
INTRODUCTION
Gliomas are the most common primary malignant tumor of the central nervous system and account for 24% of brain tumors [1]. Tumor grades range from Grade I to Grade IV and are based on histopathological and clinical criteria established by the World Health Organization (WHO) [1, 2]. Grade I tumors are relatively benign and are circumscribed tumors that display a favorable prognosis with 94% of patients surviving at 5 years and 91% at 10 years [1]. Grade II gliomas are diffusely infiltrative and can be divided into astrocytomas and oligodendrogliomas. These tumors have the inherent ability to progress to higher grade gliomas. In addition to Grade II and III astrocytomas and oligodendrogliomas, another subtype of glioma presents with a histological appearance of both oligodendrogliomas and astrocytomas. These “mixed histology” tumors, or oligoastrocytomas also have the ability to progress from Grade II to Grade III tumors. The Grade III astrocytomas have the ability to further progress into secondary Grade IV glioblastomas (GBM), which exhibit a poorer prognosis than the grade III astrocytomas. As first described by Scherer in 1940 [3] secondary GBM arises as a progression from Grade II and Grade III tumors, whereas primary GBM arises de novo and has a dismal median OS of 15 months [1]. The progression between grades along with the potential for mixed histology presents neuropathologists with diagnostic challenges that often rely on subjective measures. Consequently, diagnoses among different pathologists and institutions have weak correlations that may result in variable treatment and management of each tumor grade [2, 4]. The subjective nature of these analyses stresses the importance of an accurate, unbiased, and objective means of diagnosis. This is crucial for stratification of patients with biologically similar tumors in clinical trials, and could aid in the selection of targeted therapeutic regimens. The discovery of biomarkers that objectively identify each tumor’s unique molecular signature is a necessary next-step in managing patient outcomes more effectively. Genetic signatures performed on pathologically relevant tissues will be a potentially useful supplement to clinicians in refining and clarifying patient stratification.
Characterization of the genetic landscape of gliomas has been at the forefront of cancer research in order to better aid prognostication and classification of clinical outcomes [5, 6]. High-throughput screens have paid particular attention to understanding the genomic variability between each subgroup of glioma. The Cancer Genome Atlas and other groups, including ours, have begun to identify the molecular subgroups of these tumors and delineate which tumor types harbor which mutations [5-12]. For example, IDH1/2 mutations that occur frequently in secondary GBMs (>50%) are infrequent in primary GBMs (<5%) [8, 12].
Recent findings have established frequent mutations in the promoter region of telomerase reverse transcriptase (TERT) in a multitude of cancers, including melanomas, liposarcomas, bladder cancer, urinary tract cancers, and gliomas [13-19]. TERT is a subunit of the telomerase enzyme that, when expressed, allows cells to avoid senescence. This is especially noted as TERT is mutated in high frequencies in cells with low rates of self-renewal, such as melanocytes, urothelial cells, and glial cells [14-16, 20, 21]. Of interest to glioma genomics, TERT promoter mutations occur in 70-80% of primary GBMs and >70% of oligodendrogliomas, but occur less frequently in both lower grade astrocytomas and most oligoastrocytomas [16, 17, 22].
The discovery of TERT promoter mutations in these subsets of gliomas creates an opportunity for genomics to supplement histopathological analysis, especially when combined with IDH1/2 mutation status. Here, we have assessed the characteristic variance between IDH1/2 and TERT promoter mutations among several glioma subtypes that help refine the diagnosis of gliomas. The assay, based upon three polymerase chain reactions (PCR), provides pathologists with a manageable and reliable diagnostic supplement in the form of a simple, yet robust genetic signature unique to each tumor type.
RESULTS
TERT promoter mutations are frequent in primary GBMs and oligodendrogliomas but uncommon in lower grade astrocytoma.
To assess the prevalence and prognostic impact of TERT promoter mutations we sequenced the proximal TERT promoter hotspot mutations (C228T and C250T) in 473 adult gliomas. We identified TERT promoter mutations in 281 (59.4%) tumors (Fig. 1). In agreement with previous studies [16, 18, 23], we identified TERT promoter mutations in 74.2% of grade IV GBMs (178/240). TERT promoter mutations were also common in oligodendrogliomas (79.3%); however, TERT promoter mutations were less frequently identified in Grade II-III astrocytomas (18.2%, 16/88). Furthermore, we observed a moderate frequency of TERT promoter mutations in oligoastrocytomas (31.0%, 18/58). As expected, GBMs were diagnosed in older patients when compared to other histologic subtypes studied here (Table 1). Within each tumor type, TERT promoter mutations were associated with an older age at diagnosis (Table 2).
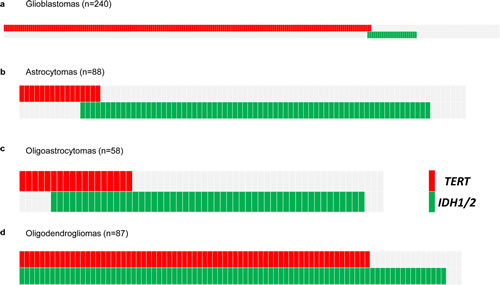
Fig 1: Distribution of TERT promoter and IDH1/2 mutations in a panel of 473 adult gliomas. Mutational analysis of 473 adult gliomas for TERT promoter and IDH1/2 mutations. Data are from 240 Grade IV GBM (A), 88 Grade II-III astrocytomas (B), 58 Grade II-III oligoastrocytomas (C), and, 87 Grade II-III oligodendrogliomas (D). Mutation status is indicated by color shading, with gray coloring indicating wild type sequence, red indicating mutations in the TERT promoter, and green indicating mutations in IDH1/2.
Co-occurring mutations in TERT promoter and IDH1/2.
IDH1/2 mutations are a well-established molecular feature of gliomas [12]. To define the co-occurrence of IDH1/2 mutations and the presence of TERT promoter mutations, we determined the status of IDH1 and IDH2 mutations in the same cohort of 473 gliomas and identified mutations in 47.9% (227/473) of tumors (Fig. 1 and Table 1). IDH1/2 mutations were much less prevalent among GBMs (10%), and much more common in Grade II-III astrocytomas (78.4%), oligoastrocytomas (86.2%) and oligodendrogliomas (96.5%). TERT mutations occurred in the absence of IDH1/2 mutations in GBMs (73.3%, 176/240). However, in oligodendrogliomas, the TERT promoter mutation always occurred in the setting of the IDH1/2 mutation, which is frequent in both oligodendrogliomas and astrocytomas (Fig. 1) [12]. The cross-tabulation of TERT promoter and IDH1/2 mutations aligned with three of the four histologic subtypes. GBMs were characterized as primarily TERT promoter mutant/IDH wildtype (73.3%), Grade II-III astrocytomas were predominantly TERT promoter wildtype/IDH mutant (73.9%), and the majority of oligodendrogliomas mainly harbored mutations in both the TERT promoter and IDH1/2 (79.3%). A majority of oligoastrocytomas (63.8%) exhibited the IDH mutation in the absence of TERT promoter mutations, much like Grade II-III astrocytomas; however, a fraction (22.4%) of oligoastrocytomas presented with both TERT promoter and IDH1/2 mutations, similar to oligodendrogliomas (Fig. 1).
TERT promoter and IDH1/2 mutations have distinct tumor distributions and are associated with OS.
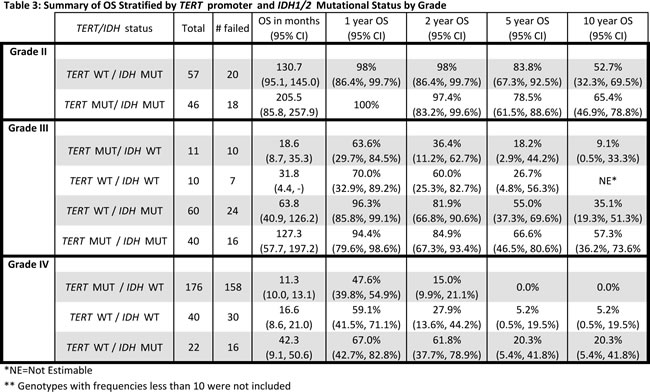
We next sought to determine whether the combination of TERT promoter and IDH1/2 mutations are associated with OS. Clinical information (survival, age at diagnosis, and histopathological diagnosis) was available for our cohort of 473 adult gliomas in both treated and untreated patients (Table 1). As grade is a well-known prognostic factor in glioma patients, we first investigated whether distinct tumor subgroups could be distinguished using only TERT promoter and IDH1/2 mutation status within each grade (Fig. 2, Table 3). Among the 112 Grade II gliomas, 103 were characterized by either mutations in both TERT and IDH or IDH alone. The median OS of those tumors harboring mutations in both TERT promoter and IDH1/2, the predominant genetic signature in oligodendrogliomas, was longer than those tumors with an IDH1/2 mutation only, typically seen in Grade II-III astrocytomas (206 months vs. 131 months), but this difference was not statistically significant (log-rank p=0.1754) (Fig. 2A). When stratified by histologic diagnosis, oligodendrogliomas had the best median OS among Grade II astrocytomas, oligodendrogliomas, and oligoastrocytomas, as expected (median OS 205 months).
Among the 121 Grade III tumors, 60 (50%) had IDH1/2 mutations alone and 40 (34%) had mutations in both the TERT promoter and IDH1/2. Those with mutations in both the TERT promoter and IDH1/2 had the largest median OS (127 months), followed by those with an IDH1/2 mutation only (median OS 64 months), and those with neither mutation (median OS 32 months). Tumors with mutations in the TERT promoter alone, which was the predominant signature present in primary GBMs had the poorest OS (median OS 19 months). Four distinct subgroups of Grade III gliomas were identified when stratified by the combination of the TERT promoter and IDH1/2 mutation status (log-rank p=0.0008) (Fig. 2B). Oligodendrogliomas again had the best median OS when Grade III tumors were stratified by histologic subtypes (median OS 125 months), but OS did not significantly differ among the three histologic subtypes, which were astrocytomas, oligodendrogliomas, and oligoastrocytomas (log-rank p=0.1626).
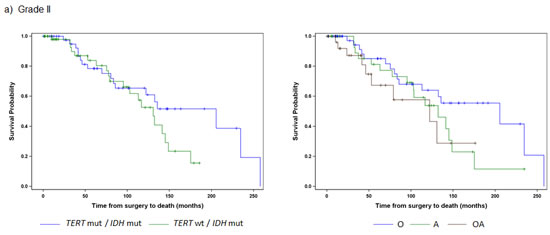
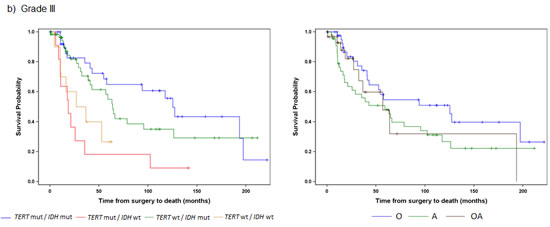
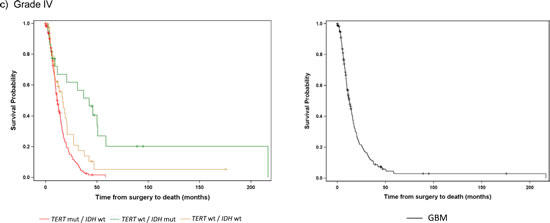
Fig 2: Overall Survival stratified by TERT promoter and IDH1/2 mutational status and histology within each tumor grade. Overall survival was represented by Kaplan Meier plots for individual WHO tumor grade: a) Grade II (n=103), b) Grade III (n=121), c) Grade IV (n=218). Only subgroups with at least 10 patients were included in the analyses. Tumors were represented by mutations status on the left (TERT promoter status / IDH1/2 status) and histology on the right (A represents Astrocytomas, O represents Oligodendrogliomas, and OA represents Oligoastrocytomas).
A majority of the GBMs were characterized by mutations in the TERT promoter alone (73%), and this genetic signature also had the worst prognosis (median OS 11.3 months) (Fig. 2, Table 3). Those without mutation in either marker had only a slightly better outcome (median OS 17 months), while those with an IDH1/2 mutation alone, the signature characteristic of Grade II-III astrocytomas and Grade IV secondary GBMs, had the best outcome among the Grade IV tumors (median OS 42 months). Within the primary and secondary GBMs, using the TERT promoter and IDH1/2 alone, we were able to distinguish three significantly different subgroups (log-rank p<0.0001), and these associations remained when adjusting for the factors of age and diagnosis (Table 4). The TERT promoter mutation is associated with poorer OS in GBMs, and as shown in the multivariable model, this association was also evident among tumors without an IDH1/2 mutation (HR: 1.9, 95% CI: 1.2-2.9).

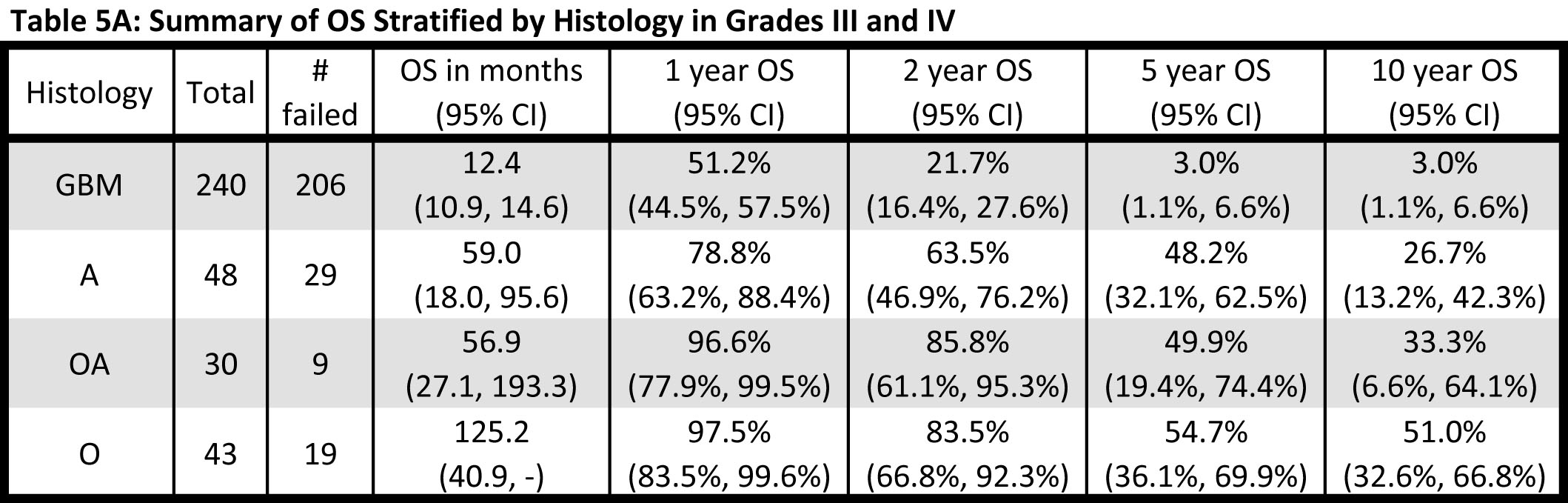
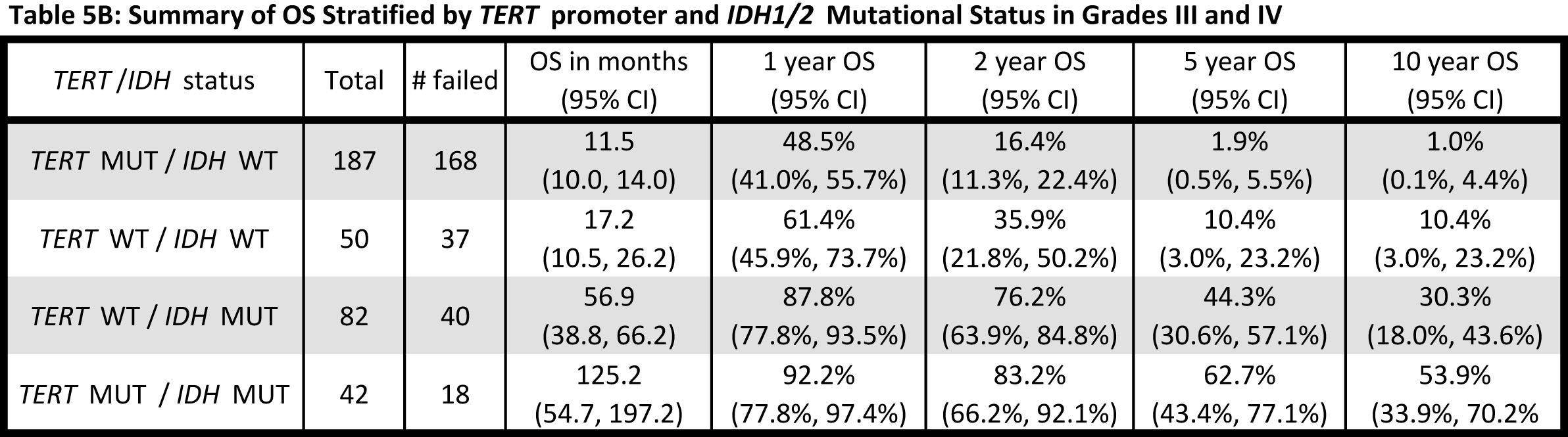
Given that both Grade III and Grade IV gliomas were successfully stratified into distinct subgroups based on TERT promoter and IDH1/2 mutational status, and that each signature was associated with a similar median OS within grade, the effect of histology and genetic signature on OS was also examined across the Grade III and IV gliomas together (Fig. 3, Table 5A). When Grade III and IV gliomas were examined based on histology, GBMs predictably had by far the worst prognosis, and oligodendrogliomas experienced the best survival outcome; however, OS among the Grade III astrocytomas and oligoastrocytomas was similar and difficult to distinguish (Fig. 3A and Table 5A). Nevertheless, when genetic signatures were applied to the same cohort of tumors, four distinct clinical subgroups emerged (Fig 3B). As observed, within Grade III and IV gliomas separately, tumors with mutations in both TERT and IDH1/2 had the best median OS (oligodendroglioma signature), followed by those with an IDH1/2 mutation only (Grade II-III astrocytoma and secondary GBM signature). Both tumors without mutation in either marker and those tumors with a TERT promoter mutation alone had a poorer prognosis, with the latter signature having the worst median OS. The strength of the association between OS and TERT/IDH1/2 mutational status is similar to that of OS and histology (Generalized R2: 0.3132 and 0.2704, respectively).
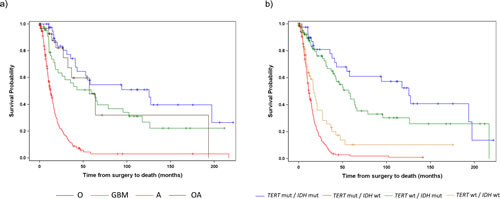
Fig 3: Overall Survival stratified by TERT promoter and IDH1/2 mutational status and histology among Grade III and IV patients. Overall survival was represented by Kaplan Meier plots stratified by a) histology (A represents Astrocytomas, O represents Oligodendrogliomas, OA represents Oligoastrocytomas, and GBM represents Glioblastoma) and b) TERT promoter / IDH1/2 mutation status for all Grade III and Grade IV gliomas analyzed in this study.
DISCUSSION
Our analysis of this tumor cohort expands upon previous reports identifying frequent TERT promoter mutations in gliomas [16-18, 22, 23], examines the association between TERT promoter and IDH1/2 mutations in glioma, and assesses their joint influence on OS. Utilizing a combined analysis of IDH1/2 and TERT promoter mutations in adult glioma, we have derived a greatly expedited and simplified genetic signature of three common glioma subtypes, namely Grade II-III astrocytomas, oligodendrogliomas, and GBMs. Additionally, we show that oligoastrocytomas can be further classified.
Among patients with GBMs, we showed that the largest fraction of GBMs present with TERT promoter mutations. IDH1/2 mutations are infrequent in these tumors and cluster within secondary GBMs. Three distinct subgroups were defined by the presence or absence of TERT promoter and IDH1/2 mutations. Where patients harboring tumors with TERT promoter mutations alone had the poorest OS (median 11.3 months), patients with tumors bearing no mutations in either TERT or IDH1/2 had a slightly better survival (median 16.6 months), and GBMs with IDH1/2 mutation alone resulted in the best survival (median 42.3 months). Furthermore, these associations remained after adjustment for factors such as age. TERT promoter mutations predicted poorer OS outcome in a multivariate model even in GBMs without IDH1/2 mutations. This finding is in contrast with previous studies that did not report a significant difference in OS between TERT promoter-mutated and TERT promoter-wildtype non IDH mutated GBMs [23]. This finding will be of particular interest to clinicians as it may provide a tool to stratify non IDH1/2 mutant GBMs and suggests that combined IDH1/2 and TERT promoter genotyping will be useful for patient management. Because of variable treatment among these histological brain tumor groups, further analyses must include large cohorts of standardized treatment arms and measurements of other genetic features such as MGMT status, EGFR wildtype amplification, and the presence of EGFRvIII to confirm the validity of our findings. At a minimum, our current findings warrant further investigation and confirmation by other investigators. Also, genetic alterations of the TERT promoter may be particularly relevant given the development of therapeutics targeted against telomerase. Telomerase inhibitors have shown promise for treating GBM in preclinical models and are currently under investigation in clinical trials for several types of cancer [24-27].
Conversely, IDH1/2 mutations in Grade II-III astrocytomas are frequent while TERT promoter mutations are uncommon. Grade II-III oligodendrogliomas have frequent co-occurring mutations in the TERT promoter and IDH1/2. We provide evidence that over 86% of oligoastrocytomas in this cohort contain genetic signatures representative of either astrocytoma (IDH1/2 mutations alone) or oligodendroglioma (TERT promoter/IDH1/2), signatures that we show are associated with OS. Reproducibility of oligoastrocytoma diagnosis by histology alone displays variable diagnoses between neuropathologists within and among different institutions [2, 4, 28]. The presence of the TERT promoter and IDH1/2 mutational status may be particularly useful to refine the classification of “mixed” oligoastrocytomas.
In addition to demonstrating the robust nature of these mutational patterns, we have further established that these genetic signatures are reliable when compared to the OS of patients derived from conventional histopathological diagnosis. As shown in Figures 2 and 3, mutations in the TERT promoter and IDH1/2 effectively stratify patients into reproducible subgroups based on survival. This phenomenon was independent of grade among high grade astrocytomas as Grade III and Grade IV tumors mimicked this relationship when analyzed independently (Fig 2). Furthermore, the strength of these genetic signatures and their association with OS is illustrated by a slightly higher R2 (0.3132 vs. 0.2704) than by histology alone.
Two clinical subgroups exist among Grade II tumors in the current cohort, as the power of the survival analysis was limited due to the smaller number of low grade gliomas. The Grade II tumors exhibited genetic signatures with mutations in IDH1/2 alone, and tumors with mutations in the TERT promoter and IDH1/2. Both subgroups had a more favorable prognosis, with a median OS of 130.7 months in tumors with IDH1/2 mutations alone, and median OS of 205.5 months among patients whose tumors harbored TERT promoter and IDH1/2 mutations. No Grade II tumors exhibited TERT promoter mutations alone.
Within Grade III-IV gliomas, those patients with the TERT promoter mutations alone had the poorest prognosis (median 11.5 months), while tumors bearing the events typically representative of astrocytomas (IDH1/2 mutation) had a more favorable prognosis (median 56.9 months). Tumors harboring mutations typically seen in oligodendroglioma (both TERT promoter and IDH1/2 mutation) had a more favorable prognosis (median 125.2 months). Tumors that did not harbor mutations in either the TERT promoter or IDH1/2 comprised a unique clinical group with a short OS (median OS 17.2 months) that was distinct from TERT promoter mutated gliomas (median OS 11.5 months) (Table 5B). As these gliomas, wildtype for both TERT promoter and IDH1/2 mutations, represented a clinically distinct unit (Fig. 2B and 2C) further investigation is required to delineate critical driver mutations in this subset of gliomas.
It is of interest to note that within each tumor type, a minority of tumors bore the genetic signature typically associated with other histological subtypes. In particular, 13.6% (12/88) of Grade II-III astrocytomas bore TERT promoter mutations alone and occasional Grade II-III astrocytomas harbored both TERT promoter and IDH1/2 mutations (4/88, 4.6%). This suggests that at least genetically, these tumors may be more similar to GBM and oligodendroglioma, respectively. Oligodendrogliomas were almost exclusively TERT promoter and IDH1/2 mutated (79.3%, 69/87), but a fraction, 17.2% (15/87) harbored mutations in IDH1/2 alone. In our cohort, no oligodendroglioma cases harbored TERT promoter mutations alone. A minor fraction of GBMs (0.8%, 2/240) contained mutations in both the TERT promoter and IDH1/2 suggesting they were treated oligodendrogliomas that were diagnosed as small cell GBMs.
Loss of chromosomal arms 1p and 19q is a well-known genetic event associated with oligodendrogliomas that many neuropathologists use as a reliable test for diagnosing oligodendroglioma, a tumor generally associated with favorable prognosis and response to chemotherapy [29-32]. As a secondary analysis, the 69 oligodendrogliomas with 1p/19q status available were analyzed for an association with TERT promoter/IDH1/2 mutational status. All 44 oligodendrogliomas with TERT promoter and IDH1/2 mutations also had the 1p/19q allelic deletions and all but 3 of the 47 tumors with 1p/19q allelic losses also contained both TERT promoter and IDH1/2 mutations, indicating that IDH1/2 and TERT promoter mutational analysis may be a comparable prognostic markers to 1p and 19q in oligodendrogliomas (Fisher exact p<0.0001).
This study supports genotyping of TERT promoter and IDH1/2 in gliomas as a rapid economical test requiring little tumoral DNA that could help inform clinicians as to the predicted OS of these tumors that may differ from their predicted outcomes based on conventional histology alone. The TERT promoter mutations analyzed in this study lay only 22 base pairs apart, allowing for PCR amplification in a single amplicon. Additionally, the most frequent mutations in IDH1 and IDH2 occur in hotspot residues located at resides R132 and R172, respectively. Combined together, these three PCR amplicons allow for expedient turnaround, objective interpretation, and vast economic advantages to glioma patients.
The TERT promoter/IDH1/2 mutational profiles of each tumor type can be used in several aspects of the clinical process including stratification of patients, examination of therapeutic response, and selection of treatment, among others. Given the background genes previously discovered in glioma, we hypothesize TERT promoter and IDH1/2 mutations as the major driver genes that are consistently found in low-grade and high-grade adult gliomas. These gene mutation assays will support and expedite the diagnosis of brain tumors while supplementing histopathological evaluation. Measurement of these biomarkers could further increase the fidelity of glioma diagnosis in a rapid and cost-effective manner. Furthermore, the simplicity and affordability of these tests underscore their importance as a tool to aid neuropathologists in glioma diagnosis. Notably, these signatures can be applied to cases that present atypical morphologic features in standard histopathological analysis. Taken together these findings simplify the genetic classification of glioma. The ability of these genetic signatures to stratify patients will refine and clarify the diagnostic accuracy of pathologists by supplementing standard histopathological criteria with genetic mutational analysis.
METHODS
Sample Collection, Processing, and Sequencing
Adult glioma (18 ≥ years old) and corresponding clinical information were obtained with consent and Institutional Review Board approval from the Preston Robert Tisch Brain Tumor Center BioRepository at Duke University in accordance with the Health Insurance Portability and Accountability Act. Newly diagnosed versus recurrent glioma status and vital status were determined by clinical chart review. Fresh frozen tissue sections (first and last sections from the block, stained with hematoxyline and eosin) were reviewed by a board-certified neuropathologist (REM) to confirm original clinical histopathologic diagnosis and to ensure intervening studied sections contain ≥ 80% tumor cells. DNA was extracted from 240 Grade IV GBMs, 88 Grade II and Grade III astrocytomas, 58 Grade II and Grade III oligoastrocytomas, and 87 Grade II and Grade III oligodendrogliomas. Of the 473 tumors, 160 gliomas had been analyzed in our previous studies of the TERT promoter [16]. Isolated DNAs were PCR amplified for the TERT promoter, exon 4 of IDH1, and analyzed via Sanger sequencing for 473 tumors as described previously [12, 16, 33]. Additionally, on those cases that did not harbor mutations in IDH1 we amplified exon 4 of IDH2 and analyzed them via Sanger sequencing. 1p and 19q copy number was evaluated by microsatellite marker analysis and via 1p and 19q FISH testing in a certified clinical laboratory as described previously [12, 29, 34].
Statistical Methods
Clinical and demographic characteristics at the time of diagnosis were summarized for all patients and stratified by histologic tumor type. Means and standard deviations were used to describe interval variables, whereas frequency distributions were used to describe categorical variables. Unpaired t-tests were used to compare the mean age of patients with and without TERT promoter mutations. The Kaplan-Meier estimator was used to describe OS. OS was defined from time of surgery to death or last follow-up. Multivariable Cox models were used to assess the effect of TERT promoter and IDH1/2 mutations on OS adjusting for baseline tumor characteristics. The generalized R2 statistic was used to assess the strength of association between covariates. Associations between categorical variables were analyzed using Fisher exact tests.
ACKNOWLEDGEMENTS
The authors would like to thank Diane Satterfield, Lisa Ehinger, Merrie Thomas, and David Lister for assistance in collection of clinical samples. Hai Yan receives royalties from Agios, Personal Genome Diagnostics, Eli Lilly and Company, and Sanofi, and research funds from Sanofi and Gilead Sciences on IDH related licensing and research. The authors would like to thank Allen Zhou for assistance in data collection. This project was supported by The V Foundation, the Accelerate Brain Cancer Cure Foundation, The Slomo and Cindy Silvian Foundation, the Voices Against Brain Cancer Foundation, the Pediatric Brain Tumor Foundation Institute at Duke, the James S. McDonnell Foundation, American Cancer Society Research Scholar Award RSG-10-126-01-CCE, and National Cancer Institute Grants 5R01-CA140316, 5 P50 NS020023-30, and 5 P01 CA154291-01.
References
1. Dolecek TA, Propp JM, Stroup NE and Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-oncology. 2012; 14 Suppl 5:v1-49.
2. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007; 114(2):97-109.
3. Scherer HJ. Cerebral Astrocytomas and Their Derivatives. The American Journal of Cancer. 1940; 40(2):159-198.
4. Wen PY and Kesari S. Malignant gliomas in adults. The New England journal of medicine. 2008; 359(5):492-507.
5. Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008; 455(7216):1061-1068.
6. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321(5897):1807-1812.
7. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013; 155(2):462-477.
8. Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012; 3(7):709-722.
9. Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, Fouse SD, Yamamoto S, Ueda H, Tatsuno K, Asthana S, Jalbert LE, Nelson SJ, Bollen AW, Gustafson WC, Charron E, et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science. 2013.
10. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG and Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer cell. 2006; 9(3):157-173.
11. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, Tamayo P, Weir BA, Gabriel S, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010; 17(1):98-110.
12. Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009; 360(8):765-773.
13. Griewank KG, Murali R, Schilling B, Scholz S, Sucker A, Song M, Susskind D, Grabellus F, Zimmer L, Hillen U, Steuhl KP, Schadendorf D, Westekemper H and Zeschnigk M. TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. British journal of cancer. 2013; 109(2):497-501.
14. Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D and Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013; 339(6122):959-961.
15. Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L and Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013; 339(6122):957-959.
16. Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Jr., Friedman AH, Friedman H, Gallia GL, Giovanella BC, Grollman AP, He TC, He Y, Hruban RH, Jallo GI, Mandahl N, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(15):6021-6026.
17. Koelsche C, Sahm F, Capper D, Reuss D, Sturm D, Jones DT, Kool M, Northcott PA, Wiestler B, Bohmer K, Meyer J, Mawrin C, Hartmann C, Mittelbronn M, Platten M, Brokinkel B, et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta neuropathologica. 2013.
18. Liu X, Wu G, Shan Y, Hartmann C, von Deimling A and Xing M. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell cycle. 2013; 12(10):1637-1638.
19. Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, Melo M, da Rocha AG, Preto A, Castro P, Castro L, Pardal F, et al. Frequency of TERT promoter mutations in human cancers. Nature communications. 2013; 4:2185.
20. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL and Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266(5193):2011-2015.
21. Shay JW and Bacchetti S. A survey of telomerase activity in human cancer. European journal of cancer. 1997; 33(5):787-791.
22. Arita H, Narita Y, Fukushima S, Tateishi K, Matsushita Y, Yoshida A, Miyakita Y, Ohno M, Collins VP, Kawahara N, Shibui S and Ichimura K. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta neuropathologica. 2013; 126(2):267-276.
23. Nonoguchi N, Ohta T, Oh JE, Kim YH, Kleihues P and Ohgaki H. TERT promoter mutations in primary and secondary glioblastomas. Acta neuropathologica. 2013.
24. Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ, Mickey BE, Wright WE, Shay JW and Bachoo RM. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(1):154-163.
25. Ruden M and Puri N. Novel anticancer therapeutics targeting telomerase. Cancer treatment reviews. 2013; 39(5):444-456.
26. Xia W, Wang P, Lin C, Li Z, Gao X, Wang G and Zhao X. Bioreducible polyethylenimine-delivered siRNA targeting human telomerase reverse transcriptase inhibits HepG2 cell growth in vitro and in vivo. Journal of controlled release : official journal of the Controlled Release Society. 2012; 157(3):427-436.
27. Zhang PH, Zou L and Tu ZG. RNAi-hTERT inhibition hepatocellular carcinoma cell proliferation via decreasing telomerase activity. J Surg Res. 2006; 131(1):143-149.
28. Ohgaki H and Kleihues P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer science. 2009; 100(12):2235-2241.
29. Bigner SH, Matthews MR, Rasheed BK, Wiltshire RN, Friedman HS, Friedman AH, Stenzel TT, Dawes DM, McLendon RE and Bigner DD. Molecular genetic aspects of oligodendrogliomas including analysis by comparative genomic hybridization. The American journal of pathology. 1999; 155(2):375-386.
30. Cairncross G and Jenkins R. Gliomas with 1p/19q codeletion: a.k.a. oligodendroglioma. Cancer journal. 2008; 14(6):352-357.
31. Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, Shaw EG and Buckner JC. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer research. 2006; 66(20):9852-9861.
32. Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW and Jenkins RB. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000; 18(3):636-645.
33. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006; 314(5797):268-274.
34. Reifenberger J, Reifenberger G, Liu L, James CD, Wechsler W and Collins VP. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. The American journal of pathology. 1994; 145(5):1175-1190.