
INTRODUCTION
Immune check point inhibitors such as anti-programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1) antibody are regarded as the “magic bullet” for refractory solid cancer [1, 2]. They inhibit the interaction between activated T cell immune check point receptors (PD-1) and tumor cell ligand (PD-L1), which facilitates tumor immune evasion [3]. Multiple phase III trials for advanced melanoma, renal cell carcinoma and non-small cell lung cancer (NSCLC) have shown better clinical outcomes for anti-PD-1 antibody than conventional treatment [4–6].
Tumor PD-L1 expression is used as a surrogate predictive marker of PD-1 treatment [1, 6]. In NSCLC, PD-L1 expression in > 50% of tumor cells is correlated with treatment efficacy. In addition, a recent study suggested that mutation profiles can be used to determine the treatment sensitivity of anti PD-1 antibody [7]. High non-synonymous mutation, neoantigen burden and DNA repair pathway mutations were correlated with good objective responses [7, 8]. However, PD-L1 expression and mutation signature have not been formally accepted as biomarkers for immunotherapy.
Immunotherapy for breast cancer (BC) has also been studied. The phase Ib KEYNOTE-012 trial with the PD-1 antibody, pembrolizumab, for heavily-treated triple-negative BC (TNBC) demonstrated an 18.5% overall response rate and three patients persisted on this treatment regime for at least 11 months [9]. In this trial, PD-L1 positivity was defined as over 1% PD-L1 expression in cancer cells or any staining in the tumor stroma and increasing expression of PD-L1 was associated with anti-PD-1 antibody response. In addition, a phase I trial of an anti-PD-L1 antibody, atezolizumab, for metastatic TNBC was performed and showed a 33% overall response [10]. A further biomarker study for atezolizumab suggested that tumor responses were correlated with high levels of PD-L1 expression in both tumor cells and tumor-infiltrating immune cells. This study also showed that T-helper type 1 gene expression and CTLA4 expression were positively correlated with response and fractalkine (CX3CL1) was negatively done [11].
With the exception of TNBC, the benefits of immunotherapy for hormone receptor-positive BC and HER2-positive BC have not been clearly defined. Advanced estrogen receptor-positive BC with PD-L1 expression achieved a 12% overall response in a phase Ib clinical trial of pembrolizumab [12]. However, only 5% of patients exhibited a response to anti-PD-L1 antibody regardless of PD-L1 expression status [13].
Here, we sought to identify the gene expression profile associated with immune check points that were potential targets of cancer immunotherapy using advanced BC specimens. Whole transcriptome sequencing analysis could provide useful clues as to which advanced BC populations would benefit from immunotherapy and could help identify predictive markers of immune check point inhibitors.
RESULTS
Immune gene expression and pathway analysis
Thirty-seven metastatic BC samples were analyzed in 91 gene sets associated with the immune pathway (Figure 1A). These 91 gene sets were selected as below: First, we selected 51 immune pathways, 8 pathways including IL-17 and 20 pathways including IRF7 using MSigDB gene sets. In addition, we performed literature review and selected other 16 gene sets [14]. Finally, previous our research showed that KEGG_LEISHMANIA_INFECTION gene set was probably associated to immune signature of BC and this gene set was included to analysis. After removal of duplicated gene sets, 91 gene sets were selected and analyzed (Supplementary Data).
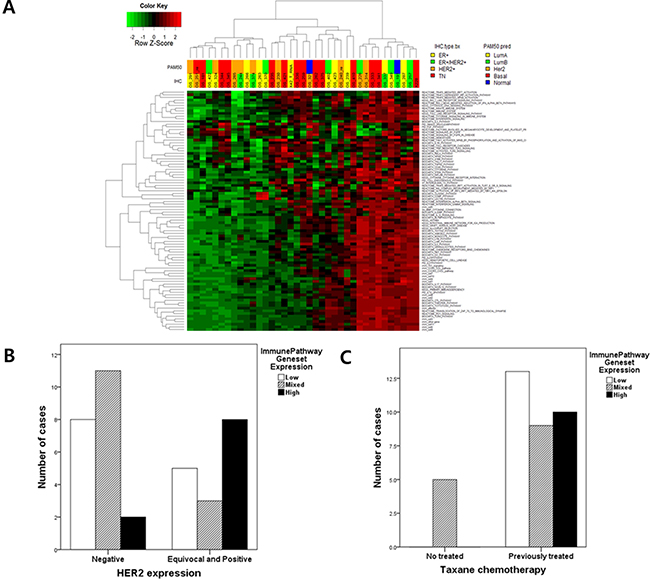
Figure 1: (A) Ninety-one immune pathway gene set enrichment analysis (GSEA) in 37 metastatic BCs : the information of 91 genesets were described in supplementary Data; (B) The level of immune pathway gene set expression according to HER2 expression (p = 0.070); According to data of GSEA of 91 immune pathway gene sets, metastatic BCs were divided into 3 groups (highly activated, mixed and inactivated immune gene sets) And then the association between the level of immune pathway gene set activation and HER2 expression were analyzed. (C) The level of immune pathway gene set expression according to previous taxane chemotherapy (p = 0.008).
This analysis was divided into three subgroups. One group consisting of 10 BCs showed high expression in nearly all immune pathway gene sets, while thirteen BCs showed no immune pathway activation. The third group exhibited = Of 91 gene sets, TRF3, SMAD2, TLR4, CD40 and the TOLL endogenous pathway were related to survival duration in metastatic BC. However, these gene sets did not interact with one another (Supplementary Figure 2).
Among these subgroups, we found that the HER2 immunohistochemical expression score was marginally associated with immune signature clustering (p = 0.070, Fischer’s exact test) (Figure 1B). Other clinical factors did not affect immune pathway activation with the exception of previous taxane chemotherapy (p = 0.008) (Figure 1C, Table 1 and Supplementary Table 1).
Table 1: Impact of clinicopathological characteristics on immune signature (N = 37)
(A) Immune pathway |
Low |
Mixed |
High |
p-value |
|---|---|---|---|---|
Immunohistochemistry |
||||
Estrogen receptor (ER) |
.441 |
|||
Positive |
7 (18.9) |
4 (10.8) |
4 (10.8) |
|
Negative |
6 (16.2) |
10 (27.0) |
6 (16.2) |
|
Progesterone receptor (PgR) |
.107 |
|||
Positive |
3 (8.1) |
3 (8.1) |
6 (16.2) |
|
Negative |
10 (27.0) |
11 (29.7) |
4 (10.8) |
|
HER2 score |
.070 |
|||
0 |
5 (13.5) |
6 (16.2) |
2 (5.4) |
|
1 |
3 (8.1) |
5 (13.5) |
0 |
|
2 |
1 (2.7) |
0 |
4 (10.8) |
|
3 |
4 (10.8) |
3 (8.1) |
4 (10.8) |
|
Ki-67 |
.957 |
|||
Low |
4 (10.8) |
2 (5.4) |
1 (2.7) |
|
High |
9 (24.3) |
12(32.4) |
9 (24.3) |
|
Chemotherapeutic regimen |
||||
Taxane |
.008 |
|||
Previously treated |
13 (35.1) |
9 (24.3) |
10 (27.0) |
|
Untreated |
0 |
5 (13.5) |
0 |
|
Anthracycline |
.999 |
|||
Previously treated |
12 (35.1) |
13 (35.1) |
9 (21.6) |
|
Untreated |
1 (2.7) |
1 (2.7) |
1 (2.7) |
(B) Immune checkpoint gene |
Low |
Mixed |
High |
p-value |
|---|---|---|---|---|
Immunohistochemistry |
||||
Estrogen receptor (ER) |
.999 |
|||
Positive |
6 (16.2) |
5 (13.5) |
4 (10.8) |
|
Negative |
8 (21.6) |
8 (21.6) |
6 (16.2) |
|
Progesterone receptor (PgR) |
.247 |
|||
Positive |
5 (13.5) |
2 (5.4) |
5 (13.5) |
|
Negative |
9 (24.3) |
11 (29.7) |
5(13.5) |
|
HER2 score |
.044 |
|||
0 |
6 (16.2) |
5 (13.5) |
2 (5.4) |
|
1 |
3 (8.1) |
5 (13.5) |
0 |
|
2 |
1 (2.7) |
0 |
4 (10.8) |
|
3 |
4 (10.8) |
3 (8.1) |
4 (10.8) |
|
Ki-67 |
.915 |
|||
Low |
4 (10.8) |
2 (5.4) |
1 (2.7) |
|
High |
10 (27.0) |
11 (29.7) |
9 (24.3) |
|
Chemotherapeutic regimen |
||||
Taxane |
.105 |
|||
Previously treated |
13 (35.1) |
9 (24.3) |
10 (27.0) |
|
Untreated |
1 (2.7) |
4 (10.8) |
0 |
|
Anthracycline |
.999 |
|||
Previously treated |
13 (35.1) |
12 (32.4) |
9 (24.3) |
|
Untreated |
1 (2.7) |
1 (2.7) |
1 (2.7) |
(C) TIL markers |
Low |
Mixed |
High |
p-value |
|---|---|---|---|---|
Immunohistochemistry |
||||
Estrogen receptor (ER) |
.762 |
|||
Positive |
2 (5.4) |
5 (13.5) |
7(18.9) |
|
Negative |
6 (16.2) |
8 (21.6) |
9(24.3) |
|
Progesterone receptor (PgR) |
.182 |
|||
Positive |
1 (2.7) |
3 (8.1) |
8 (21.6) |
|
Negative |
7 (18.9) |
10 (27.0) |
8 (21.6) |
|
HER2 score |
.357 |
|||
0 |
5 (13.5) |
3 (8.1) |
5 (13.5) |
|
1 |
2 (5.4) |
4 (10.8) |
2 (5.4) |
|
2 |
0 |
1 (2.7) |
4 (10.8) |
|
3 |
1 (2.7) |
5 (13.5) |
5 (13.5) |
|
Ki-67 |
.553 |
|||
Low |
1 (2.7) |
4 (10.8) |
2 (5.4) |
|
High |
7 (18.9) |
9 (24.3) |
14 (37.8) |
|
Chemotherapeutic regimen |
||||
Taxane |
.695 |
|||
Previously treated |
8 (21.6) |
11 (29.7) |
13 (35.1) |
|
Untreated |
0 |
2 (5.4) |
3 (8.1) |
|
Anthracycline |
.999 |
|||
Previously treated |
7 (18.9) |
12 (32.4) |
15 (40.5) |
|
Untreated |
1(2.7) |
1(2.7) |
1(2.7) |
Immune checkpoint gene expression
Immune checkpoint gene expression, which is associated with treatment responses of immune check point inhibitors, was also analyzed. We evaluated nine genes previously reported to be targets of immune checkpoint inhibitors: PDCD1 (PD-1), CD274 (PD-L1), CD276 (B7-H3), CTLA-4, IDO1, LAG3, VTCN1, HAVCR2 and TNFRSF4 (OX40) (Figure 2A). Six of 9 genes, CD274, CTLA4, IDO1, LAG3 and HAVCR2, were similarly expressed in each metastatic BC sample. The expression patterns of TNFRSF4 and PDCD1 were also similar to one another.
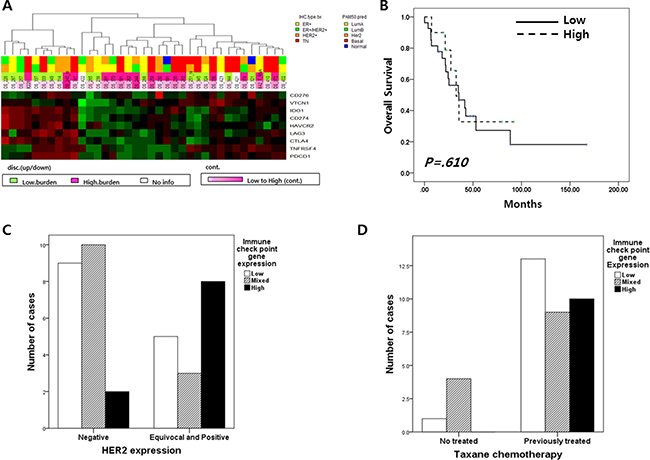
Figure 2: (A) Nine immune checkpoint gene (CD276, CD274, VTCN1, IDO1, HAVCR2, LAG, CTLA4, TNFRSF4 and PDCD1) expression profile in 37 metastatic BC; 10 breast cancer had high expression of nine immune check point genes and 27 did not. (B) Overall survival according to the level of immune checkpoint gene expression;(C) The level of immune checkpoint gene expression according to HER2 expression(p = 0.044); (D) The level of immune checkpoint gene expression according to previous taxane chemotherapy (p = 0.105).
Subgroups divided according to expression pattern of the 9 immune checkpoint genes did not have any distinct clinical characteristics, including survival duration (Figure 2B). However, HER2 immunohistochemical expression scores were related to immune check point gene expression (p = 0.044; Table 1 and Figure 2C) and previous taxane treatment was marginally affected to these gene expression ((p = 0.105; Table 1 and Figure 2D).
Tumor-infiltrating lymphocytes
Tumor-infiltrating lymphocyte markers: CD3, CD4, CD8, CD20 and CD163 were evaluated using RNA-Seq data. Because CD3 was composed of CD3E, CD3G and CD3D, we analyzed seven TIL markers (CD3D, CD3E, CD3G, CD4, CD8, CD20 and CD163) [15].
In this analysis, 37MBCs were divided into two groups according to gene expression pattern (Revised Figure 3A). One group included 16 MBCs had high expression of CD8, CD20, CD3E, CD3D and CD3G and the other did remains of MBCs. (Figure 3A). Based on this gene expression pattern, survival analysis showed that the expression of TIL markers did not influence into BC prognosis (p = 0.947) (Figure 3B).
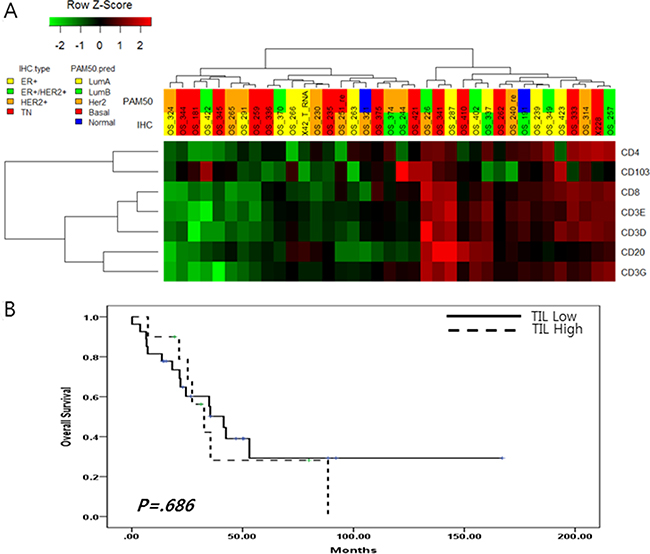
Figure 3:(A) Tumor infiltrating lymphocyte markers expression in 37 metastatic BC; (B) Overall survival according to the level of tumor infiltrating lymphocyte markers.
Further analysis suggested that these groups were not associated with BC subtype or any clinical characteristics of metastatic BC, but no baseline characteristics were found to have a impact on lymphocyte infiltration (Table 1C).
We analyzed the relationship between the expression of nine immune check point genes and seven tumor-infiltrating lymphocyte markers. In this analysis, BC with high expression of immune check point genes also highly expressed CD8, CD20 and CD3 (p < 0.001) (Figure 2A and Figure 3A).
Relationship between mutation burden and immune checkpoint gene expression
Mutation burden, defined as the number of non-synonymous mutations, was checked in 34 metastatic BC samples by analyzing whole-exome sequencing data (Figure 4). The median number of non-synonymous mutations was 72.5 and this was used as the cut-off value for mutation burden (Table 2). In this analysis, non-synonymous single nucleotide variants (SNVs) were most commonly detected in metastatic BC. Low frequency frameshift deletion and stop gain SNVs were also observed.
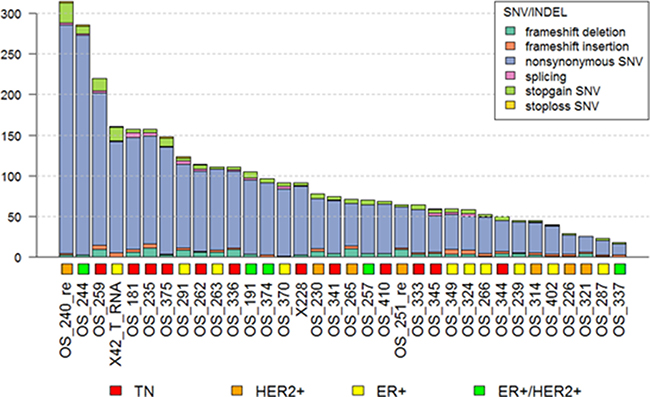
Figure 4: Mutation burden in metastatic BCs.
Table 2: Impact of clinicopathological characteristics on mutation burden (N = 34)
Mutation burden |
Low |
High |
p-value |
|---|---|---|---|
Age (median) 45.1 ± 11.0, range 26.5–75.7 |
.807 |
||
< 40 years old |
8(23.5) |
6(17.6) |
|
≥ 40 years old |
9(26.5) |
11(32.4) |
|
Estrogen receptor (ER) |
.481 |
||
Positive |
9(26.5) |
12(35.3) |
|
Negative |
8(23.5) |
5(14.7) |
|
Progesterone receptor (PgR) |
.999 |
||
Positive |
12(35.3) |
12(35.3) |
|
Negative |
5(14.7) |
5(14.7) |
|
HER2 score |
.379 |
||
0 |
5(14.7) |
6(17.6) |
|
1 |
2(5.9) |
5(14.7) |
|
2 |
4(11.8) |
1(2.9) |
|
3 |
6(17.6) |
5(14.7) |
|
Subtype |
.404 |
||
HR*+HER2- |
6(17.6) |
4(11.8) |
|
HR+HER2+ |
2(5.9) |
3(8.8) |
|
HR-HER2- |
4(11.8) |
8(23.5) |
|
HR-HER2+ |
5(14.7) |
2(5.9) |
|
Intrinsic subtype |
.404 |
||
Luminal A |
2(5.9) |
3(8.8) |
|
Luminal B |
6(17.6) |
4(11.8) |
|
Basal-like |
4(11.8) |
8(23.5) |
|
HER2-enriched |
5(14.7) |
2(5.9) |
|
Ki-67 |
.842 |
||
Low |
4(10.8) |
1(2.7) |
|
High |
10(27.0) |
8(21.6) |
|
Visceral metastasis |
.999 |
||
Yes |
10(29.4) |
10(29.4) |
|
No |
7(20.6) |
7(20.6) |
|
Chemotherapeutic regimen |
|||
Taxane |
.601 |
||
Previously treated |
16(47.0) |
14(41.2) |
|
Untreated |
1(2.9) |
3(17.6) |
|
Anthracycline |
.999 |
||
Previously treated |
15(44.1) |
14(41.2) |
|
Untreated |
2(5.9) |
1(2.9) |
|
Number of chemotherapy |
.728 |
||
≤ 3 |
9(26.5) |
11(32.5) |
|
> 3 |
8(23.5) |
6(17.6) |
|
* Hormone receptor
Mutation burden was not associated with clinical characteristics such as subtype, intrinsic subtype, previous treatment history or BRCA1/2 mutation status (Table 2). Neither gene expression nor gene set pathway score was associated with mutation burden (Supplementary Figure 3). Gene expression of immune checkpoint genes did not have any association with mutation burden. Also, immune checkpoint pathway score did not interact with mutation burden (Figure 2A).
Clinical characteristics of metastatic BC
We enrolled 54 patients with metastatic BC. RNA sequencing was performed on 37 patients and WES was on 34 patients. DNA and RNA extraction failure was the cause of RNA-Seq and WES failure.
The demographic and clinical features of 37 patients are summarized in Supplementary Table 2. The median age of the enrolled patients was 45.1 years, and 35.1% (13 patients) had TNBC. With regard to intrinsic subtype, 14 of 37 patients (37.8%) had basal-like subtype BC. Testing for BRCA1/2 mutation was performed in five patients, and a germline BRCA1 and/or BRCA2 mutation was detected in three patients. Visceral metastasis was found in 15 patients; 8 patients had brain metastasis and the others had liver metastasis. On average, patients with metastatic BC received more than three chemotherapeutic agents for palliative treatment. Thirty-six of 37 patients received anthracycline-containing cytotoxic chemotherapy and 32 were treated with taxane chemotherapy. All patients with ER-positive BC were treated with tamoxifen and/or non-steroidal aromatase inhibitor. Anti-HER2 treatment was administered to all patients with HER2-positive BC. Time to RNA-Seq from diagnosis with metastatic BC varied according to BC subtype (Supplementary Table 2). For ER-HER2-positive BC, mean time to RNA-Seq was 29.3 months (range 5.5–69.7) compared with 4.3 months (range, 0.0–36.7) for ER-HER2-negative BC.
DISCUSSION
This study sought to identify an immune signature that would facilitate the development of therapeutic strategies for metastatic BC. We suggested that the level of HER2 expression was positively correlated to the expression level of immune check point genes in metastatic BC. Moreover, previous taxane treatment might influence immune signature. However, hormone receptor expression and intrinsic subtype did not affect the immune signature of metastatic BC.
Traditionally, tissue-infiltrating lymphocytes (TILs) have been investigated as immune markers of BC. CD8-positive cytotoxic T-lymphocytes provide useful information regarding patient survival and response to therapy and were more frequently observed in ER-negative BC [16]. In addition, CD45 and CD163, pan-leukocyte markers and M2 macrophage markers were neutral or poor prognostic markers in BC [17, 18]. In this study, the expression of CD4, CD8, PTPRC (CD45) and CD163 interacted with one another; we were able to divide groups into two categories according to whether all four genes were highly expressed. This categorization indicated that there was no difference in survival between the two groups. Moreover, clinical characteristics did not influence this categorization, while HER2 expression has a marginal impact on lymphocyte infiltration (p = 0.077). Accordingly, we suggested that TIL in metastatic BC might not inform.
The expression of immune check point, a potential therapeutic target of BC, was also analyzed in this study. The immune check point signaling pathway, including PD-L1, CTLA-4 and IDO1, was consistently upregulated in highly immune-activated BCs. These findings indicated that expression of new immunotherapy targets was correlated to expression of PD-L1 and CTLA-4; therefore, we suggested that BCs that showed clinical response to anti-PD-1 or PD-L1 antibody might also respond well to new immune check point inhibitors, such as TNFSF40 antibody and IDO1 inhibitor. Moreover, these results suggested that a combination of immune check point inhibitors could be an effective therapeutic strategy in metastatic BC.
Hormone receptor expression did not affect immune signature in this study. TNBC immunotherapy has been focused on in prior research, rather than other subtypes of BC. According to preliminary data from clinical trials, immune check point inhibitor is more effective in TNBC. However, up to 10% of BCs with hormone receptor expression shrank in size in response to immune checkpoint inhibitor. Therefore, other stratification factors beyond hormone receptor expression need to be explored to more accurately predict response to immunotherapy. Our study suggested that HER2 expression was related to immune signature, and thus HER2 expression might be a predictive marker for immune check point inhibitor.
Lastly, previous taxane treatment might be associated with the expression of immune check point genes. Although only 5 patients were not treated with taxane-containing regimens, all 5 of these patients did not have high immune check point gene expression. All five BCs which did not expose taxane had been treated using three therapeutic agents and under. Therefore, we performed subgroup analysis using two clinical characteristics; taxane treatment and number of previously exposed therapeutic agents. In BC treated by under three agents, taxane treatment impacted on immune check point gene expression (p = 0.080) and immune pathway gene expression (p = 0.016) (Supplementary Figure 4).
Most clinical trials on cytotoxic chemotherapy combined with immune check point inhibitor have used nab-paclitaxel. However, previous taxane chemotherapy might induce immune check point gene expression; thus, a new strategy for improving the treatment efficacy of cytotoxic chemotherapy combination for metastatic BC might be needed.
Mutation burden did not influence the immune signature of metastatic BC in this study. Melanoma [19], lung cancer [7] which were known to be correlated between mutation burden and response of immune check point inhibitor. Melanoma and lung cancer retain non-synonymous mutations the most frequently among all cancers; in contrast, BCs have only one tenth the somatic mutations of the abovementioned cancers [20]. Accordingly, mutation burden did not appear to play a significant role in the immune signature of BC, but other factors may affect immune signature and response to immunotherapy.
Tissue-infiltrating lymphocytes, immune check point gene expression were positively correlated (p < 0.001). BCs with high TILs had high immune check point gene expression. Therefore, TIL might be a predictive marker of immunotherapy in metastatic BC.
We sought to demonstrate the immune signature of metastatic BC in this study. The limitations of this study included a small sample size and heterogeneous sample collection. Another limitation was that all of the specimens could not be treated with immune check point inhibitor. In terms of breast cancer, the effect of immune check point inhibitor was not revealed yet. Currently, phase III clinical trials of anti-PD-L1 antibodies in breast cancer patients are ongoing [10] and these antibodies are not approved by Food and Drug Administration (FDA) for metastatic breast cancer. Therefore, we could not have any data of metastatic breast cancer patients treated with immune check point inhibitors. Therefore, our study might be the first step to understand the status of immune biomarkers in breast cancer that previously came up in other cancers in spite of some limitations.
However, translational analysis with regard to whole exome sequencing and RNA-Seq is the first step to understanding the immune signature of metastatic BC and immunotherapy response patterns in metastatic BC. Thus, this study could determine which BC types would respond to immune check point inhibitor.
In conclusion, we suggest that metastatic BC with HER2 expression and previous taxane treatment might express immune check point genes, immune pathway gene sets and tissue-infiltrating lymphocytes at high levels. Further investigation of immune signature in BC with large-scale translational studies is warranted.
MATERIALS AND METHODS
Patients
This study was conducted as a prospective explorative analysis of patients with metastatic BC at Samsung Medical Center. Women diagnosed with stage IV BC or recurrent BC after curative treatment on diagnostic examination and a staging work-up (breast magnetic resonance imaging [MRI], chest computed tomography [CT] scan, abdominal CT scan, bone scan, and/or positron emission tomography [PET]-CT scans if indicated) were included.
All patients provided written informed consent, and study approval was obtained from the Institutional Review Board of Samsung Medical Center, Seoul, Korea (IRB No: SMC 2012-08-065).
Immunohistochemical (IHC) staining
Two experienced pathologists reviewed all pathology specimens to determine IHC staining for ER, PgR, and HER2. ER and PgR positivity were defined using Allred scores ranging from 3 to 8 based on IHC using antibodies to ER (Immunotech, Marseille, France) and PgR (Novocastra Laboratories Ltd., Newcastle upon Tyne, UK). HER2 status was evaluated using a specific antibody (Dako, Glostrop, Denmark) and/or silver n situ hybridization (SISH). Grades 0 and 1 for HER2, as assessed by IHC, were defined as a negative result, and grade 3 was defined as a positive result. Amplification of HER2 rated as 2+ by IHC was confirmed by SISH. Ki67 IHC analyses were performed by independent semiquantitative and quantitative methods (Dako). Triple negativity was defined as a lack of expression of ER, PgR, and HER2.
DNA and RNA extraction
Tumors consisting of over 75% malignant cells were dissected under microscopy from 4-mm unstained sections by comparison with an H&E-stained slide, and genomic DNA was extracted using a Qiagen DNA FFPE Tissue kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After extraction, concentration as well as 260/280 and 260/230 nm ratios were measured by spectrophotometry (ND1000, NanoDrop Technologies, ThermoFisher Scientific, MA, USA). Each sample was then quantified using a Qubit fluorometer (Life Technologies, Carlsbad, CA, USA). Genomic DNA with a total yield > 10 ng was used for library preparation.
Areas containing representative invasive breast carcinoma were outlined on the slide. Total RNA was then extracted using a High Pure RNA Paraffin kit (Roche Diagnostic, Mannheim, Germany) and the RNA concentration and 260/280- and 260/230-nm ratios were measured using a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Rockland, DE, USA). Samples with less than 1 mg/μL total RNA even after concentration with a SpeedVacTM concentrator (Thermo Scientific™, Waltham, MA, USA) were excluded from downstream analysis.
Whole-exome sequencing
Poor quality reads were filtered out and aligned to the human reference genome (hg19) using Burrows-Wheeler Alignment tool (BWA, version 0.7.5a). In order to convert Sequence Alignment and Mapping (SAM) files into Binary Alignment and Mapping files (BAM), we used SAMtools (version 0.1.19). Polymerase chain reaction (PCR) duplicates were removed from the BAM files by Picard (version 1.93, http://broadinstitute.github.io/picard/) and SAMtools before variant calling. The Genome Analysis Toolkit (GATK, version 2.4.7) was used to recalibrate base quality and optimize local realignment. Single nucleotide variants (SNVs) and indels were called using muTect (version 1.1.4) and Varscan2 (version 2.3.5) using default parameter settings. Copy number variations were detected by CONTRA (version 2.0.4). Variants were annotated using ANNOVAR, with gene, chromosomal information, exonic function function (synonymous, non-synonymous, stop gain, non-frameshift or frameshift indel), amino acid change, allele frequency in frequency in public databases such as 1000 Genomes Project (February 2012 version) and dbSNP version (version 132).
Variants chosen for further statistical analyses were located in the exonic regions with sufficient coverage (minimum depth of coverage ≥ 8) and variant allele frequency (VAF ≥ 0.1). Synonymous variants were filtered out. Read alignments were manually investigated using the Integrative Genomic Viewer (http://www.broadinstitute.org/igv/).
Fisher’s exact test was used for the analysis of mutations and polymorphic variants, to discover variants that were enriched in patients with favorable outcomes. P-values < 0.05 were considered significantly different. All statistical analyses, plots and heatmaps were conducted using R version 3.0.2 (http://www.R-project.org/).
RNA-Seq analysis and normalization
After trimming poor quality bases from the FASTQ files for whole-transcriptome sequencing, we aligned the reads to human reference genome hg19 with Tophat (version 2.0.6) and performed reference-guided assembly of transcripts with Cufflinks (version 2.1.1). The alignment quality was verified with SAMtools (version 0.1.19). Transcript abundance was estimated using a count-based method with a htseq-count. Gene counts were used as input for Trimmed Mean of M values (TMM) normalization i the R package edgeR [21], and normalized counts were transformed to log2-counts per million (logCPM) by applying voom from the R package limma [22] to account for higher variability at low expression levels. Genes with zero read counts across all samples were removed for more powerful statistical analysis (Supplementary Figure 1).
Intrinsic subtyping
We performed intrinsic subtyping with log-scaled normalized expression values using the 50-gene Prediction Analysis of Microarray (PAM50) subtype predictor as described by Parker et al. [23]. The PAM50 subtype predictor classified tumors into the following groups: Luminal A, Luminal B, HER2-enriched, basal-like, and normal-like (Supplementary Figure 1).
Gene set enrichment analysis
To examine how overall survival-associated genes share predefined gene sets representing common processes, pathways, and underlying biological themes, we investigated sub-collections in the Molecular Signatures Database (MSigDB, version 5.0) with overall survival-associated genes using the Gene Set Enrichment Analysis (GSEA) website. We also calculated Gene Set Enrichment (GSE) scores for canonical pathways in MSigDB and several AR-related gene sets from the literature [24, 25] using the R package Gene Set Variation Analysis (GSVA). GSVA is a nonparametric method that provides sample-wise gene set enrichment scores to identify differential gene set activity. A two-sample t-test was then performed, and gene sets with a false discovery rate less than 0.05 were considered to show significantly different activity between the two groups. All normalization, statistical analyses, and visualization were conducted within the R statistical system (version 3.0.2).
Survival analysis
We evaluated the association between gene expression and overall survival using the R package. Overall survival was defined as the elapsed time between the date of stage IV BC diagnosis and the date of death. For each gene, patients were grouped based on the normalized expression value of the gene, with the top 50% and the bottom 50% representing high and low expression groups, respectively. Survival curves for the two groups were estimated with the Kaplan-Meier method, and the log-rank test was used to compare overall survival curves between the two groups (p < 0.05). Fisher’s exact test was used to identify the pathways with significant enrichment of associated genes in terms of overall survival (p < 0.05).
ACKNOWLEDGMENTS AND FUNDING
J-K performed statistical analyses of genetic and clinical sata and drafted and revised the manuscript. EL and KP handled whole exome and transcriptome data analysis. HHJ carried out the sample preparation. J-K and HHJ participated in interpreting the genetic and clinical data. JSA, YHP and Y-I participated in sample preparation and coordination to analyze clinical data. YHP conceived and designed of this study. All authors read and approved the final manuscript. The authors have no conflicts of interest to declare.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–2454.
2. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012; 366:2455–2465.
3. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002; 99:12293–12297.
4. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015; 372:2521–2532.
5. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015; 373:1803–1813.
6. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–2028.
7. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–128.
8. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–2520.
9. Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, Karantza V, Buisseret L. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol. 2016; 34:2460–7.
10. Cha E, Wallin J, Kowanetz M. PD-L1 inhibition with MPDL3280A for solid tumors. Semin Oncol. 2015; 42:484–487.
11. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563–567.
12. Rugo HS, Delord JP, Im SA, Ott PA, Piha-Paul SA, Bedard PL, Sachdev J, Le Tourneau C, van Brummelen E, Varga A, Saraf S, Pietrangelo D, Karantza V, Tan A. Abstract S5–07: Preliminary efficacy and safety of pembrolizumab (MK-3475) in patients with PD-L1–positive, estrogen receptor-positive (ER+)/HER2-negative advanced breast cancer enrolled in KEYNOTE-028. Cancer Res. 2016; 76:S5–07. doi: 10.1158/1538-7445.SABCS15-S5-07.
13. Dirix LY, Takacs I, Nikolinakos P, Jerusalem G, Arkenau HT, Hamilton EP, von Heydebreck A, Grote HJ, Chin K, Lippmann ME. Abstract S1–04: Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase Ib JAVELIN solid tumor trial. Cancer Res. 2016; 76:S1–04; doi: 10.1158/1538-7445.SABCS15-S1-04.
14. Bedognetti D, Hendrickx W, Marincola FM, Miller LD. Prognostic and predictive immune gene signatures in breast cancer. Curr Opin Oncol. 2015; 27:433–444.
15. Savas P, Salgado R, Denkert C, Sotiriou C, Darcy PK, Smyth MJ, Loi S. Clinical relevance of host immunity in breast cancer: from TILs to the clinic. Nat Rev Clin Oncol. 2016; 13:228–241.
16. Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol. 2011; 29:1949–1955.
17. Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, Budczies J, Darb-Esfahani S, Kronenwett R, Hanusch C, von Torne C, Weichert W, Engels K, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010; 28:105–113.
18. Shabo I, Stal O, Olsson H, Dore S, Svanvik J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int J Cancer. 2008; 123:780–786.
19. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–2199.
20. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421.
21. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012; 40:4288–4297.
22. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47.
23. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009; 27:1160–1167.
24. Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene. 2006; 25:3994–4008.
25. Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI, Osborne CK, Hilsenbeck SG, Chang JC, Mills GB, Lau CC, Brown PH. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015; 21:1688–1698.