) higher “depth” and genome-wide detection of chromosomal aberrations for the patients. (2) improved sensitivity which can detect mutations that are present in only ~1% cells. (3) potential to monitor clonal aberrations during treatment [111]. A recent study has selectively sequenced a small portion of human genome termed Selected Target Regions (SeTRs), in order to identify genome-wide CNV, LOH and UPD. They found that SeTRs are covered by 99.73%~99.95% with adequate depth. This new technique can identify chromosomal aberrations exempt from using a matched sample or familial information [112]. Furthermore, another study has found that NGS is highly-sensitive for accurate testing and quantification of various RUNX1 abnormalities with subsequent personalized monitoring of disease progression and therapeutic efficacy [113]. In addition, NGS can also detect chromosomal inversions and intra-chromosomal rearrangements which were not identified by SNP arrays [114]. For example, NGS data can partially simulate a large amount of breakpoints in chromosome 5. The complex structural rearrangement in chromothripsis and intra-chromosomal breakpoints were confined to a localized region of the genome [115]. So the breakpoints of deletions could be mapped with single-nucleotide resolution by NGS.
However, limitations of the sequencing-based technologies should be concerned. These methods are expensive and time-consuming, also require complicated bioinformatic analysis [116]. Owing to the inherent sequencing error rate of NGS, it is tough to reliably detect low-frequency variants [117].
Furthermore, RNA-Seq techniques can also identify novel mutation or aberrations in MDS. For instance, tRNA fragments can be accurately detected through miRNA sequencing data, the expression of these species may be useful in the diagnosis of MDS and the prediction of response to therapy [118]. Large expression differences were found for MDS-associated and novel miRNAs, which were predicted to regulate disease stage specific molecular functions and pathways, including apoptosis and response to DNA damage. Extensive post-translation editing via transfer RNAs (tRNAs) in high-grade MDS may provide a potential link for reduced apoptosis, a hallmark for this disease stage [119]. Another study has applied RNA-seq technology to study the transcriptome on 20 MDS patients and 5 age-matched controls. They identified 38 mutated genes contributing to MDS pathogenesis, including 37 genes that haven’t been reported previously. Hence RNA-seq is critical for identifying novel mutated genes in MDS. The most recurrent mutation happened in gene IFRD1 [120]. These results provide us new insights into the pathogenesis of MDS, which may inspire further investigations of diagnostic biomarkers and targeted therapies for MDS patients.
In general, sequencing-based technologies can detect a full spectrum of genomic aberrations, including single nucleotide variant (SNV), small insertion/deletion (indel), CNV, UPD, translocation, and novel mutations in MDS. The limitations of NGS have to be considered.
Combination of multiple techniques
Given the application region, advantages and shortcomings of different techniques for detecting chromosomal aberrations in MDS, it is better to combine multiple techniques if necessary. MC in conjunction with FISH proved to be powerful to better identify additional chromosomal aberrations in MDS patients [88]. SNP-A is able to scan the whole genome and cryptic chromosomal aberrations in MDS, yet MC also can reveal balanced translocation and inversion, so the diagnostic information from MC and SNP-A are complementary, combined application of MC and SNP-A may maximize the detection rate of chromosomal abnormalities [102]. Combined aCGH with SNP-A could simultaneously detect CNV and UPD at high-resolution in a single experiment. It also provides allelic information on deletions, duplications, and amplifications [80]. CGH+SNP microarray could reveal different genetic profiles that may underlie differences in phenotypes and genetic aberrations with potential prognostic impact on MDS patients [85].
CONCLUSIONS AND PERSPECTIVES
Although chromosomal aberrations, such as Del(5q), trisomy 8, del(20q), del(7q), monosomy 7, and complex karyotypes are prevalent in MDS, the rapid technological progress in recent years has enabled a more precise detection of multiple chromosomal abnormalities. The new findings may enhance our understanding of the molecular mechanisms underlying the pathogenesis and malignant evolution of MDS. In the future, for the multiple techniques to enter clinical application, efforts should be made to standardize the assays and refine the bioinformatic analysis for data interpretation. Further technological advance should also be made to overcome the limitations of diverse techniques.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by National Natural Science Foundation of China (NO. 81372407 and NO. 81670123).
CONFLICTS OF INTEREST
None of the authors declare any relevant conflicts of interest.
REFERENCES
1. Fozza C, Crobu V, Isoni MA, Dore F. The immune landscape of myelodysplastic syndromes. Crit Rev Oncol Hematol. 2016; 107:90–99.
2. Ganguly BB, Kadam NN. Mutations of myelodysplastic syndromes (MDS): an update. Mutat Res Rev Mutat Res. 2016; 769:47–62.
3. Schlegelberger B, Göhring G, Thol F, Heuser M. Update on cytogenetic and molecular changes in myelodysplastic syndromes. Leuk Lymphoma. 2012; 53:525–36.
4. Neukirchen J, Schoonen WM, Strupp C, Gattermann N, Aul C, Haas R, Germing U. Incidence and prevalence of myelodysplastic syndromes: data from the Düsseldorf MDS-registry. Leuk Res. 2011; 35:1591–96.
5. Almeida A, Fenaux P, List AF, Raza A, Platzbecker U, Santini V. Recent advances in the treatment of lower-risk non-del(5q) myelodysplastic syndromes (MDS). Leuk Res. 2017; 52:50–57.
6. Myelodysplastic Syndromes - MDS. Subtypes and Classification. Approved by the Cancer. Net Editorial Board, 10/2016. http://www.cancer.net/cancer-types/myelodysplastic-syndromes-mds/subtypes-and-classification.
7. van Spronsen MF, Ossenkoppele GJ, Westers TM, van de Loosdrecht AA. Prognostic relevance of morphological classification models for myelodysplastic syndromes in an era of the revised International Prognostic Scoring System. Eur J Cancer. 2016; 56:10–20.
8. Giagounidis A, Haase D. Morphology, cytogenetics and classification of MDS. Best Pract Res Clin Haematol. 2013; 26:337–53.
9. Arenillas L, Mallo M, Ramos F, Guinta K, Barragán E, Lumbreras E, Larráyoz MJ, De Paz R, Tormo M, Abáigar M, Pedro C, Cervera J, Such E, et al. Single nucleotide polymorphism array karyotyping: a diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer. 2013; 52:1167–77.
10. Tiu RV, Gondek LP, O’Keefe CL, Elson P, Huh J, Mohamedali A, Kulasekararaj A, Advani AS, Paquette R, List AF, Sekeres MA, McDevitt MA, Mufti GJ, Maciejewski JP. Prognostic impact of SNP array karyotyping in myelodysplastic syndromes and related myeloid malignancies. Blood. 2011; 117:4552–60.
11. Nolte F, Hofmann WK. Myelodysplastic syndromes: molecular pathogenesis and genomic changes. Ann Hematol. 2008; 87:777–95.
12. Slovak ML, Smith DD, Bedell V, Hsu YH, O’Donnell M, Forman SJ, Gaal K, McDaniel L, Schultz R, Ballif BC, Shaffer LG. Assessing karyotype precision by microarray-based comparative genomic hybridization in the myelodysplastic/myeloproliferative syndromes. Mol Cytogenet. 2010; 3:23.
13. Svobodova K, Zemanova Z, Lhotska H, Novakova M, Podskalska L, Belickova M, Brezinova J, Sarova I, Izakova S, Lizcova L, Berkova A, Siskova M, Jonasova A, et al. Copy number neutral loss of heterozygosity at 17p and homozygous mutations of TP53 are associated with complex chromosomal aberrations in patients newly diagnosed with myelodysplastic syndromes. Leuk Res. 2016; 42:7–12.
14. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, Wheatley K, Harrison CJ, Burnett AK, National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010; 116:354–65.
15. Gao S, Li Z, Fu JH, Hu XH, Xu Y, Jin ZM, Tang XW, Han Y, Chen SN, Sun AN, Wu DP, Qiu HY. Decitabine in the Treatment of Acute Myeloid Leukemia and Myelodysplastic Syndromes, Which Combined with Complex Karyotype Respectively. Asian Pac J Cancer Prev. 2015; 16:6627–32.
16. da Silva FB, Traina F. Metaphase cytogenetics and single nucleotide polymorphism arrays in myeloid malignancies. Rev Bras Hematol Hemoter. 2015; 37:71–72.
17. Nazha A, Sekeres MA, Gore SD, Zeidan AM. Molecular Testing in Myelodysplastic Syndromes for the Practicing Oncologist: Will the Progress Fulfill the Promise? Oncologist. 2015; 20:1069–76.
18. Komrokji RS, Padron E, Ebert BL, List AF. Deletion 5q MDS: molecular and therapeutic implications. Best Pract Res Clin Haematol. 2013; 26:365–75.
19. Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, Kundgen A, Lübbert M, Kunzmann R, Giagounidis AA, Aul C, Trümper L, Krieger O, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007; 110:4385–95.
20. Schanz J, Steidl C, Fonatsch C, Pfeilstöcker M, Nösslinger T, Tuechler H, Valent P, Hildebrandt B, Giagounidis A, Aul C, Lübbert M, Stauder R, Krieger O, et al. Coalesced multicentric analysis of 2,351 patients with myelodysplastic syndromes indicates an underestimation of poor-risk cytogenetics of myelodysplastic syndromes in the international prognostic scoring system. J Clin Oncol. 2011; 29:1963–70.
21. Zemanova Z, Michalova K, Buryova H, Brezinova J, Kostylkova K, Bystricka D, Novakova M, Sarova I, Izakova S, Lizcova L, Ransdorfova S, Krejcik Z, Merkerova MD, et al. Involvement of deleted chromosome 5 in complex chromosomal aberrations in newly diagnosed myelodysplastic syndromes (MDS) is correlated with extremely adverse prognosis. Leuk Res. 2014; 38:537–44.
22. Boultwood J, Pellagatti A, McKenzie AN, Wainscoat JS. Advances in the 5q- syndrome. Blood. 2010; 116:5803–11.
23. Eisenmann KM, Dykema KJ, Matheson SF, Kent NF, DeWard AD, West RA, Tibes R, Furge KA, Alberts AS. 5q- myelodysplastic syndromes: chromosome 5q genes direct a tumor-suppression network sensing actin dynamics. Oncogene. 2009; 28:3429–41.
24. Wu L, Li X, Xu F, Zhang Z, Chang C, He Q. Low RPS14 expression in MDS without 5q - aberration confers higher apoptosis rate of nucleated erythrocytes and predicts prolonged survival and possible response to lenalidomide in lower risk non-5q- patients. Eur J Haematol. 2013; 90:486–93.
25. Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C, Beier F, Brümmendorf TH, Germing U, Platzbecker U, Büsche G, Knüchel R, Chen MC, et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016; 22:288–97.
26. Ear J, Hsueh J, Nguyen M, Zhang Q, Sung V, Chopra R, Sakamoto KM, Lin S. A Zebrafish Model of 5q-Syndrome Using CRISPR/Cas9 Targeting RPS14 Reveals a p53-Independent and p53-Dependent Mechanism of Erythroid Failure. J Genet Genomics. 2016; 43:307–18.
27. Hosono N, Makishima H, Mahfouz R, Przychodzen B, Yoshida K, Jerez A, LaFramboise T, Polprasert C, Clemente MJ, Shiraishi Y, Chiba K, Tanaka H, Miyano S, et al. Recurrent genetic defects on chromosome 5q in myeloid neoplasms. Oncotarget. 2017; 8:6483–95. https://doi.org/10.18632/oncotarget.14130.
28. Mortensen TB, Frederiksen H, Marcher CW, Preiss B. Refractory primary immune thrombocytopenia with subsequent del(5q) MDS: complete remission of both after lenalidomide. BMJ Case Rep. 2017 Jan 4;2017.
29. Leitch HA, Buckstein R, Shamy A, Storring JM. The immunomodulatory agents lenalidomide and thalidomide for treatment of the myelodysplastic syndromes: a clinical practice guideline. Crit Rev Oncol Hematol. 2013; 85:162–92.
30. Butrym A, Lech-Maranda E, Patkowska E, Kumiega B, Bieniaszewska M, Mital A, Madry K, Torosian T, Wichary R, Rybka J, Warzocha K, Mazur G. Polish experience of lenalidomide in the treatment of lower risk myelodysplastic syndrome with isolated del(5q). BMC Cancer. 2015; 15:508.
31. Konuma T, Miyazaki Y, Uchida N, Ohashi K, Kondo T, Nakamae H, Takahashi S, Mori T, Ozawa Y, Kato C, Iwato K, Fukuda T, Ichinohe T, et al, and Adult Myelodysplastic Syndrome Working Group of the Japan Society for Hematopoietic Cell Transplantation. Outcomes of Allogeneic Hematopoietic Stem Cell Transplantation in Adult Patients with Myelodysplastic Syndrome Harboring Trisomy 8. Biol Blood Marrow Transplant. 2017; 23:75–80.
32. Paulsson K, Johansson B. Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol (Paris). 2007; 55:37–48.
33. Irons RD, Wang X, Gross SA, Bao L, Ryder J, Chen Y, Chen H, Sun H, Zhou J, Ji M, Du X, Fu H, Lin G. Prevalence of MDS subtypes in Shanghai, China: a comparison of the World Health Organization and French American British classifications. Leuk Res. 2006; 30:769–75.
34. Xiao Y, Wei J, Chen Y, Zhang K, Zhou J, Zhang Y. Trisomy 8 is the most frequent cytogenetic abnormality in de novo myelodysplastic syndrome in China. Onkologie. 2012; 35:100–06.
35. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F, Bennett JM, Bowen D, Fenaux P, Dreyfus F, Kantarjian H, Kuendgen A, Levis A, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012; 120:2454–65.
36. Caramazza D, Lasho TL, Finke CM, Gangat N, Dingli D, Knudson RA, Siragusa S, Hanson CA, Pardanani A, Ketterling RP, Tefferi A. IDH mutations and trisomy 8 in myelodysplastic syndromes and acute myeloid leukemia. Leukemia. 2010; 24:2120–22.
37. Chen TC, Hou HA, Chou WC, Tang JL, Kuo YY, Chen CY, Tseng MH, Huang CF, Lai YJ, Chiang YC, Lee FY, Liu MC, Liu CW, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014; 4:e177.
38. Saumell S, Solé F, Arenillas L, Montoro J, Valcárcel D, Pedro C, Sanzo C, Luño E, Giménez T, Arnan M, Pomares H, De Paz R, Arrizabalaga B, et al. Trisomy 8, a Cytogenetic Abnormality in Myelodysplastic Syndromes, Is Constitutional or Not? PLoS One. 2015; 10:e0129375.
39. Yue QF, Chen L, She XM, Hu B, Hu Y, Zou P, Liu XY. Clinical Prognostic Factors in 86 Chinese Patients with Primary Myelodysplastic Syndromes and Trisomy 8: A Single Institution Experience. Yonsei Med J. 2016; 57:358–64.
40. Qu S, Xu Z, Zhang Y, Qin T, Zhang T, Cui R, Xiao Z. Impacts of cytogenetic categories in the Revised International Prognostic Scoring System on the prognosis of primary myelodysplastic syndromes: results of a single-center study. Leuk Lymphoma. 2012; 53:940–46.
41. Langemeijer SM, Mariani N, Knops R, Gilissen C, Woestenenk R, de Witte T, Huls G, van der Reijden BA, Jansen JH. Apoptosis-related gene expression profiling in hematopoietic cell fractions of MDS patients. PLoS One. 2016; 11:e0165582.
42. Sloand EM, Mainwaring L, Fuhrer M, Ramkissoon S, Risitano AM, Keyvanafar K, Lu J, Basu A, Barrett AJ, Young NS. Preferential suppression of trisomy 8 compared with normal hematopoietic cell growth by autologous lymphocytes in patients with trisomy 8 myelodysplastic syndrome. Blood. 2005; 106:841–51.
43. Valli R, Pressato B, Marletta C, Mare L, Montalbano G, Curto FL, Pasquali F, Maserati E. Different loss of material in recurrent chromosome 20 interstitial deletions in Shwachman-Diamond syndrome and in myeloid neoplasms. Mol Cytogenet. 2013; 6:56.
44. Jana B, Khanfar A, Ninan M. Durable hematological and major cytogenetic response in a patient with isolated 20q deletion myelodysplastic syndrome treated with lenalidomide. Case Rep Oncol Med. 2014; 2014:949515.
45. Kanagal-Shamanna R, Yin CC, Miranda RN, Bueso-Ramos CE, Wang XI, Muddasani R, Medeiros LJ, Lu G. Therapy-related myeloid neoplasms with isolated del(20q): comparison with cases of de novo myelodysplastic syndrome with del(20q). Cancer Genet. 2013; 206:42–46.
46. Ganguly BB, Dolai TK, De R, Kadam NN. Spectrum of complex chromosomal aberrations in a myelodysplastic syndrome and a brief review. J Cancer Res Ther. 2016; 12:1203–06.
47. Huh J, Tiu RV, Gondek LP, O’Keefe CL, Jasek M, Makishima H, Jankowska AM, Jiang Y, Verma A, Theil KS, McDevitt MA, Maciejewski JP. Characterization of chromosome arm 20q abnormalities in myeloid malignancies using genome-wide single nucleotide polymorphism array analysis. Genes Chromosomes Cancer. 2010; 49:390–99.
48. Saberwal G, Broderick E, Janssen I, Shetty V, Alvi S, Lisak L, Venugopal P, Raza A, Mundle SD. Involvement of cyclin D1 and E2F1 in intramedullary apoptosis in myelodysplastic syndromes. J Hematother Stem Cell Res. 2003; 12:443–50.
49. Saberwal G, Lucas S, Janssen I, Deobhakta A, Hu WY, Galili N, Raza A, Mundle SD. Increased levels and activity of E2F1 transcription factor in myelodysplastic bone marrow. Int J Hematol. 2004; 80:146–54.
50. Braun T, de Botton S, Taksin AL, Park S, Beyne-Rauzy O, Coiteux V, Sapena R, Lazareth A, Leroux G, Guenda K, Cassinat B, Fontenay M, Vey N, et al. Characteristics and outcome of myelodysplastic syndromes (MDS) with isolated 20q deletion: a report on 62 cases. Leuk Res. 2011; 35:863–67.
51. Courville EL, Singh C, Yohe S, Linden MA, Naemi K, Berger M, Ustun C, McKenna RW, Dolan M. Patients With a History of Chemotherapy and Isolated del(20q) With Minimal Myelodysplasia Have an Indolent Course. Am J Clin Pathol. 2016; 145:459–66.
52. Bacher U, Haferlach T, Schnittger S, Zenger M, Meggendorfer M, Jeromin S, Roller A, Grossmann V, Krauth MT, Alpermann T, Kern W, Haferlach C. Investigation of 305 patients with myelodysplastic syndromes and 20q deletion for associated cytogenetic and molecular genetic lesions and their prognostic impact. Br J Haematol. 2014; 164:822–33.
53. Wu C, Pan J, Qiu H, Xue Y, Chen S, Wu Y, Zhang J, Bai S, Wang Y, Shen J, Gong Y. Microarray CGH analysis of hematological patients with del(20q). Int J Hematol. 2015; 102:617–25.
54. van Spronsen MF, Witte BI, Ossenkoppele GJ, Westers TM, van de Loosdrecht AA. Response to letter commenting on: prognostic relevance of morphological classification models for myelodysplastic syndromes in an era of the revised International Prognostic Scoring System. Eur J Cancer. 2017; 72:269–71.
55. Elias HK, Schinke C, Bhattacharyya S, Will B, Verma A, Steidl U. Stem cell origin of myelodysplastic syndromes. Oncogene. 2014; 33:5139–50.
56. Wong JC, Weinfurtner KM, Alzamora MP, Kogan SC, Burgess MR, Zhang Y, Nakitandwe J, Ma J, Cheng J, Chen SC, Ho TT, Flach J, Reynaud D, et al. Functional evidence implicating chromosome 7q22 haploinsufficiency in myelodysplastic syndrome pathogenesis. eLife. 2015; 4:4.
57. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, Stock W, LeBeau MM, Shannon KM, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014; 25:652–65.
58. McNerney ME, Brown CD, Wang X, Bartom ET, Karmakar S, Bandlamudi C, Yu S, Ko J, Sandall BP, Stricker T, Anastasi J, Grossman RL, Cunningham JM, et al. CUX1 is a haploinsufficient tumor suppressor gene on chromosome 7 frequently inactivated in acute myeloid leukemia. Blood. 2013; 121:975–83.
59. Hasegawa N, Oshima M, Sashida G, Matsui H, Koide S, Saraya A, Wang C, Muto T, Takane K, Kaneda A, Shimoda K, Nakaseko C, Yokote K, Iwama A. Impact of combinatorial dysfunctions of Tet2 and Ezh2 on the epigenome in the pathogenesis of myelodysplastic syndrome. Leukemia. 2017; 31:861–71.
60. Hosono N, Makishima H, Jerez A, Yoshida K, Przychodzen B, McMahon S, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Sanada M, Gómez-Seguí I, Verma AK, et al. Recurrent genetic defects on chromosome 7q in myeloid neoplasms. Leukemia. 2014; 28:1348–51.
61. Hemmat M, Chen W, Anguiano A, Naggar ME, Racke FK, Jones D, Wang Y, Strom CM, Chang K, Boyar FZ. Submicroscopic deletion of 5q involving tumor suppressor genes (CTNNA1, HSPA9) and copy neutral loss of heterozygosity associated with TET2 and EZH2 mutations in a case of MDS with normal chromosome and FISH results. Mol Cytogenet. 2014; 7:35.
62. Kayser S, Zucknick M, Döhner K, Krauter J, Köhne CH, Horst HA, Held G, von Lilienfeld-Toal M, Wilhelm S, Rummel M, Germing U, Götze K, Nachbaur D, et al, and German-Austrian AML Study Group. Monosomal karyotype in adult acute myeloid leukemia: prognostic impact and outcome after different treatment strategies. Blood. 2012; 119:551–58.
63. Hasle H, Niemeyer CM. Advances in the prognostication and management of advanced MDS in children. Br J Haematol. 2011; 154:185–95.
64. Hasle H. Myelodysplastic and myeloproliferative disorders of childhood. Hematology Am Soc Hematol Educ Program. 2016; 2016:598–604.
65. Dimitriou M, Woll PS, Mortera-Blanco T, Karimi M, Wedge DC, Doolittle H, Douagi I, Papaemmanuil E, Jacobsen SE, Hellström-Lindberg E. Perturbed hematopoietic stem and progenitor cell hierarchy in myelodysplastic syndromes patients with monosomy 7 as the sole cytogenetic abnormality. Oncotarget. 2016; 7:72685–98. https://doi.org/10.18632/oncotarget.12234.
66. Xing R, Li C, Gale RP, Zhang Y, Xu Z, Qin T, Li B, Fang L, Zhang H, Pan L, Hu N, Qu S, Xiao Z. Monosomal karyotype is an independent predictor of survival in patients with higher-risk myelodysplastic syndrome. Am J Hematol. 2014; 89:E163–68.
67. Zhang T, Xu Y, Pan J, Qiu H, Wu D, Chen S, Sun A. Monosomal karyotype of chromosome 5/7 was an independent poor prognostic factor for Chinese myelodysplastic syndrome patients. Cancer Genet. 2016; 209:423–29.
68. McQuilten ZK, Sundararajan V, Andrianopoulos N, Curtis DJ, Wood EM, Campbell LJ, Wall M. Monosomal karyotype predicts inferior survival independently of a complex karyotype in patients with myelodysplastic syndromes. Cancer. 2015; 121:2892–99.
69. Strahm B, Nöllke P, Zecca M, Korthof ET, Bierings M, Furlan I, Sedlacek P, Chybicka A, Schmugge M, Bordon V, Peters C, O’Marcaigh A, de Heredia CD, et al, and EWOG-MDS study group. Hematopoietic stem cell transplantation for advanced myelodysplastic syndrome in children: results of the EWOG-MDS 98 study. Leukemia. 2011; 25:455–62.
70. Göhring G, Karow A, Steinemann D, Wilkens L, Lichter P, Zeidler C, Niemeyer C, Welte K, Schlegelberger B. Chromosomal aberrations in congenital bone marrow failure disorders—an early indicator for leukemogenesis? Ann Hematol. 2007; 86:733–39.
71. Hong M, Hao S, Patel KP, Kantarjian HM, Garcia-Manero G, Yin CC, Medeiros LJ, Lin P, Lu X. Whole-arm translocation of der(5;17)(p10;q10) with concurrent TP53 mutations in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): A unique molecular-cytogenetic subgroup. Cancer Genet. 2016; 209:205–14.
72. Tang YL, Chia WK, Yap EC, Julia MI, Leong CF, Salwati S, Wong CL. Dismal outcome of therapy-related myeloid neoplasm associated with complex aberrant karyotypes and monosomal karyotype: a case report. Malays J Pathol. 2016; 38:315–19.
73. Nomdedeu M, Calvo X, Pereira A, Carrió A, Solé F, Luño E, Cervera J, Vallespí T, Muñoz C, Gómez C, Arias A, Such E, Sanz G, et al, and Spanish Group of Myelodysplastic Syndromes. Prognostic impact of chromosomal translocations in myelodysplastic syndromes and chronic myelomonocytic leukemia patients. A study by the spanish group of myelodysplastic syndromes. Genes Chromosomes Cancer. 2016; 55:322–27.
74. Tuna M, Knuutila S, Mills GB. Uniparental disomy in cancer. Trends Mol Med. 2009; 15:120–28.
75. Abáigar M, Robledo C, Benito R, Ramos F, Díez-Campelo M, Hermosín L, Sánchez-Del-Real J, Alonso JM, Cuello R, Megido M, Rodríguez JN, Martín-Núñez G, Aguilar C, et al. Chromothripsis Is a Recurrent Genomic Abnormality in High-Risk Myelodysplastic Syndromes. PLoS One. 2016; 11:e0164370.
76. Huh YO, Tang G, Talwalkar SS, Khoury JD, Ohanian M, Bueso-Ramos CE, Abruzzo LV. Double minute chromosomes in acute myeloid leukemia, myelodysplastic syndromes, and chronic myelomonocytic leukemia are associated with micronuclei, MYC or MLL amplification, and complex karyotype. Cancer Genet. 2016; 209:313–20.
77. Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, Granada I, Hildebrandt B, Slovak ML, Ohyashiki K, Steidl C, Fonatsch C, Pfeilstöcker M, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012; 30:820–29.
78. Eclache V, Lafage-Pochitaloff M, Lefebvre C, Penther D, Raynaud S, Tigaud I. Cytogenetic place in managing myelodysplastic syndromes: an update by the Groupe francophone de cytogénétique hématologique (GFCH). Ann Biol Clin (Paris). 2016; 74:525–34.
79. da Silva FB, Machado-Neto JA, Bertini VH, Velloso ED, Ratis CA, Calado RT, Simões BP, Rego EM, Traina F. Single-nucleotide polymorphism array (SNP-A) improves the identification of chromosomal abnormalities by metaphase cytogenetics in myelodysplastic syndrome. J Clin Pathol. 2017; 70:435–42.
80. Ahmad A, Iqbal MA. Significance of genome-wide analysis of copy number alterations and UPD in myelodysplastic syndromes using combined CGH - SNP arrays. Curr Med Chem. 2012; 19:3739–47.
81. Visconte V, Selleri C, Maciejewski JP, Tiu RV. Molecular pathogenesis of myelodysplastic syndromes. Transl Med UniSa. 2014; 8:19–30.
82. Fenaux P. Chromosome and molecular abnormalities in myelodysplastic syndromes. Int J Hematol. 2001; 73:429–37.
83. Tiu RV, Visconte V, Traina F, Schwandt A, Maciejewski JP. Updates in cytogenetics and molecular markers in MDS. Curr Hematol Malig Rep. 2011; 6:126–35.
84. Evans AG, Ahmad A, Burack WR, Iqbal MA. Combined comparative genomic hybridization and single-nucleotide polymorphism array detects cryptic chromosomal lesions in both myelodysplastic syndromes and cytopenias of undetermined significance. Mod Pathol. 2016; 29:1183–99.
85. Koh KN, Lee JO, Seo EJ, Lee SW, Suh JK, Im HJ, Seo JJ. Clinical significance of previously cryptic copy number alterations and loss of heterozygosity in pediatric acute myeloid leukemia and myelodysplastic syndrome determined using combined array comparative genomic hybridization plus single-nucleotide polymorphism microarray analyses. J Korean Med Sci. 2014; 29:926–33.
86. Jonas BA, Greenberg PL. MDS prognostic scoring systems – past, present, and future. Best Pract Res Clin Haematol. 2015; 28:3–13.
87. Park JH, Kim M, Kong SY, Yoon SS, Lee DS. Monitoring of the Clonal Fraction by Fluorescence In Situ Hybridization in Myelodysplastic Syndrome: Comparison With International Working Group Treatment Response Criteria. Arch Pathol Lab Med. 2016; 140:560–69.
88. Rathnayake AJ, Goonasekera HW, Dissanayake VH. Phenotypic and cytogenetic characterization of mesenchymal stromal cells in de novo myelodysplastic syndromes. Anal Cell Pathol (Amst). 2016; 2016:8012716.
89. Mishima T, Watari M, Iwaki Y, Nagai T, Kawamata-Nakamura M, Kobayashi Y, Fujieda S, Oikawa M, Takahashi N, Keira M, Yoshida H, Tonoki H. Miller-Dieker Syndrome with unbalanced translocation 45, X, psu dic(17;Y)(p13;p11.32) detected by fluorescence in situhybridization and G-banding analysis using high resolution banding technique. Congenit Anom (Kyoto). 2017; 57:61–63.
90. Göhring G, Giagounidis A, Büsche G, Hofmann W, Kreipe HH, Fenaux P, Hellström-Lindberg E, Schlegelberger B. Cytogenetic follow-up by karyotyping and fluorescence in situhybridization: implications for monitoring patients with myelodysplastic syndrome and deletion 5q treated with lenalidomide. Haematologica. 2011; 96:319–22.
91. Platzbecker U, Santini V, Mufti GJ, Haferlach C, Maciejewski JP, Park S, Solé F, van de Loosdrecht AA, Haase D. Update on developments in the diagnosis and prognostic evaluation of patients with myelodysplastic syndromes (MDS): consensus statements and report from an expert workshop. Leuk Res. 2012; 36:264–70.
92. Mundle SD, Sokolova I. Clinical implications of advanced molecular cytogenetics in cancer. Expert Rev Mol Diagn. 2004; 4:71–81.
93. Stevens-Kroef MJ, Hebeda KM, Verwiel ET, Kamping EJ, van Cleef PH, Kuiper RP, Groenen PJ. Microarray-based genomic profiling and in situ hybridization on fibrotic bone marrow biopsies for the identification of numerical chromosomal abnormalities in myelodysplastic syndrome. Mol Cytogenet. 2015; 8:33.
94. He R, Wiktor AE, Durnick DK, Kurtin PJ, Van Dyke DL, Tefferi A, Patnaik MS, Ketterling RP, Hanson CA. Bone Marrow Conventional Karyotyping and Fluorescence In SituHybridization: Defining an Effective Utilization Strategy for Evaluation of Myelodysplastic Syndromes. Am J Clin Pathol. 2016; 146:86–94.
95. Imataka G, Arisaka O. Chromosome analysis using spectral karyotyping (SKY). Cell Biochem Biophys. 2012; 62:13–17.
96. Cohen N, Trakhtenbrot L, Yukla M, Manor Y, Gaber E, Yosef G, Amariglio N, Rechavi G, Amiel A. SKY detection of chromosome rearrangements in two cases of tMDS with a complex karyotype. Cancer Genet Cytogenet. 2002; 138:128–32.
97. Guo B, Han X, Wu Z, Da W, Zhu H. Spectral karyotyping: an unique technique for the detection of complex genomic rearrangements in leukemia. Transl Pediatr. 2014; 3:135–39.
98. Xu LX, Holland H, Kirsten H, Ahnert P, Krupp W, Bauer M, Schober R, Mueller W, Fritzsch D, Meixensberger J, Koschny R. Three gangliogliomas: results of GTG-banding, SKY, genome-wide high resolution SNP-array, gene expression and review of the literature. Neuropathology. 2015; 35:148–57.
99. Afable MG 2nd, Wlodarski M, Makishima H, Shaik M, Sekeres MA, Tiu RV, Kalaycio M, O’Keefe CL, Maciejewski JP. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. 2011; 117:6876–84.
100. Hu Q, Chu Y, Song Q, Yao Y, Yang W, Huang S. The prevalence of chromosomal aberrations associated with myelodysplastic syndromes in China. Ann Hematol. 2016; 95:1241–48.
101. Hahm C, Mun YC, Seong CM, Han SH, Chung WS, Huh J. Single nucleotide polymorphism array-based karyotyping in acute myeloid leukemia or myelodysplastic syndrome with trisomy 8 as the sole chromosomal abnormality. Acta Haematol. 2013; 129:154–58.
102. Huh J, Jung CW, Kim HJ, Kim YK, Moon JH, Sohn SK, Kim HJ, Min WS, Kim DH. Different characteristics identified by single nucleotide polymorphism array analysis in leukemia suggest the need for different application strategies depending on disease category. Genes Chromosomes Cancer. 2013; 52:44–55.
103. Volkert S, Haferlach T, Holzwarth J, Zenger M, Kern W, Staller M, Nagata Y, Yoshida K, Ogawa S, Schnittger S, Haferlach C. Array CGH identifies copy number changes in 11% of 520 MDS patients with normal karyotype and uncovers prognostically relevant deletions. Leukemia. 2016; 30:257–60.
104. Tauscher M, Praulich I, Steinemann D. Array-CGH in childhood MDS. Methods Mol Biol. 2013; 973:267–78.
105. Lukackova R, Gerykova Bujalkova M, Majerova L, Mladosievicova B. Molecular genetic methods in the diagnosis of myelodysplastic syndromes. A review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2014; 158:339–45.
106. Kjeldsen E. Oligo-based High-resolution aCGH Analysis Enhances Routine Cytogenetic Diagnostics in Haematological Malignancies. Cancer Genomics Proteomics. 2015; 12:301–37.
107. Smetana J, Frohlich J, Zaoralova R, Vallova V, Greslikova H, Kupska R, Nemec P, Mikulasova A, Almasi M, Pour L, Adam Z, Sandecka V, Zahradová L, et al. Genome-wide screening of cytogenetic abnormalities in multiple myeloma patients using array-CGH technique: a Czech multicenter experience. Biomed Res Int. 2014; 2014:209670.
108. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010; 86:749–64.
109. Link DC, Schuettpelz LG, Shen D, Wang J, Walter MJ, Kulkarni S, Payton JE, Ivanovich J, Goodfellow PJ, Le Beau M, Koboldt DC, Dooling DJ, Fulton RS, et al. Identification of a novel TP53 cancer susceptibility mutation through whole-genome sequencing of a patient with therapy-related AML. JAMA. 2011; 305:1568–76.
110. Welch JS, Westervelt P, Ding L, Larson DE, Klco JM, Kulkarni S, Wallis J, Chen K, Payton JE, Fulton RS, Veizer J, Schmidt H, Vickery TL, et al. Use of whole-genome sequencing to diagnose a cryptic fusion oncogene. JAMA. 2011; 305:1577–84.
111. Kuo FC, Steensma DP, Dal Cin P. Conventional cytogenetics for myeloid neoplasms in the era of next-generation-sequencing. Am J Hematol. 2017; 92:227–29.
112. Li W, Xia Y, Wang C, Tang YT, Guo W, Li J, Zhao X, Sun Y, Hu J, Zhen H, Zhang X, Chen C, Shi Y, et al. Identifying Human Genome-Wide CNV, LOH and UPD by Targeted Sequencing of Selected Regions. PLoS One. 2015; 10:e0123081.
113. Kohlmann A, Grossmann V, Haferlach T. Integration of next-generation sequencing into clinical practice: are we there yet? Semin Oncol. 2012; 39:26–36.
114. Jacoby MA, Walter MJ. Detection of copy number alterations in acute myeloid leukemia and myelodysplastic syndromes. Expert Rev Mol Diagn. 2012; 12:253–64.
115. Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, McLaren S, Lin ML, McBride DJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011; 144:27–40.
116. Duncavage EJ, Tandon B. The utility of next-generation sequencing in diagnosis and monitoring of acute myeloid leukemia and myelodysplastic syndromes. Int J Lab Hematol. 2015; 37:115–21.
117. Spencer DH, Tyagi M, Vallania F, Bredemeyer AJ, Pfeifer JD, Mitra RD, Duncavage EJ. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J Mol Diagn. 2014; 16:75–88.
118. Guo Y, Bosompem A, Mohan S, Erdogan B, Ye F, Vickers KC, Sheng Q, Zhao S, Li CI, Su PF, Jagasia M, Strickland SA, Griffiths EA, Kim AS. Transfer RNA detection by small RNA deep sequencing and disease association with myelodysplastic syndromes. BMC Genomics. 2015; 16:727.
119. Beck D, Ayers S, Wen J, Brandl MB, Pham TD, Webb P, Chang CC, Zhou X. Integrative analysis of next generation sequencing for small non-coding RNAs and transcriptional regulation in Myelodysplastic Syndromes. BMC Med Genomics. 2011; 4:19.
120. Liu L, Wang H, Wen J, Tseng CE, Zu Y, Chang CC, Zhou X. Mutated genes and driver pathways involved in myelodysplastic syndromes—a transcriptome sequencing based approach. Mol Biosyst. 2015; 11:2158–66.