
INTRODUCTION
In T cell-mediated anti-tumor immunity, CD8+ cytotoxic T cells recognize MHC class I antigens on the tumor cell surface via T cell receptor to trigger the release of granzyme A (GZMA) and perforin (PRF1) to kill tumor cells. Thus, presentation of MHC class I antigens on the tumor cell surface plays a crucial role in T cell-mediated tumor immune surveillance and in immune checkpoint blockage therapy, which depends on the activated cytotoxic T cells to kill tumor cells [1–5]. MHC class I antigen presentation is regulated by IFNγ, which binds to its receptors (IFNGR1/IFNGR2) to activate JAK1/JAK2 and STAT1 and induces IRF1 [6]. IRF1 then controls expression of antigen presentation machinery (APM) proteins that include immune proteasomes low molecular weight protein-2 (LMP2)/LMP7/LMP10 and transporter associated with antigen processing-1 (TAP1)/TAP2. Immune proteasomes and TAP1/TAP2 produce and load MHC class I antigens onto MHC class I molecules for translocation to the cell surface [2]. Defects in this IFNγ-IRF1 regulated APM pathway allow tumors to escape immune surveillance [2], facilitate tumor development in animal models that include lung tumors [5, 7], and are associated with resistance to anti-PD-1 immunotherapy in melanoma [8, 9].
Anti-PD-1 and anti-PD-L1 immunotherapies have resulted in superior clinical responses in non-small cell lung cancer (NSCLC) [10–16]. However, responses are generally limited to ≤20% of NSCLC cases [14, 15, 17]. There is an urgent need to understand the susceptibility and resistance mechanisms in order to better predict responders/non-responders and to increase the response rate through combination treatments [3, 17–19]. Deficiency in the IFNγ-regulated MHC class I antigen presentation has been recognized as a plausible mechanism of resistance to immune checkpoint therapy [1, 20].
We previously found somatic loss-of-function (LOF) JAK1 mutations in 9.5% of uterine cancer in Total Cancer Care (TCC@) tumors [21]. Majority of these JAK1 mutations occurred as the result of frameshift mutations in polyhomonucleotide regions. In parallel, Kim and colleagues found 30% of endometrial cancer in TCGA were microsatellite instability-high (MSI-H) and 30% of TCGA MSI-H endometrial cancer cases had JAK1 frame-shift truncating mutations [22]. This showed that 9% (30% x 30% = 9%) of endometrial cancer cases in TCGA had frame-shift truncating mutations. Moreover, missense LOF JAK1 mutations were reported in uterine leiomyosarcoma [23]. JAK1 truncating mutations impaired IFNγ-induced IRF1 and MHC class I antigen presentation in endometrial and ovarian cancer cells [21].
NSCLC and small cell lung cancer are two major forms of lung cancer [24]. Approximately 85% of lung cancer cases are NSCLC, which is comprised of adenocarcinoma (40%), squamous-cell carcinoma (25-40%), and large-cell carcinoma (10-15%) subtypes. Since NSCLC is a cancer type that anti-PD-1/anti-PD-L1 antibody therapies are effective and that the response/resistance mechanisms to immune checkpoint therapy remains incompletely understood, we focused our examination of the IFNγ-regulated MHC class I antigen presentation pathway in NSCLC in this study. We found that the genetic defects in the IFNγ receptor-IRF1 pathway genes [21] in NSCLC occurred predominantly via a mechanism distinct from that in endometrial cancer. Specifically, we identified JAK2 deletion on chromosome 9p as the predominant mechanism of genetic defects in the IFNγ receptor-IRF1 pathway genes. Deletions of PD-L1 (CD274) and PD-L2 (PDCD1LG2) genes were always accompanied by JAK2 deletion, suggesting that JAK2 deletion may be a mechanism to safeguard tumor cells from activated cytotoxic T cells in the absence of negative regulators PD-L1/PD-L2. Knocking out JAK2 or inhibition of JAK2 kinase activity prevented presentation of MHC class I molecules on NSCLC cell surface. While chromosome 9p deletion was observed frequently in NSCLC in previous studies, its role has not been attributed to JAK2. Our analysis suggests that JAK2 deletion offers tumor cells an advantage of evading immune surveillance and reveals a previously unknown functional significance of chromosome 9p deletion.
RESULTS
Genetic deficiencies of IFNγ-IRF1 signaling pathway genes in NSCLC occur prevalently in JAK2
In T cell-mediated tumor immune surveillance, recognition of MHC class I antigens on the tumor cells by the T cell receptor of CD8+ cytotoxic T cells is mandatory for the effector T cells to kill tumor cells [2]. The IRF1-mediated IFNγ signaling pathway regulates MHC class I antigen presentation in tumor cells. Thus, a deficiency in the IFNγ-IRF1 signaling pathway would impair the T cell-mediated cytolytic activity. To assess the spectrum and prevalence of genetic defects in the IFNγ-IRF1 signaling pathway in NSCLC, we examined TCGA lung adenocarcinoma (LuAd) and lung squamous cell carcinoma (LuSc) data [25–28] for apparent loss-of-function (LOF) gene alterations (homozygous deletion or truncating mutation) of the IFNγ-IRF1 signaling pathway genes. These included IFNGR1, IFNGR2, JAK2, JAK1, STAT1, and IRF1. Among 515 cases of LuAd, 30 cases (5.8%) had a LOF alterations in one of the above 6 genes (Figure 1A). These 30 cases were mutually exclusive. LOF alterations of the JAK2 gene occurred most often with 16 cases (2.5%), which included 13 homozygous deletion cases and 3 truncating mutation cases. Similarly, 34 of 501 LuSc cases (7.0%) had LOF alterations in one of the IFNγ signaling pathway genes (Figure 1B). LOF alterations in 33 of these 34 cases were mutually exclusive. JAK2 LOF occurred most frequently in 17 cases (3.4%). Thus, genetic defects in the IFNγ-IRF1 signaling pathway in NSCLC occur prevalently in the JAK2 gene.
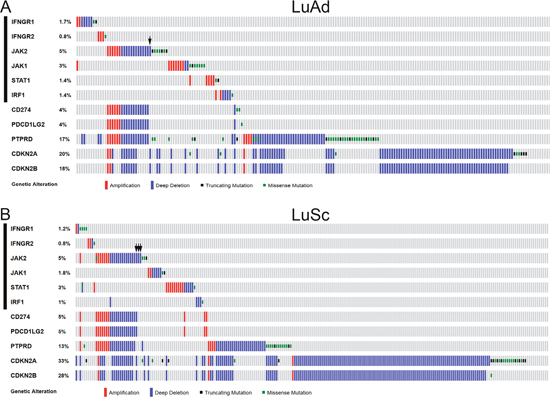
Figure 1: Genetic alterations of IFNγ-IRF1 pathway and selected chromosome 9p genes in NSCLC. CNAs and mutations of the list genes were examined in the TCGA LuAd tumor samples (515 patients/517 samples RNA Seq V2 data) (A) and TCGA LuSc tumor samples (501 patients/501 samples RNA Seq V2 data) (B) through cBioPortal for Cancer Genomics (www.cbioportal.org) [27, 28]. Oncoprints of the genetic alternations in the list genes are shown. Arrows, non-continuous chromosome deletions involving JAK2 or JAK2 only deletion cases among the list genes. Black bars on the left indicate six IFNγ pathway genes examined in this study.
Co-occurrence of JAK2 deletion with other chromosome 9p genes
Among the 33 cases of JAK2 LOF alterations, 29 cases (88%) were chromosomal deletion. Thus, unlike our previous finding in endometrial cancer where JAK1 frameshift was the predominant mechanism of the IFNγ-IRF1 pathway genetic defects [21], JAK2 gene deletion was the predominant mechanism of the IFNγ-IRF1 pathway genetic defects in NSCLC. Interestingly, 39 of 42 JAK2 copy number alternation (CNA) cases, including both amplification and deletion, coincided with CNAs of CD274 and PDCD1LG2 that encode PD-L1 and PD-L2, respectively, in LuAd and LuSc (Figure 1). JAK2 was deleted in all of CD274 and PDCD1LG2 deletion cases. Examination of chromosomal locations of JAK2, CD274, and PDCD1LG2 genes indicated that these genes are co-localized at human chromosome 9p24.1 (Supplementary Figure 1).
Chromosome 9p21 and 9p23-9p24 were identified previously as frequently deleted regions in NSCLC genomes [29–35]. Tumor suppressor genes CDKN2A/CDKN2B and PTPRD are located at chromosome 9p21.3 and 9p23-9p24.1, respectively (Figure 2). Loss of CDKN2A/CDKN2B or PTPRD tumor suppressor was often used to explain the frequent chromosome 9p deletion in human cancer [29, 30, 33, 34, 36]. The TCGA data showed that JAK2 deletion in NSCLC was often associated with deletions of either PTPRD and/or CDKN2A/CDKN2B genes (Figure 1). Thus, the mechanism of frequent JAK2 deletion in NSCLC may be a bystander event due to deletions of the tumor suppressor genes PTPRD and CDKN2A/CDKN2B because the proximity of these genes co-located at the chromosome 9p21.3-9p24.1 region.
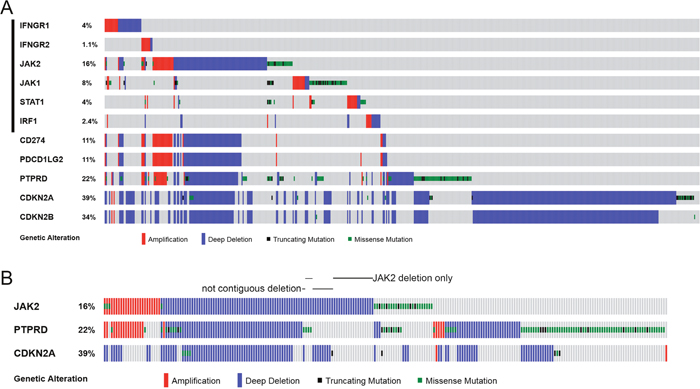
Figure 2: Genetic alterations of IFNγ-IRF1 pathway and selected chromosome 9p genes in cancer cell lines. (A) CNAs and mutations of the listed 11 genes were examined in 881 cancer cell lines in Cancer Cell Line Encyclopedia (CCLE) [37] through cBioPortal. Oncoprints of the genetic alterations are shown. (B) Oncoprints JAK2, PTPRD, and CDKN2A genetic alterations in CCLE cells are shown. Cell lines with non-continuous chromosome 9p deletions or have JAK2 deletion in the absence of PTPRD and CDKN2A deletion are indicated.
However, it was observed previously in NSCLC that deletions in 9p24, 9p23, and 9p21 were not contiguous [29]. Similarly, chromosomal deletions of JAK2, PTPRD, and CDKN2A/CDKN2B genes did not always occur in a contiguous manner in TCGA NSCLC tumors (Figure 1, see cases indicated by black arrows). JAK2 and CDKN2A/CDKN2B loci were deleted while PTPRD was not changed in the TCGA-50-930 and TCGA-22-1002 tumors. In TCGA-21-1082 and TCGA-77-7335 tumors, only the JAK2 locus was deleted.
Examination of TCGA data of 881 cancer cell lines in the cancer cell line encyclopedia (CCLE) cohorts [37] again showed that the IFNγ-IRF1 pathway genetic defects in these cancer cells occurred most often through JAK2 deletion (Figure 2). CD274 and PDCD1LG2 deletions were always accompanied by JAK2 deletion (Figure 2A). JAK2 deletion was often, but not always, associated with PTPRD and/or CDKN2A/CDKN2B deletions (Figure 2B). Among the 90 CCLE cancer cell lines with JAK2 homozygous deletion, 17 were lung cancer cell lines.
The non-contiguity of CDKN2A/CDKN2B-PTPRD-JAK2 deletion on chromosome 9p and the fact that JAK2 deletion occurred in the absence of CDKN2A/CDKN2B or PTPRD deletion suggest that JAK2 deletion is not merely attributed to deletion of CDKN2A/CDKN2B or PTPRD loci that accidently extended beyond these tumor suppressor genes. Rather, it suggests that JAK2 deficiency by itself offers an advantage to certain cancer cells.
IRF1 expression correlates with granzyme A and perforin expression in NSCLC
After binding to MHC class I antigens on tumor cells via T cell receptor, activated CD8+ cytotoxic T cells release granzyme A (GZMA) and perforin (PRF1) to destroy tumor cells. mRNA levels of these two cytolytic effectors are markers of immune cytolytic activity of CD8+ cytolytic T cells and clinical responsiveness to immune checkpoint therapies [3, 19, 38]. We used IRF1 mRNA as a marker of IFNγ-regulated MHC class I antigen presentation pathway in tumor cells, CD8 as activated cytotoxic T cell marker, and GZMA and PRF1 as immune cytolytic activity markers to analyze if the IFNγ signaling pathway activity is correlated with cytotoxic T cell-associated cytolytic activity.
Analyses of TCGA RNA-seq data from 515 cases of LuAd and 501 cases of LuSc showed that the IRF1 mRNA levels in these NSCLC cohorts were positively correlated with GZMA, PRF1, and CD8 mRNA levels (Figure 3). The Spearman correlation coefficients ranged from 0.639 to 0.792. Therefore, the IRF1 level was well correlated with the GZMA and PRF1 levels and GZMA and PRF1 levels were mainly associated with activated T cells in these 1,016 cases of NSCLC.
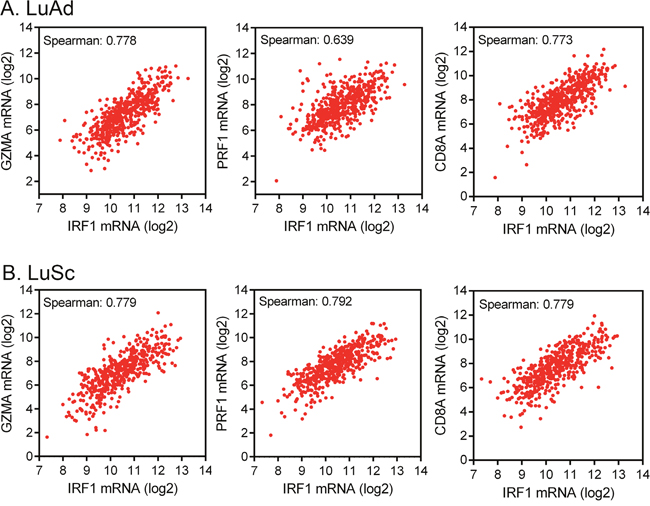
Figure 3: Correlation of IRF1 expression with immune cytolytic activity markers. (A) Co-expression of IRF1 mRNA and immune cytolytic activity markers [3] granzyme A (GZMA) and perforin (PRF1) and cytotoxic T cell marker CD8 in 515 TCGA cases of LuAd. (B) Co-expression of IRF1 with granzyme A, perforin, and cytotoxic T cell markers in 501 TCGA cases of LuSc. Data were obtained via cBioPortal.
Inhibition of JAK2 prevents IFNγ-induced IRF1 expression and cell surface of HLA-ABC
A549, H661, H292, and H2172 are NSCLC cell lines without homozygous JAK2 deletions or mutation based on CCLE data. HLA-A, -B, and -C (HLA-ABC) are human MHC class I molecules. MHC class I antigens are normally loaded into HLA-ABC/β-2 microglobulin (B2M) and transported to cell surface for presentation to cytotoxic T cells. When HLA-ABC/β-2 microglobulin are not loaded with antigens, they become unstable and are degraded more rapidly in the cells, resulting in a lower level of HLA-ABC molecules on the cell surface [21, 39, 40]. IFNγ stimulated IRF1 expression and increased cell surface HLA-ABC in these cells (Figure 4), indicative of MHC class I antigen presentation [21]. To determine if the JAK2 protein tyrosine kinase activity is required for IFNγ-stimulated IRF1 expression and MHC class I antigen presentation, we treated A549, H661, H292, and H2172 NSCLC cells with the JAK2 inhibitor ruxolitinib. Complete inhibition of IFNγ-induced IRF1 was observed at 1 μM ruxolitinib (Figure 4B). Consistently, while IFNγ increased the cell surface HLA-ABC level in all four cell lines significantly (p < 0.05), pretreatment with ruxolitinib (1 μM) blocked the IFNγ-induced cell surface HLA-ABC, indicating that the JAK2 protein kinase activity is necessary for these IFNγ-induced responses.
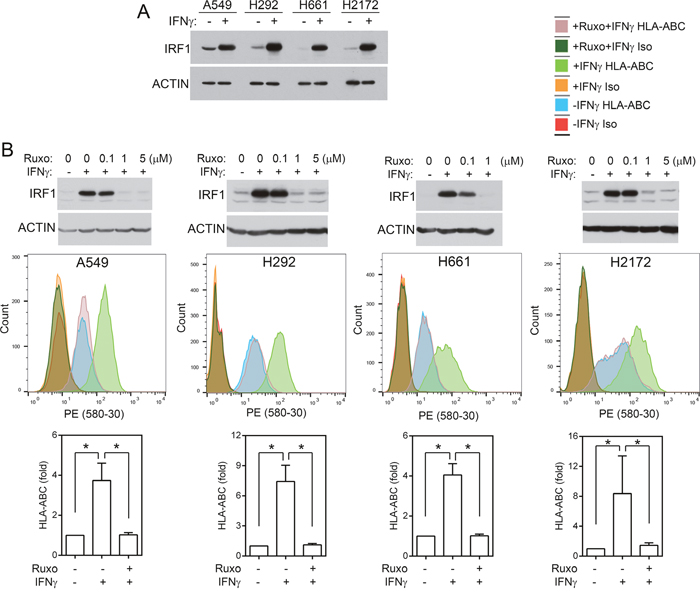
Figure 4: JAK2 protein tyrosine kinase activity is required for IFNγ-induced IRF1 and cell surface MHC class I molecules. (A) Cells were treated with IFNγ (20 ng/ml) for 6 h. An equal amount of cell lysates were analyzed by immunoblotting with antibodies to IRF1 and ACTIN. (B) Upper panels, cells were pretreated with indicated concentrations of ruxolitinib for 1h and then stimulated with IFNγ for 6h. Cell lysates were analyzed by immunoblotting as in (A). Lower panels, cells were pre-treated with ruxolitinib for 1 h and then mock-treated or treated with IFNγ (100 ng/ml) for 24 h and processed for immunostaining with a PE-conjugated anti-HLA-ABC antibody or an isotype control antibody followed by flow cytometric analysis. Examples of spectra from representative experiments are shown. Graphs: fold changes between cells with and without IFNγ stimulation. n = 6 for all conditions in all cell lines performed in three or more separate experiments. *, p < 0.05 (Wilcoxon signed-rank test).
JAK2 deficiency impairs IFNγ-stimulated IRF1 expression and MHC class I antigen presentation on NSCLC cells
To determine if loss of JAK2 impairs the IFNγ-induced MHC class I antigen presentation pathway, we knockout JAK2 in A549 cells using lentiviral CRISPR/Cas9 sgRNAs. Two stable cell lines (A549/KO1, A549/KO2) that did not have detectable JAK2 protein were isolated (Figure 5A). While IFNγ activated STAT1 and induced IRF1, TAP1, and LMP2 expression in A549 cells, it could not activate STAT1 or induce IRF1, TAP1, or LMP2 in the JAK2 knockout A549/KO1 and A549/KO2 cells (Figure 5A). Flow cytometric analysis of cell surface HLA-ABC expression indicated that IFNγ could not increase cell surface HLA-ABC in A549/KO1 and A549/KO2 cells (Figure 5B).
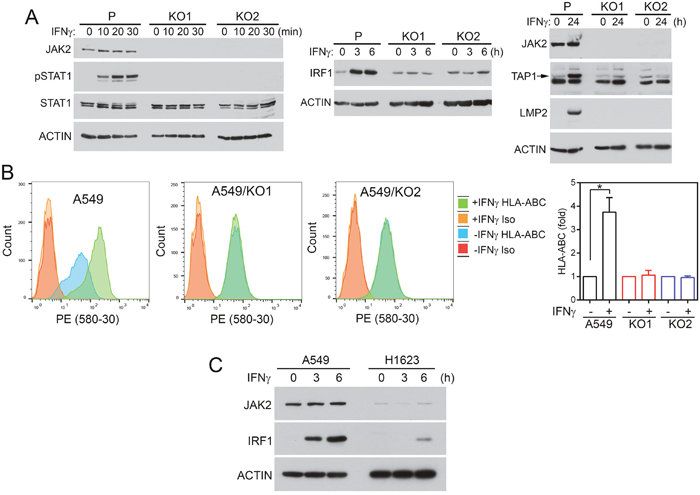
Figure 5: JAK2 deficiency impairs responses to IFNγ in NSCLC cells. (A, B) JAK2 was knocked out using CRISPR/Cas9 as described in Materials and Methods. Two JAK2 knockout cell lines (KO1, KO2) were isolated and compared with the parental cells (P). (A) Cells were treated with IFNγ as indicated. Equal amounts of cell lysate proteins were analyzed by immunoblotting with the indicated antibodies. (B) Cells were treated with or without IFNγ (100 ng/ml) for 24 h. The amounts of HLA-ABC on the cell surface were analyzed by flow cytometry following immunostaining with PE-conjugated anti-HLA-ABC or isotype control antibodies. n = 6 for all conditions in all three cell lines performed in two or more separate experiments. *, p < 0.05 between cells treated with or without IFNγ, Wilcoxon signed-rank test. (C) A549 and H1623 cells were stimulated with IFNγ and cell lysates were analyzed by immunoblotting as indicated.
Next, H661 cells were infected with lentiviral JAK2 CRISPR/Cas9 sgRNAs. The pool of puromycin-resistant cells (H661/KO) were used in subsequent analyses. As shown in Supplementary Figure 2, JAK2 was not detected in H661/KO cells. IFN-γ induced pSTAT1, IRF1, TAP1, and LMP2 in H661 but not in the JAK2 knockout H661/KO cells. Furthermore, IFNγ could not increase cell surface HLA-ABC in H661/KO cells (Supplementary Figure 2B).
Based on CCLE data (cbioportal.org), the NSCLC cell line H1623 has JAK2 deep deletion. Compared with A549 cells, H1623 cells had a minimal amount of JAK2 protein (Figure 5C). While IRF1 was markedly increased in A549 cells after IFNγ stimulation, a very low level of IRF1 was detected in H1623 cells.
Restoration of JAK2 in H1573 cells rescues IFN-γ signaling
H1573 human NSCLC cells harbor JAK2 S507* nonsense mutation that results in truncation of both pseudo kinase and tyrosine kinase domain of JAK2. IFNγ could not increase STAT1 tyrosine phosphorylation or increase IRF1, TAP1, or LMP2 level in H1573, while parallel experiments in the JAK2 wildtype A549 cells showed readily detectable IFNγ-induced responses. (Figure 6A), indicating that H1573 cells are defective in IFNγ signaling. We established a H1573/JAK2 cell line by infecting H1573 cells with retrovirus-encoded JAK2. As shown in Figure 6B, the control H1573/V cells did not have detectable JAK2 protein and were unresponsive to IFNγ-stimulation in STAT1 tyrosine phosphorylation and expression of IRF1, TAP1, and LMP2. In the JAK2-restored H1573/JAK2 cells, IFNγ was able to induce pSTAT1, IRF1, TAP1, and LMP2. Moreover, IFNγ could not increase the cell surface HLA-ABC level in H1573/V cells, whereas treatment of H1573/JAK2 cells with IFNγ significantly increased the cell surface HLA-ABC (Figure 6C, p < 0.05). These results show that JAK2 deficiency is responsible for the defect in the IFNγ-regulated MHC class I antigen presentation in H1573 cells.
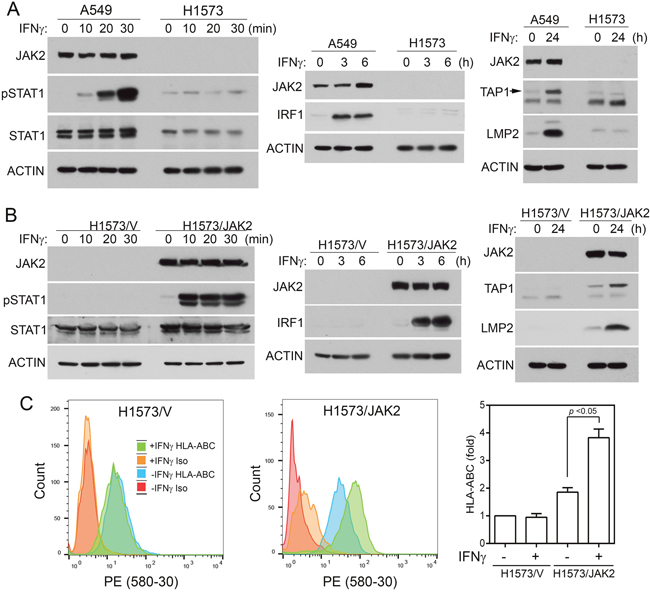
Figure 6: Expression of JAK2 in H1573 cells restores IFNγ responses. (A) H1573 cells had no detectable JAK2 and were defective in IFNγ-induced IRF1 axis. A549 (positive control cells) and H1573 cells were treated with IFNγ as indicated. Cell lysates were analyzed by immunoblotting with indicated antibodies. (B) H1573/V and H1573/JAK2 cells were treated with IFNγ as shown and cell lysates were analyzed by immunoblotting with indicated antibodies. (C) H1573/V and H1573/JAK2 cells were treated with or without IFNγ for 24 h and cell surface HLA-ABC were analyzed by flow cytometry after immunostaining with HLA-ABC. Left, representative spectra from flow cytometric analysis. Right, fold changes. n = 6 for all conditions in both cells (*, p < 0.05, Wilcoxon signed-rank test).
DISCUSSION
We found here that genetic defects in the IFNγ-IRF1 signaling pathway genes occurred in 5.8% of LuAd and 7.0% of LuSc among 1016 NSCLC cases in TCGA. Approximately 52% of these defects occurred in JAK2, of which chromosome deletion of JAK2 accounting for 88% of the cases. JAK2 resides on chromosome 9p24.1. Interestingly, CD274, PDCD1LG2, PTPRD, and CDKN2A/CDKN2B are also located in the nearby chromosome 9p region (Supplementary Figure 1). Chromosome 9p deletions have been observed frequently in NSCLC [25, 26, 29-31, 33] and in some other types of tumors such as glioma [34, 41]. CDKN2A/CDKN2B at chromosome 9p21.3 and PTPRD at chromosome 9p23-p921.1 were identified as tumor suppressor genes in the chromosome 9p arm to give the functional explanation of chromosome 9p deletions [25, 26, 29, 30, 32-34, 41]. The high incidence of JAK2 deletion in NSCLC was likely due in part to the frequent deletion of tumor suppressors CDKN2A/CDKN2B and PTPRD in the chromosome 9p arm in NSCLC.
However, it appears unlikely that JAK2 deletion was merely a bystander effect in which chromosome 9p deletion accidently extended beyond the CDKN2A/CDKN2B and PTPRD gene regions. First, as previously reported [29], deletions of chromosome 9p were not contiguous (Figures 1, 2), suggesting that deletion of JAK2 conferred a functional selection advantage to the cancer cells. Second, there were cases in TCGA tumors and CCLC cancer cell lines in which JAK2 was deleted in the absence of CDKN2A/CDKN2B or PTPRD deletion (Figures 1, 2), suggesting that JAK2 deletion alone may have a functional benefit to cancer cells. Third, CD274 and PDCD1LG2 were deleted in some cases of TCGA NSCLC and CCLC cancer cell lines. CD274 and PDCD1LG2 deletions were always accompanied by JAK2 deletion. Since PD-L1/PD-L2 suppress T cell-based immunity against tumor cells, deletions of PD-L1/PD-L2 could result in constitutively active tumor-targeting cytotoxic T cells. In fact, blocking PD-1/PD-L1 interaction is the principle of immune checkpoint blockage therapies using anti-PD-1/anti-PD-L1 antibodies. Loss of PD-L1/PD-L2 T cell suppressors while maintaining the IFNγ-regulated MHC class I antigen presentation pathway that allows cytotoxic T cells to recognize tumor cells would be disadvantageous to the tumor cells. Thus, JAK2 deletion is a potential mechanism to hide tumor cells from recognition by cytotoxic T cells, especially in the cases of CDC274 and PDCD1LG2 deletions.
While anti-PD-1/anti-PD-L1 antibodies therapies have shown remarkable efficacies in a subset of NSCLC patients, the majority of NSCLC are unresponsive to these immune checkpoint blockage therapies [12, 14, 15]. Various therapy resistance mechanisms may exist. In the absence of PD-L1, the anti-PD-L1 antibody atezolizumab would lack the target on tumor cells while anti-PD-1 antibodies nivolumab and pembrolizumab would lack one of bases of their action. More importantly, recognition of MHC class I antigens by T cell receptor of CD8+ cytotoxic T cells is required for the effector T cells to destroy tumor cells. The well-correlated mRNA levels of IRF1 with that of immune cytotoxic markers GZMA/PRF1 presented in this study support the important role of the IFNγ-IRF1 signaling axis in modulation of the T cell-based immune cytotoxic activities. Consistent with this notion, NSCLC patients with pre-existing high T-effector/IFNγ-associated gene expression had improved overall survival in a recent atezolizumab clinical trial [16].
Using knockout and a chemical inhibitor, our data show that JAK2 was necessary for IFNγ-induced IRF1 and cancer cell surface expression of HLA-ABC MHC class I molecules. On the other hand, expression of exogenous JAK2 in a cell line lacking endogenous JAK2 was sufficient to restore IFNγ-induced IRF1 and cell surface expression of HLA-ABC. These results demonstrate that JAK2 deficiency would impair IFNγ-regulated MHC class I antigen presentation. We thereby postulate that JAK2 genetic defects (deletion and truncating mutations) in NSCLC represent a likely mechanism underlying the resistance of cancer cells to anti-PD-1/anti-PD-L1 immune checkpoint therapy.
Gain-of-function JAK2 mutant JAK2V617F is an established oncoprotein associated with myeloproliferative neoplasms (MPNs) polycythemia vera, essential thrombocythemia, and primary myelofibrosis [42]. Moreover, JAK2 activated STAT3 pathway has tumor promoting activity [43–45]. JAK1/JAK2 inhibitor such as ruxolitinib is being used to treat MPNs and other JAK2 inhibitors are in various stages of development [46]. Unorthodoxically, this study in NSCLC and the previous studies of JAK1 in uterine cancer [21–23] also suggest that JAK1 and JAK2 have tumor suppressor function, deficiencies of them could allow tumor cells to escape T-cell mediated immune surveillance. Caution should be taken when considering JAK inhibitors for combination treatments involving tumor immunity.
MATERIALS AND METHODS
Reagents
Human IFNγ was from Peprotech. Antibodies to STAT1 (SC79877), IRF1 (SC497), and LMP2 (SC271354) were from Santa Cruz Biotechnologies. STAT1-pTyr701 (pSTAT1) antibodies were from Santa Cruz (SC7988) or Cell Signaling Technologies (#7649). Antibodies to JAK2 (#3230) was from Cell Signaling Technologies. Anti-TAP1 antibody (ADI-CSA-620-E) was from ENZO. PE-conjugated mouse anti-human HLA-ABC antibody (clone G46-2.6) and the isotype control antibody were from BD Pharmingen. LentiCRISPRv2 [47] was obtained from Addgene. Ruxolitinib was from LC laboratories.
Genomic data analysis
CNAs, mutations, and mRNA expression of lung adenocarcinoma [tumor samples with mRNA data (RNA seq V2), 515 patients, 517 tumor samples] and lung squamous cell carcinoma [tumor samples with mRNA data (RNA Seq V2), 501 patients, 501 tumor samples] were from TCGA [25, 26] and was down loaded via www.cBioPortal.org [27, 28]. CNA and mutation of Cancer Cell line encyclopedia [37] cell lines (881 cell lines with sequencing and CNA data) were based on cBioPortal. Oncoprint visualization of CNAs and mutations were adapted from cBioPortal. Human chromosome 9p information and gene locations were obtained from Ensembl (www.ensembl.org) [48] and was based on human genome assembly GRCh38.
Cell lines, cell cultures, and immunoblotting
Human NSCLC cell lines A549, H661, H292, and H2172 were from a central repository at the H. Lee Moffitt Cancer Center and Research Institute and had been authenticated by STR analysis (ACTG Inc, Wheling, IL) as of September 2010 [49]. Cells in the central repository had been routinely tested and were negative for mycoplasma (PlasmoTest, InvivoGen, San Diego, CA). H1593 cells were obtained from American Type Culture Collection (ATCC) in 2015. H1623 cells were obtained from ATCC in 2017. A549 cells were cultured in DMEM/10% fetal bovine serum (FBS). H611, H292, H2172, H1573, and H1623 cells were cultured in RPMI-1640/10% FBS. Cell lysate preparation and immunoblotting were performed as described previously [21, 49, 50].
Flow cytometry
Cells were treated with or without IFNγ (100 ng/ml) for 24 h, detached from plates using Accutase, and suspended in PBS containing 0.5% BSA. Cells were stained with a PE-conjugated mouse anti-human HLA-ABC antibody (clone G46-2.6) or the isotype control antibody according to the supplier’s instruction as described previously [21]. Stained cells were fixed with 4% paraformaldehyde. Data were acquired on a FACSCanto II flow cytometer at the Moffitt Cancer Center or a Stratedigm S1000 flow cytometer at University of Oklahoma Health Sciences Center. Statistical analysis was performed using Wilcoxon signed-rank test. p < 0.05 was considered significantly different.
CRISPR/Cas9-mediated JAK2 knockout
Three human JAK2 targeting single guide RNAs (sgRNAs) were selected based on the Zhang Lab Cas9 target design tool (http://crispr.mit.edu:8079/) [51]. The target sites were located on JAK2 exon 7. The targeting sequences were: G1: TTTTGACAAGGAAGCGAATA; G2: ATTATAATAACTGGAAACGG; G3: TCACCTGAAGGACCACTTCC. 5’-phosphorylated oligonucleotides were annealed and cloned into BsmBI digested lentiCRISPRv2 by standard procedure to generate lentiCRISPRv2-JAK2sgRNA-1, -2, and -3 plasmids. Lentiviruses containing JAK2 sgRNAs CRISPR/Cas9 were prepared by transfection of 293T cells with a mixture of equal amounts of the lentiCRISPRv2-JAK2sgRNA-1, -2, and -3 plasmids plus PMD2 and PSPAX. A549 cells were infected with the JAK2 sgRNAs CRISPR/Cas lentiviruses. Infected cells were selected by puromycin. Puromycin-resistant cell lines were isolated and screened for the absence of JAK2 protein by immunoblotting. H661 cells were similarly infected with the lentiviruses but puromycin-resistant pool of cells were used in subsequent analysis without clone selection since JAK2 was not detectable in the pool of cells.
Expression of exogenous JAK2
JAK2 coding sequence was cloned into a retroviral vector containing puromycin-resistant gene. Retroviruses were produced by transfection of empty or JAK2-encoding retroviral plasmids into Ampho-293 cells. H1573 cells were infected with retroviruses containing the JAK2 coding sequence. Approximately 80 puromycin-resistant cell lines (clones) were screened for the expression of JAK2 protein by immunoblotting and a cell line with detectable JAK2 (H1573/JAK2) was isolated. Control H1573 cells (H1573/V) were made by infected with retroviruses without the JAK2 cDNA insert and selected with puromycin (1 μg/ml).
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health grant R01CA178456 (to J. Wu) and in part by a pilot project grant from Stephenson Cancer Center (to J. Wu). We thank Moffitt Cancer Center and University of Oklahoma Health Sciences Center flow cytometry core facility staffs for assistance. We have used publically available data from TCGA and analyzed them through cBioPortal (www.cbioportal.org). We also utilized Ensembl (www.ensembl.org) in our examination of human chromosome 9.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Garrido F, Aptsiauri N, Doorduijn EM, Garcia Lora AM, van Hall T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016; 39:44–51.
2. Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res. 2006; 12:3890–95.
3. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61.
4. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–99.
5. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA. 1998; 95:7556–61.
6. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005; 5:375–86.
7. Hayashi T, Faustman DL. Development of spontaneous uterine tumors in low molecular mass polypeptide-2 knockout mice. Cancer Res. 2002; 62:24–27.
8. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet Moreno B, Mezzadra R, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016; 375:819–29.
9. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, Palaskas N, Rodriguez GA, Parisi G, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017; 7:188–201.
10. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54.
11. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366:2455–65.
12. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al, and KEYNOTE-001 Investigators. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–28.
13. Rizvi NA, Mazières J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, Otterson GA, Campos LT, Gandara DR, et al. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015; 16:257–65.
14. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhäufl M, Arrieta O, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015; 373:1627–39.
15. Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015; 373:123–35.
16. Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J, Park K, Smith D, Artal-Cortes A, Lewanski C, Braiteh F, Waterkamp D, He P, et al, and POPLAR Study Group. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet. 2016; 387:1837–46.
17. Morgensztern D, Herbst RS. Nivolumab and Pembrolizumab for Non-Small Cell Lung Cancer. Clin Cancer Res. 2016; 22:3713–17.
18. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–28.
19. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563–67.
20. Wang X, Schoenhals JE, Li A, Valdecanas DR, Ye H, Zang F, Tang C, Tang M, Liu CG, Liu X, Krishnan S, Allison JP, Sharma P, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Res. 2017; 77:839–50.
21. Ren Y, Zhang Y, Liu RZ, Fenstermacher DA, Wright KL, Teer JK, Wu J. JAK1 truncating mutations in gynecologic cancer define new role of cancer-associated protein tyrosine kinase aberrations. Sci Rep. 2013; 3:3042.
22. Kim TM, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013; 155:858–68.
23. Hayashi T, Horiuchi A, Sano K, Hiraoka N, Kasai M, Ichimura T, Sudo T, Tagawa Y, Nishimura R, Ishiko O, Kanai Y, Yaegashi N, Aburatani H, et al. Potential role of LMP2 as tumor-suppressor defines new targets for uterine leiomyosarcoma therapy. Sci Rep. 2011; 1:180.
24. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008; 359:1367–80.
25. Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ, Danilova L, Ding L, Getz G, Hammerman PS, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–50.
26. Hammerman PS, Lawrence MS, Voet D, Jing R, Cibulskis K, Sivachenko A, Stojanov P, McKenna A, Lander ES, Gabriel S, Getz G, Sougnez C, Imielinski M, et al, and Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012; 489:519–25.
27. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
28. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–04.
29. Sato M, Takahashi K, Nagayama K, Arai Y, Ito N, Okada M, Minna JD, Yokota J, Kohno T. Identification of chromosome arm 9p as the most frequent target of homozygous deletions in lung cancer. Genes Chromosomes Cancer. 2005; 44:405–14.
30. Kohno T, Otsuka A, Girard L, Sato M, Iwakawa R, Ogiwara H, Sanchez-Cespedes M, Minna JD, Yokota J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer. 2010; 49:342–52.
31. Nagayama K, Kohno T, Sato M, Arai Y, Minna JD, Yokota J. Homozygous deletion scanning of the lung cancer genome at a 100-kb resolution. Genes Chromosomes Cancer. 2007; 46:1000–10.
32. Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, Richards WG, Sugarbaker D, Chen F, Rubin MA, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005; 65:5561–70.
33. Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, et al. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007; 450:893–98.
34. Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, Paty PB, Rohle D, Vivanco I, Chmielecki J, Pao W, Ladanyi M, Gerald WL, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci USA. 2009; 106:9435–40.
35. Peyser ND, Du Y, Li H, Lui V, Xiao X, Chan TA, Grandis JR. Loss-of-Function PTPRD Mutations Lead to Increased STAT3 Activation and Sensitivity to STAT3 Inhibition in Head and Neck Cancer. PLoS One. 2015; 10:e0135750.
36. Ortiz B, White JR, Wu WH, Chan TA. Deletion of Ptprd and Cdkn2a cooperate to accelerate tumorigenesis. Oncotarget. 2014; 5:6976–82. https://doi.org/10.18632/oncotarget.2106.
37. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603–07.
38. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, Jackson JR, Shahabi V. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012; 61:1019–31.
39. de la Salle H, Hanau D, Fricker D, Urlacher A, Kelly A, Salamero J, Powis SH, Donato L, Bausinger H, Laforet M, et. Homozygous human TAP peptide transporter mutation in HLA class I deficiency. Science. 1994; 265:237–41.
40. Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4-8+ T cells. Cell. 1992; 71:1205–14.
41. Roy DM, Walsh LA, Desrichard A, Huse JT, Wu W, Gao J, Bose P, Lee W, Chan TA. Integrated Genomics for Pinpointing Survival Loci within Arm-Level Somatic Copy Number Alterations. Cancer Cell. 2016; 29:737–50.
42. Tefferi A. JAK2 mutations and clinical practice in myeloproliferative neoplasms. Cancer J. 2007; 13:366–71.
43. Gao SP, Chang Q, Mao N, Daly LA, Vogel R, Chan T, Liu SH, Bournazou E, Schori E, Zhang H, Brewer MR, Pao W, Morris L, et al. JAK2 inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to tyrosine kinase inhibitors. Sci Signal. 2016; 9:ra33.
44. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014; 26:207–21.
45. Gao SP, Mark KG, Leslie K, Pao W, Motoi N, Gerald WL, Travis WD, Bornmann W, Veach D, Clarkson B, Bromberg JF. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J Clin Invest. 2007; 117:3846–56.
46. Sonbol MB, Firwana B, Zarzour A, Morad M, Rana V, Tiu RV. Comprehensive review of JAK inhibitors in myeloproliferative neoplasms. Ther Adv Hematol. 2013; 4:15–35.
47. Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014; 11:783–84.
48. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, et al. Ensembl 2016. Nucleic Acids Res. 2016; 44:D710–16.
49. Fan C, Chen L, Huang Q, Shen T, Welsh EA, Teer JK, Cai J, Cress WD, Wu J. Overexpression of major CDKN3 transcripts is associated with poor survival in lung adenocarcinoma. Br J Cancer. 2015; 113:1735–43.
50. Schneeberger VE, Ren Y, Luetteke N, Huang Q, Chen L, Lawrence HR, Lawrence NJ, Haura EB, Koomen JM, Coppola D, Wu J. Inhibition of Shp2 suppresses mutant EGFR-induced lung tumors in transgenic mouse model of lung adenocarcinoma. Oncotarget. 2015; 6:6191–202. https://doi.org/10.18632/oncotarget.3356.
51. Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013; 31:827–32.