
INTRODUCTION
Sepsis is defined as a systemic inflammatory response to life-threatening infection with the presence of organ failure [1]. Despite progress in antibiotic treatment and prompt institution of life support, sepsis remains the major cause of death in intensive care units [2]. The mechanism involved in the host immune response to sepsis is debated. Traditionally, the immune response was considered to be typified by a hyperinflammatory status with excessive production of pro-inflammatory mediators by macrophages and neutrophils [3, 4]. Until recently, most research was focused on ameliorating the hyperinflammatory response of the disorder, however, clinical trials using anti-inflammatory treatment failed to reduce overall sepsis mortality [5]. In a postmortem study of sepsis, more than 70% of patients died after an early/acute hyperinflammatory phase and most patients enter a state of late/chronic hypoinflammation and immunosuppression [6, 7]. So far there is no effective therapy for late sepsis.
Growing evidence suggests that cardiovascular dysfunction as a major complication of sepsis is correlated with worse performance and poor prognosis, and cardiac dysfunction increases the risk of mortality rates up to 70% in sepsis/septic shock [8, 9]. Therefore, it is a critically unmet need to clarify the mechanisms of cardiac dysfunction caused by sepsis and explore effective treatment to ameliorate the sepsis prognosis.
Over the past few years, microRNAs (miRs) have proved to be critical regulators of physiological homeostasis in the cardiovascular system [10, 11]. A subset of miRs has been identified as potential prognostic indicators of heart failure following acute myocardial infarction (MI). Using a high-throughput array method, serum expression of miR-155 and miR-380 in patients who experienced cardiac death were approximately 3-4 fold higher compared with those who returned for their 1-year follow-up [12]. It has been shown that miR-155 exerts an antiangiogenic but proarteriogenic role in the regulation of neovascularization, and miR-155 down- regulation would result in impaired arteriogenesis after MI and could exacerbate further injury to the myocardium [13]. However, the role of miR-155 in the progression of cardiac dysfunction in sepsis has not yet been elucidated.
β-arrestin 2 (Arrb2), a multifunctional regulator of G protein-coupled receptors with especially high expression in cardiovascular tissues, can modulate a diverse array of biological processes [14, 15]. Increasing evidence suggests that Arrb2 plays a crucial role in regulating systemic immune responses and the kinetics of Arrb2-mediated signaling is tissue-dependent [15, 16]. Our previous studies found that miR-155 could directly target the 3′-UTR of Arrb2 mRNA in a sequence-specific manner and there is a loop pathway between Arrb2 and miR-155 to promote the transition of cardiac stem cells into cardiomyocytes [17]. However, the effect of Arrb2 on cardiac function during late sepsis remains unknown.
In the present study, we demonstrated that increased expression of miR-155 protects against cardiac dysfunction in late sepsis. We also observed that miR-155 markedly reduces the infiltration of macrophages and neutrophils into the myocardium and attenuates the inflammatory response via suppression of JNK activity. Overexpression of Arrb2 exacerbates the mortality and immunosuppression in late sepsis mice. Transfection of miR-155 mimic reduces Arrb2 expression in the myocardium, and restores mice immunocompetence and improves survival. These findings suggest that modulation of the miR-155/Arrb2 pathway is capable of preventing late sepsis-induced cardiac dysfunction.
RESULTS
In vivo transfection of miR-155 mimic improves late sepsis survival
Previous study shows that miR-155 was increased in early sepsis and returned to baseline level in late sepsis [18]. To determine whether increased expression of miR-155 in late phase could impact the sepsis outcome, miR-155 mimic was injected through the tail vein 48 h after CLP to allow the initiation of sepsis and mortality was monitored for 28 days. As shown in Figure 1A, circulating miR-155 maintain baseline level on day 12 after CLP sepsis, and miR-155 mimic transfection significantly increased circulating miR-155 expression. The Kaplan-Meier survival curve (Figure 1B) indicated that 50% of the miR-Con treated septic mice died within 10 days, and 100% mortality occurred at 24 days after CLP. However, survival in miR-155 mimic treated septic mice was improved by 67% compared with miR-Con group. Our CLP model is operated to develop into early and late sepsis phases, which produces 20-30% mortality during early sepsis (days 1 to 5). We didn’t observe the effect of miR-155 mimic on mortality until day 6 after CLP. These results suggest that increased miR-155 expression during late phase prevents sepsis mortality. In the following experiments, we intended to identify the underlying mechanism for the protective effect of miR-155 in late sepsis.
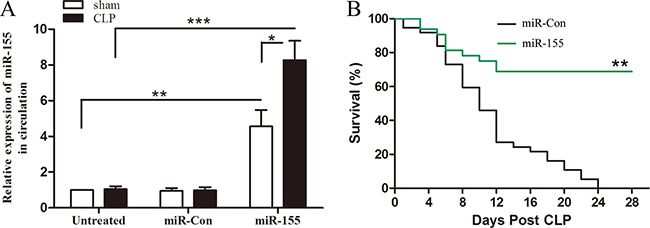
Figure 1: Increased expression of miR-155 in circulation improves late-septic mice survival outcome. Eight- to 10-weeks C57BL/6 male mice were subjected to CLP, and sham surgical operation served as sham control. miR-155 mimic or miR-Con (80 mg/kg) was injected through the tail vein 48 h after CLP. (A) After 12 days of CLP, sera were harvested and the expression of miR-155 was determined by real time PCR. There were four mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with indicated groups. (B) After 48 h of CLP, mice were transfected with miR-155 mimic or miR-Con and then monitored for survival for up to 28 days. There were 15-18/group. **p < 0.01 compared with miR-Con group.
Increased myocardium expression of miR-155 attenuates cardiac dysfunction in late sepsis
Recently, it has been shown that cardiac dysfunction plays a pivotal role in sepsis- induced mortality [8, 9]. To determine the role of miR-155 in cardiac function during late sepsis, we firstly measured its level in the myocardium of septic mice. As shown in Figure 2A, the miR-155 level was increased approximately 5-fold in early sepsis (compared to sham control, set at 1-fold), and returned to baseline level in late sepsis (day 12). After transfection with miR-155 mimic, the expression of miR-155 was increased 24.5-fold compared with sham control on day 3, and sustained increase around 20-fold on day 12. To evaluate the effect of miR-155 on cardiac function in late sepsis, we collected hemodynamic parameters by pressure-volume loop measurement on day 12 after sepsis. CLP induced significant cardiac dysfunction in late sepsis mice as evidenced by decreased heart rate (32.3%) (Figure 2B), EF% (40.6%) (Figure 2C), cardiac output (61.7%) (Figure 2D), and Pes (39.9%) (Figure 2E) compared with baseline values. In contrast, miR-155 mimic treatment significantly attenuated CLP-induced cardiac dysfunction, which increased heart rate by 29.3%, EF% by 33.5%, cardiac output by 72.8%, and Pes by 47.8% when compared with the untreated CLP group. Taken together, increased expression of miR-155 in the myocardium attenuates sepsis-reduced cardiac output and enhances left ventricular systolic function, which may improve late sepsis mice survival.
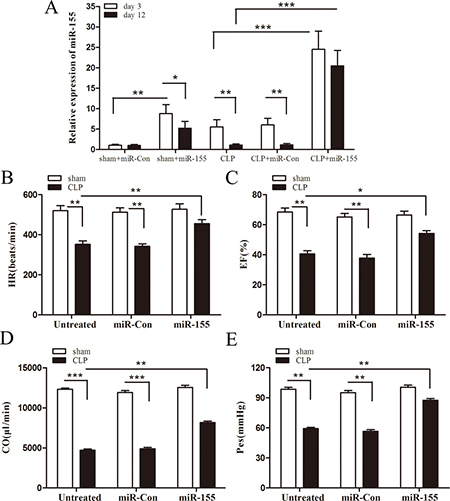
Figure 2: Increased expression of miR-155 in the myocardium attenuates cardiac dysfunction in late sepsis. Eight- to 10-weeks old C57BL/6 male mice were subjected to CLP or sham surgical operation. miR-155 mimic or miR-Con (80 mg/kg) was injected through the tail vein 48 h after CLP. (A) After 3 days or 12 days of CLP, hearts were harvested and the level of miR-155 was determined by real time PCR. There were four mice per group. After 12 days of CLP, cardiac function measurement was analysis by pressure-volume loop hemodynamic parameters. (B) HR, heart rate. (C) EF, ejection fraction. (D) CO, cardiac output. (E) Pes, left ventricular end-systolic pressure. There were five mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with indicated groups.
Transfection of miR-155 mimic attenuates infiltration of macrophages and neutrophils into the myocardium in late sepsis
It has been reported that the infiltration of macrophages and neutrophils was markedly increased early into the myocardium following CLP, which contributes to cardiac dysfunction during the development of septic shock [19]. It remains unclear whether this phenomenon occurs in late sepsis. Figure 3A–3C shows that macrophages and neutrophils accumulation were markedly increased in the myocardium during the late-septic phase. miR-155 mimic transfection significantly reduced sepsis-induced infiltration of macrophages by 51.2% and neutrophils by 56% into the myocardium when compared with untreated CLP mice. It is well known that adhesion molecules, such as ICAM-1 and VCAM-1, play a critical role in inducing inflammatory cells infiltration into the tissues [19, 20]. Next, we examined the effect of miR-155 on the expression of adhesion molecules in the myocardium. As shown in Figure 3D and 3E, there is more positive staining of ICAM and VCAM-1 in the late-septic mice myocardium compared with sham control, and miR-155 mimic transfection attenuated sepsis-mediated expression of myocardial ICAM-1 and VCAM-1. CLP-induced accumulation of macrophages and neutrophils, or the expression of ICAM-1 or VCAM-1 in the myocardium was not affected when treated with miR-Con.
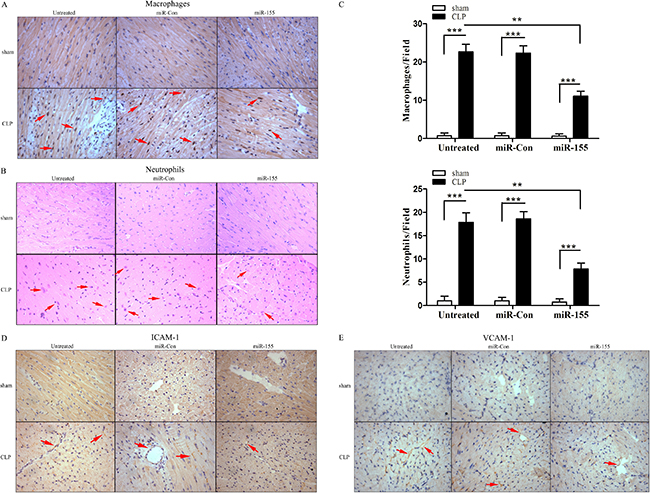
Figure 3: Transfection of miR-155 mimic prevents infiltration of macrophages and neutrophils into the myocardium and reduces adhesion molecule expression in late sepsis. miR-155 mimic or miR-Con (80 mg/kg) was transfected through the tail vein 48 h after CLP. Hearts were harvested 12 days after CLP and sectioned for immunohistochemical staining to identify macrophages (A) and neutrophils (B). The bar graphs indicate the numbers of infiltrating macrophages and neutrophils in five heart fields (C). Transfection of miR-155 mimic attenuates late sepsis-induced ICAM-1 (D) and VCAM-1 (E) expression in the myocardium. There were five mice per group. **p < 0.01, ***p < 0.001 compared with indicated groups.
Transfection of miR-155 mimic suppresses myocardium pro-inflammatory responses in late sepsis
Increased infiltration of inflammatory cells into the myocardium and persistence of an inflammatory microenvironment contribute to cardiac dysfunction [21]. Then we examined the effect of increased expression of miR-155 on inflammatory cytokine production in late-septic mice myocardium. As shown in Figure 4A, CLP promoted significantly high expression of pro-inflammatory cytokine TNF-α (3-fold) and IFN-γ (2.5-fold) compared with sham control (set at 1-fold) on day 12 after sepsis. miR-155 mimic transfection reduced TNF-α, IFN-γ production by 48.9%, 45.6% respectively, and increased anti-inflammatory cytokine IL-10 (1.8-fold) in the myocardium compared with untreated CLP mice.
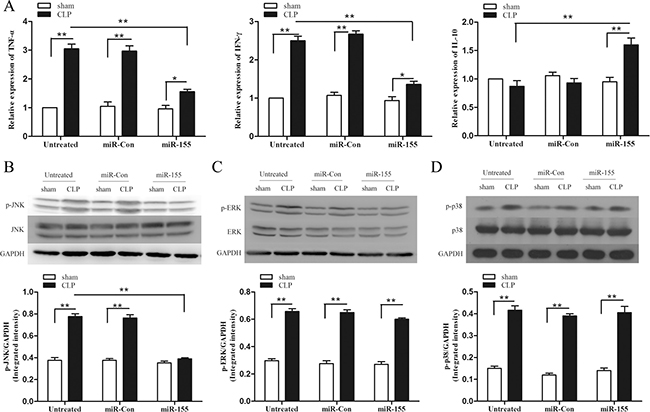
Figure 4: Transfection of miR-155 mimic attenuates inflammatory responses in late sepsis mice and suppresses p-JNK expression. miR-155 mimic or miR-Con (80 mg/kg) was transfected through the tail vein 48 h after CLP. (A) Hearts were harvested for assay of cytokine TNF-α, IFN-γ and IL-10 by qPCR 12 days after CLP. (B–D) Levels of p38, phospho-p38, ERK, phospho-ERK, JNK and phospho-JNK in the myocardium were examined by immunoblotting with specific antibodies 12 days after CLP. GAPDH expression was used as an internal control. There were five mice per group. *p < 0.05, **p < 0.01 compared with indicated groups.
MAPK signaling pathway is an important component which regulates the expression of inflammatory cytokines and adhesion molecules [22]. To investigate the mechanisms responsible for the immunomodulation, we evaluated the role of miR-155 in activation of MAPKs. Figure 4B–4D shows that MAPKs were activated in the late-septic myocardium on day 12 after CLP. Interestingly, CLP-enhanced JNK phosphorylation in miR-155 mimic transfected heart was significantly decreased by 49.8% compared with untreated CLP mice. Whereas the dynamic of ERK1/2 and p38 activation was not altered after miR-155 mimic transfection. Our findings reveal in late sepsis, JNK is a downstream effector of miR-155. Transfection of miR-Con did not alter sepsis-induced MAPKs activity in the myocardium.
Increased miR-155 inhibits cardiac Arrb2 expression in late sepsis
Using luciferase report assay, our previously research finds miR-155 could target Arrb2 and inhibit its expression in vitro [17]. In this study, we further explored the effect of miR-155 on Arrb2 expression in the late-septic myocardium. As shown in Figure 5A, CLP increased Arrb2 expression (2-fold) compared to sham control on day 10 after sepsis. Transfection of miR-155 mimic reduced the level of Arrb2 by 60.5% compared with untreated CLP mice. Using Arrb2 TG mice (Figure 5B), we detected the expression of Arrb2 in sham Arrb2 TG mice was 1.8-fold higher than in sham WT mice. miR-155 mimic transfection in Arrb2 TG mice decreased Arrb2 expression by 33.9% compared with sham Arrb2 TG mice. After CLP in Arrb2 TG mice, transfection of miR-155 mimic suppressed myocardial Arrb2 expression by 55.6% compared with untreated Arrb2 TG mice in late sepsis. Transfection of miR-Con did not alter the expression of Arrb2 in the presence and absence of sepsis.
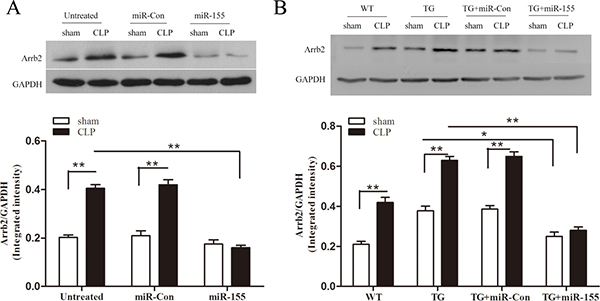
Figure 5: Transfection of miR-155 mimic inhibits myocardium Arrb2 expression in late sepsis. miR-155 mimic or miR-Con (80 mg/kg) was injected through the tail vein 48 h following CLP. The level of Arrb2 expression in the myocardium was examined by immunoblotting in WT (A) and Arrb2 TG (B) mice 10 days following CLP. There were five mice per group. *p < 0.05, **p < 0.01 compared with indicated groups. WT, wild-type; Arrb2 TG, Arrb2 overexpression.
miR-155 improves Arrb2 TG mice survival and cardiac dysfunction in late sepsis
To further elucidate the relationship between miR-155 and Arrb2, and their effect on late-septic mice survival, we tested miR-155 expression in Arrb2 TG mice myocardium, meanwhile observed Arrb2 TG mice survival in late sepsis model. As shown in Figure 6A, miR-155 expression in CLP Arrb2 TG mice was reduced by 56.9% compared with in CLP WT mice, meaning that Arrb2 inhibits the expression of miR-155 and there may be a loop pathway between Arrb2 and miR-155. Transfection of miR-155 mimic increased miR-155 expression in the presence and absence of sepsis, when compared with untreated Arrb2 TG mice. Figure 6B indicated Arrb2 TG mice were susceptive to late sepsis. Death occurred with the highest frequency on day 6–10 after sepsis, and 100% mortality occurred at 20 days after CLP. Survival in miR-155 mimic transfection septic Arrb2 TG mice was improved by 40% compared with miR-Con group. We also evaluate the effect of miR-155 on cardiac function in Arrb2 TG mice at day 10 after sepsis. CLP induced significant cardiac dysfunction in Arrb2 TG mice by decreased heart rate (25.1%) (Figure 6C), EF% (48.3%) (Figure 6D), cardiac output (70.9%) (Figure 6E), and Pes (46%) (Figure 6F) compared with baseline values. miR-155 mimic transfection significantly attenuated sepsis-induced cardiac dysfunction, which increased heart rate by 25.9%, EF% by 39.1%, cardiac output by 52.4%, and Pes by 49.6% when compared with the untreated CLP TG group. Our results indicate that miR-155 may attenuate cardiac dysfunction through downregulation of the Arrb2 expression level.
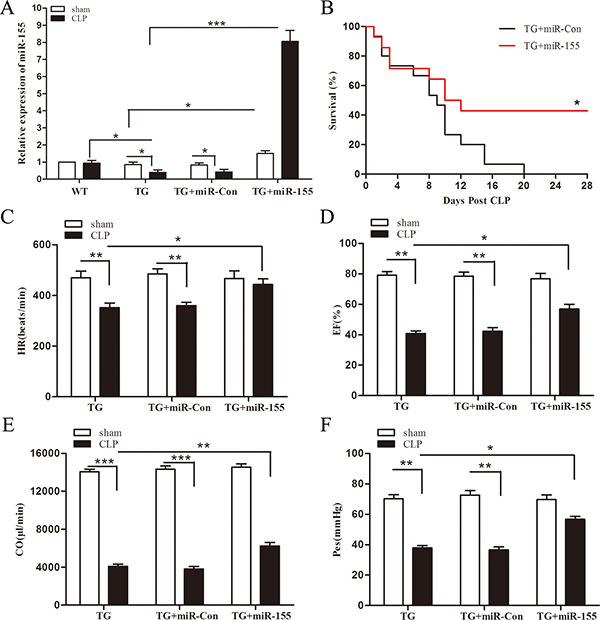
Figure 6: Increased expression of miR-155 attenuates Arrb2 overexpression (TG) mice cardiac dysfunction and improves survival in late sepsis. Eight- to 10-weeks Arrb2 TG male mice were subjected to CLP or sham surgical operation. miR-155 mimic or miR-Con (80 mg/kg) was injected through the tail vein 48 h after CLP. (A) Hearts were harvested for assay of miR-155 by qPCR 10 days after CLP. There were four mice per group. (B) Arrb2 overexpression (TG) mice were transfected with miR-155 mimic or miR-Con 48 h after CLP and then monitored for survival for up to 28 days. There were 15-18/group. *p < 0.05 compared with Arrb2 TG + miR-Con group. Ten days after CLP, cardiac function measurement of Arrb2 TG mice was analysis by pressure-volume loop hemodynamic parameters. (C) HR, heart rate. (D) EF, ejection fraction. (E) CO, cardiac output. (F) Pes, left ventricular end-systolic pressure. There are five mice per group. *p < 0.05, **p < 0.01, ***p < 0.001 compared with indicated groups.
Increased miR-155 restores responsiveness of late-septic Arrb2 TG mice to LPS
Late-septic mice are shown in a state of immunosuppression, indicated by producing less pro-inflammatory but more anti-inflammatory cytokines in our previous study [23]. We hypothesized that mice which survived late sepsis due to miR-155 mimic transfection should be immunoreactive, and then they should resist to a challenge with secondary bacterial LPS. TG mice that survived (the miR-155 mimic group) or moribund (the miR-Con group) were pooled at days 6 to 16 after CLP and infected with LPS (10 μg, i.p). Sham-operated Arrb2 TG mice also received LPS and served as a positive control. The inflammatory response was determined by measuring the sera levels of the pro-inflammatory (TNF-α, INF-γ) and anti-inflammatory (IL-10, IL-4) cytokines 6 h after LPS challenge. As shown in Figure 7, the miR-155 mimic transfection in Arrb2 TG mice significantly increased TNF-α and INF-γ expression and reduced IL-10 and IL-4 production compared with miR-Con group. We also observed that the cytokine profiles in the miR-155 mimic transfection Arrb2 TG mouse were similar to those in sham-operated control group, suggesting that increased expression of miR-155 restored immunoreactivity to LPS, while Arrb2 TG mice transfection with miR-Con remained immunosuppressed.
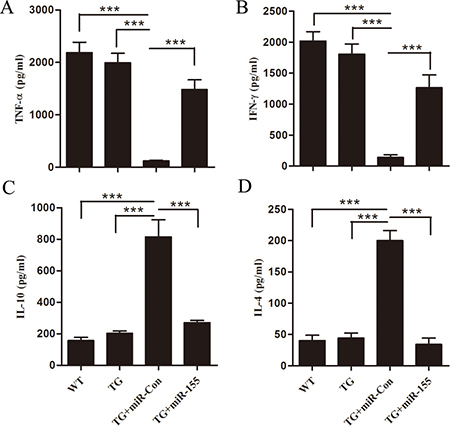
Figure 7: miR-155 mimic restores Arrb2 overexpression (TG) mice responsiveness to bacterial LPS in late sepsis. At days 6 to 16 after CLP, mice were moribund (TG+miR-Con group) and a corresponding number of surviving, healthy-appearing mice (TG+miR-155 group) were challenged with 10 μg of bacterial LPS (i.p.). Sham-operated TG mice and wild-type mice also received LPS and served as positive controls. After 6 h, sera were obtained and cytokine levels of TNF-α, INF-γ, IL-10 and IL-4 were examined by ELISA. There are five mice per group. ***p < 0.001 compared with indicated groups.
DISCUSSION
Sepsis is now the leading cause of death in the intensive care unit. Clinical observation finds that sepsis has biphasic death distribution, with an initial early peak at several days and a late peak at several weeks due to persistent organ injury or failure [24, 25]. However, the precise mechanisms of late sepsis mortality are still uncertain. Several studies suggest that immune dysfunction, immune suppression, and persistent catabolism and inflammation contribute to the deleterious state of chronic disease [26, 27]. Though some experimental agents, such as thymosin (NCT00711620) and granulocyte-macrophage colony stimulating factor (NCT00252915), are being tested in clinical trials to reconstitute immune responsiveness in patients with severe sepsis, an effective treatment is still unavailable until now. Using the late-septic mice model, our present study demonstrates that transfection of miR-155 mimic improves sepsis mice survival and attenuates inflammation-induced myocardium injury. We also found that Arrb2 exacerbated the mice mortality and immunosuppression in late sepsis. Increased expression of miR-155 inhibited cardiac Arrb2 expression, and then prevented Arrb2 TG mice cardiac dysfunction and restored late-septic Arrb2 TG mice immunoreactivity to LPS. Taken together, in vivo miR-155 mimic transfection attenuates septic sequelae and could pave the way toward new therapies.
The cardiovascular system plays an important role in the pathogenesis of sepsis. It has been shown that cardiac activation of PI3K/Akt-dependent signaling significantly attenuated myocardial dysfunction and improved survival in CLP-induced sepsis, which demonstrates a causal relationship between the protection of myocardial function and survival in early sepsis [28]. However, as recent researches suggest that benefits in early sepsis survival have been forfeited to escalating long-term sepsis mortality [25, 29], the changes of myocardial function during late-septic phase are still unclear. Here, we found CLP also induced significant cardiac dysfunction in late sepsis mice by decreased heart rate, cardiac output, EF%, and Pes. Therefore, clarifying the pathogenesis of late sepsis-induced myocardial dysfunction will provide benefits to seek specific therapy.
miRNAs are known to modulate normal physiological and pathophysiological signals within multiple organ systems [10, 11, 30] . We focused our attention on the expression of miR-155, which is closely related to a variety of cardiovascular diseases. miR-155 may be a potential prognostic indicator for heart failure [12]. miR-155 was found to repress eNOS expression, and regulate endothelium dependent vasorelaxation [31]. In our study, miR-155 level in the myocardium was increased during early sepsis and returned to baseline level in late sepsis. After transfection with miR-155 mimic, the expression of miR-155 was sustained increase during late-septic phase. Interestingly, miR-155 mimic treatment significantly attenuated CLP-induced cardiac dysfunction, and most importantly, increased miR-155 improved late-septic mice survival. A wide variety of stimuli, such as TLR ligands, can increase miR-155 expression, and then promotes both antimicrobial and antiviral responses of macrophages [32, 33]. miR-155 is required for the effective control of streptococcus pneumonia from the nasopharynx, and antipneumococcal adaptive immunity is broadly impaired in miR-155 knockout mice [34]. Mice with a deficiency of miR-155 are highly susceptible to herpes simplex encephalitis [35], suggesting that miR-155 can enhance the protective immune response to pathogens.
Endothelial cell activation is involved in septic injury in multiple organs [36]. Systolic cardiac dysfunction is directly associated with endothelial dysfunction in septic patients. Myocardium-depressing factors, such as TNF-α, can increase the expression of ICAM-1 and VCAM-1 in coronary endothelial cells and cardiomyocytes, and blockade of VCAM-1 or ICAM-1 abrogates LPS-induced cardiac dysfunction [37]. In vivo transfection of miR-155 mimic, we found miR-155 attenuated late sepsis-mediated expression of myocardial ICAM-1 and VCAM-1. Meanwhile, miR-155 significantly reduced sepsis-induced infiltration of macrophages and neutrophils into the myocardium. miR-155 is also one of the most widely studied miRNAs in regulating inflammation. We observed miR-155 mimic transfection decreased TNF-α, IFN-γ production and increased anti-inflammatory cytokine IL-10 in the myocardium, and then attenuates the inflammatory microenvironment which contributes to cardiac dysfunction.
MAPK pathways have been implicated in different aspects of cardiac regulation. The ERK1/2 pathway is found to be responsive mainly to stimulation of growth signaling, JNK and p38 are selectively responsive to pathophysiological stressors, such as infection and cytokines [15, 38]. Our study shows that levels of phospho-p38, phospho-ERK and phospho-JNK were all increased in late-septic myocardium. However, CLP-enhanced JNK activity in miR-155 mimic transfected heart was significantly decreased, whereas the dynamic of ERK1/2 and p38 activation was not altered. Our findings indicate JNK is a downstream effector of miR-155 in late sepsis. Constitutively activated JNK can result in different aspects of pathological remodeling and may contribute to systolic dysfunction and slowed conduction velocity in the heart [39], which is consistent with our detection results of cardiac function. Previous research shows that miR-155 downregulates JNK activity through reducing the expression of PAK1 in mouse CD4+ T cells [40]. However, the precise mechanisms of miR-155 in the regulation of JNK activity in late-septic myocardium need to be further clarified.
Arrb2, a multifunctional adapter protein, plays important roles in both regulations of immune activation and cardiac function [14–16]. Arrb2 interacts with the adaptor TRAF6 to negatively regulate Toll-like receptor signaling in innate immunity [41]. Arrb2 mediates the binding of Src homology-containing tyrosine phosphatases to KIR2DL1 to regulate the inhibitory signaling of NK cells, and Arrb2-deficient mice were less susceptible to cytomegalovirus infection compared with wild-type mice [42]. In our study, Arrb2 TG mice were observed to be more susceptive to late sepsis than WT mice, as death occurred with the highest frequency on day 6–10 after sepsis, which was earlier than in WT mice (day 10–12). Our previous research has shown that Arrb2 is a direct target by miR-155 [17], so we further test the hypothesis that in vivo transfection of miR-155 mimic may reverse the effect of Arrb2 overexpression. CLP increased Arrb2 expression during the late-septic phase, and miR-155 mimic transfection reduced the level of Arrb2 expression, and then improved TG mice survival rate. We also observed the changes of cardiac function in late-septic TG mice, and found miR-155 mimic transfection significantly attenuated sepsis-induced cardiac dysfunction. Taken together, miR-155 may protect cardiac dysfunction through downregulation of the expression of Arrb2.
Recent studies indicate most patients surviving the early initial hyperinflammatory phase enter a long-term immunosuppressive phase [7, 25, 26]. Deaths in this immunosuppressive phase are mainly due to the deficit in controlling the primary infection or the susceptibility of secondary infections [43]. In our present study, late-septic Arrb2 TG mice were challenged with secondary bacterial LPS. We found that sepsis Arrb2 TG mice were immunosuppressive, characterized by reduced pro-inflammatory (TNF-α, INF-γ) and increased anti-inflammatory (IL-10, IL-4) cytokines expression. Transfection of miR-155 mimic restored TG mice immunoreactivity to LPS. Interestingly, we also observed that increased expression of miR-155 in the myocardium attenuated pro-inflammatory cytokine levels in late sepsis. Wang and colleagues observed that pharmacological inhibition of miR-155 using antagomiR improved cardiac function and suppressed cardiac apoptosis induced by LPS in acute septic mice [44]. The distinct animal models with immunological differences (acute sepsis: hyperinflammatory and late sepsis: immunosuppressive) could explain these seemingly conflicting experimental results. These findings highlight the importance of fully elucidating any miRNA mechanisms in the right time and in the right cell before pursuing miRNA-based therapies, and biomarker-guided immunotherapy at the proper immune phase needs further exploration.
In summary, the data presented here demonstrated that miR-155 protects against late sepsis-induced cardiac dysfunction and improves survival by attenuation of inflammatory responses at least partially via suppressing JNK activation. Increased miR-155 also inhibits Arrb2 expression and restores Arrb2 TG mice immunocompetency. miR-155/Arrb2 may be an effective target in the management of sepsis and in the field of infectious disease, and our mice model also provides an important insight into particular mechanisms of sepsis that may have a comparable role in humans.
MATERIALS AND METHODS
Experimental animals
Wild-type (WT) C57BL/6 mice were obtained from Jackson Laboratory (Bar Harbor, ME). Arrb2 overexpression (TG) mice were reported previously [42]. Briefly, full-length human Arrb2 cDNA from brain cDNA/λphage library was cloned into pcDNA3 with HA tag under the control of a human cytomegalovirus promoter. Then the DNA constructions were injected into fertilized mice eggs on a C57BL/6 background. Real-time PCR was used to check the transgene mRNA expression in founder mice and their offspring. The primers used to identify transgenic mice were Arrb2, sense 5′-CAG CCA GGA CCA GAG GAC A-3′, antisense 5′-TGA TAA GCC GCA CAG AGT T-3′. There is no difference between physical activity, productivity and life span in WT and Arrb2 TG mice. 8–10-week old male mice were used in survival and cardiac function analysis. All mice were maintained in the Division of Laboratory Animal Resources at East Tennessee State University (ETSU), a facility accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International. The animal experimental protocols were approved by the ETSU Committee on Animal Care.
Cecal ligation and puncture (CLP) late sepsis model
Polymicrobial late sepsis was induced by CLP as described previously by us [23]. Briefly, mice were anesthetized via 5.0% isoflurane inhalation in 100% O2 in a closed chamber and then maintained with 2.5% isoflurane during surgery. A small anterior abdominal incision was made, and the cecum was ligated 1 cm proximal to the terminal of cecum with 2–0 silk, and then was punctured twice with a 23-gauge needle. A small amount of feces content was extruded into the abdominal cavity. The abdomen was then closed in layers with 3-0 silk. Sham-operated mice were processed identically, except without ligation and puncture. Immediately following surgery, 1 ml 0.9% saline was administrated by intraperitoneal (i.p.) injection for fluid resuscitation. To create the late sepsis phenotype, mice were subcutaneously administered antibiotic (imipenem, 25 mg/kg body weight) or an equivalent volume of 0.9% saline at 8 and 16 h after CLP.
Transfection and injection of miR-155 mimic
Both mirVana in vivo ready miR-155 mimic and negative control (miR-Con) (Ambion, Carlsbad, CA) were complexed with Invivofectamine 3.0 (Invitrogen) reagent according to the manufacturer’s protocol and our previous study [30], and were injected via the tail vein of male C57BL/6 mice at the dose of 80 mg/kg in 100 μl volumes. Injection was performed 48 h after CLP to allow initiation of sepsis.
Endotoxin challenge
Surviving septic Arrb2 TG mice that received miR-155 mimic or miR-Con between days 6 and 16 after CLP were challenged (i.p.) with 10 μg of Gram-negative bacterial LPS (E. coli serotype 0111: B4; Sigma-Aldrich, St. Louis, MO). Meanwhile, sham-operated Arrb2 TG mice and wild-type mice also received LPS and served as a positive control. All mice were sacrificed after 6 h. Blood was obtained via cardiac puncture and sera were collected for cytokines measurement.
Cardiac functional analysis
Cardiac function was detected by the SPR-839 instrument (Millar Instruments, Houston, TX, USA), and heart rate (HR), ejection fraction (EF), left ventricular end- systolic pressure (Pes), cardiac output (CO) were calculated as described previously [45]. Briefly, each mouse was intubated with a 22-gauge soft catheter and ventilated with a rodent ventilator (Columbus Instruments International Corp., Columbus, OH, USA), using a tidal volume of 0.3–0.5 ml and a respiratory rate of 110–120 breaths/min. After left thoracotomy, a microtip pressure-volume catheter was inserted through a 25-gauge apical stab into the left ventricular to measure the cardiac function. The signals were continuously recorded using an ARIA pressure-volume conductance system (Millar Instruments) connected to a Powerlab/4SPA/D converter (AD Instruments, Mountain View, CA, USA). All pressure-volume loop data were analyzed with PVAN3.4 (Millar Instruments). After the functional analysis, the hearts were removed and perfused for 2 min to remove the remaining blood. Portions of the midventricle were fixed for immunological studies.
Quantitative PCR assay of miRs
miRs were isolated from heart tissues using the mirVanaTM miR isolation kit (Ambion) according to the manufacturer’s protocol. miR-155 levels were quantified by quantitative PCR (qPCR) using specific primers TaqMan MicroRNA Assay (primer identification numbers: 002571 for mmu-miR-155 and 001973 for snRU6) and TaqMan Universal PCR Master Mix (Applied Biosystems) on a Bio-Rad PCR instrument. miR-155 level was calculated using the 2-ΔΔCT cycle threshold method after normalization to the snRU6 as an internal control [30].
Real-time quantitative RT-PCR
The real-time PCR detection technique was performed as described in our previous publications [30, 46]. Total RNA was isolated from mouse heart using an RNeasy Plus Mini Kit (QIAGEN Sciences, MD). One microgram of RNA from each sample was used for reverse transcription using a Reaction Ready™ first strand cDNA synthesis kit (SABiosciences, Frederick, MD). PCR was performed using real-time™ SYBR Green Fluorescein PCR Master Mix (SABiosciences). GAPDH expression was used as an internal control. The primer sequences used were as follows: TNF-α sense 5′-AGG CAC TCC CCC AAA AGA TG-3′, antisense 5′-TCA CCC CGA AGT TCA GTA GAC AGA-3′; IFN-γ sense 5′-AGG AAC TGG CAA AAG GAT GGT GAC-3′, antisense 5′-TGA CGC TTA TGT TGT TGC TGA TGG-3′; IL-10 sense 5′-TGC TAA CCG ACT CCT TAA TGC AGG AC-3′, antisense 5′-CCT TGA TTT CTG GGC CAT GCT TCT C-3′; GAPDH sense 5′-TGA CCA CAG TCC ATG CCA TC-3′, antisense 5′-GAT GGG GGT TAC ACA GGC AG-3′.
Detection tissue invading neutrophils and macrophages
Neutrophil accumulation in the myocardium was stained with naphtol AS-D Chloroacetate Esterase (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s protocol. Macrophages in heart tissues were examined with the specific Ab F4/80 (1:50 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Three slides from each block were evaluated and observed with bright-field microscopy. The results are expressed as the numbers of macrophages/field (×40 magnification).
Immunohistochemistry
Heart tissues were immersion-fixed in 4% buffered paraformaldehyde, embedded in paraffin. ICH was performed with a Diaminobenzidine (DAB) Histochemistry kit (Molecular Probes, Invitrogen, CA) according to the manufacturer’s protocol and our previous studies [14, 46]. Briefly, following antigen retrieval with 1mM EDTA (pH 8.0), deparaffinized sections were incubated in 1% Blocking Reagent solution for 1 h at 37°C and then stained with anti-ICAM-1 (1:50 dilution; Santa Cruz Biotechnology) and anti-VCAM-1 (1:50 dilution; Santa Cruz Biotechnology) respectively, overnight at 4°C. After washing, sections were incubated with biotinylated anti-mouse IgG (H+L) (Vector Laboratories, Burlingame, CA) and subsequently with HRP conjugate. Finally, the signal was shown with DAB substrate and counterstained with hematoxylin. Three different areas of each section were evaluated.
Western blot analysis
Western blot was performed as described previously [15, 47]. Samples containing equal amounts of protein extracted from heart tissue lysis were loaded into 10–15% SDS-PAGE, and then transferred to a nitrocellulose membrane. Blocking the membrane for 1 h at room temperature with 3% BSA. The membrane was incubated overnight at 4°C with the primary antibody. The signals were detected with the ECL system (Amersham Biosciences) and quantified by scanning densitometry using a Bio-Image Analysis System (Bio-Rad). Anti-phospho-p38 MAPK, anti-p38 MAPK, anti-JNK, anti-phospho-JNK, anti-ERK, anti-phospho-ERK, anti-Arrb2 and GAPDH antibodies were obtained from Cell Signaling Technology (Beverly, MA).
ELISA for cytokine assay
Blood was collected from the experimental and control mice to determine the serum level of cytokines as described by us [46]. Samples were allowed to clot for 2 h at room temperature before centrifugation for 20 min at 2000 g. Then serum was removed and stored at –80°C for subsequent ELISA assay. The quantity of cytokines (IFN-γ, TNF-α, IL-4 and IL-10) was examined using a Quantikine Mouse ELISA kit (R&D Systems, MN).
Statistical analysis
All quantitative data were presented as mean ± SD. Statistical analysis of data between groups was performed with two-tailed Student t test or one-way ANOVA for experiments with more than two subgroups. Survival curves were generated by the Kaplan-Meier method, and the log-rank test was used to compare group survival. p values < 0.05 were considered to be statistically significant.
Abbreviations
CLP, cecal ligation and puncture; Arrb2, β-arrestin 2; miR, microRNA; MI, myocardial infarction; WT, Wild-type; Arrb2 TG, Arrb2 overexpression; i.p., intraperitoneal; HR, heart rate; EF, ejection fraction; Pes, left ventricular end-systolic pressure; CO, cardiac output; qPCR, quantitative PCR; DAB, Diaminobenzidine.
CONFLICTS OF INTEREST
The authors have no financial conflicts of interest.
GRANT SUPPORT
This work was supported in part by NIH grants NIGM114716 and NIGM094740 to D. Yin. This research was also supported in part by NIH grant C06RR0306551. This work was also supported in part by NSFC81401051.
REFERENCES
1. Vincent JL, Opal SM, Marshall JC, Tracey KJ. Sepsis definitions: time for change. Lancet. 2013; 381:774–775.
2. Mayr FB, Yende S, Angus DC. Epidemiology of severe sepsis. Virulence. 2014; 5:4–11.
3. Williams DL, Ha T, Li C, Kalbfleisch JH, Schweitzer J, Vogt W, Browder IW. Modulation of tissue Toll-like receptor 2 and 4 during the early phases of polymicrobial sepsis correlates with mortality. Crit Care Med. 2003; 31:1808–1818.
4. Rialdi A, Campisi L, Zhao N, Lagda AC, Pietzsch C, Ho JS, Martinez-Gil L, Fenouil R, Chen X, Edwards M, Metreveli G, Jordan S, Peralta Z, et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science. 2016; 352:aad7993.
5. Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. 2012; 12:503–505.
6. Torgersen C, Moser P, Luckner G, Mayr V, Jochberger S, Hasibeder WR, Dünser MW. Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth Analg. 2009; 108:1841–1847.
7. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013; 13:260–268.
8. Merx MW, Weber C. Sepsis and the heart. Circulation. 2007; 116:793–802.
9. Antonucci E, Fiaccadori E, Donadello K, Taccone FS, Franchi F, Scolletta S. Myocardial depression in sepsis: from pathogenesis to clinical manifestations and treatment. J Crit Care. 2014; 29:500–511.
10. Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011; 469:336–342.
11. Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015; 116:751–762.
12. Matsumoto S, Sakata Y, Nakatani D, Suna S, Mizuno H, Shimizu M, Usami M, Sasaki T, Sato H, Kawahara Y, Hamasaki T, Nanto S, Hori M, et al. A subset of circulating microRNAs are predictive for cardiac death after discharge for acute myocardial infarction. Biochem Biophys Res Commun. 2012; 427:280–284.
13. Pankratz F, Bemtgen X, Zeiser R, Leonhardt F, Kreuzaler S, Hilgendorf I, Smolka C, Helbing T, Hoefer I, Esser JS, Kustermann M, Moser M, Bode C, et al. MicroRNA-155 Exerts Cell-Specific Antiangiogenic but Proarteriogenic Effects During Adaptive Neovascularization. Circulation. 2015; 131:1575–1589.
14. Yin D, Yang X, Li H, Fan H, Zhang X, Feng Y, Stuart C, Hu D, Caudle Y, Xie N, Liu Z, LeSage G. β-Arrestin 2 Promotes Hepatocyte Apoptosis by Inhibiting Akt Protein. J Biol Chem. 2016; 291:605–612.
15. Li H, Hu D, Fan H, Zhang Y, LeSage GD, Caudle Y, Stuart C, Liu Z, Yin D. β-Arrestin 2 negatively regulates Toll-like receptor 4 (TLR4)-triggered inflammatory signaling via targeting p38 MAPK and interleukin 10. J Biol Chem. 2014; 289:23075–23085.
16. Smith JS, Rajagopal S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J Biol Chem. 2016; 291:8969–8977.
17. Zhao J, Feng Y, Yan H, Chen Y, Wang J, Chua B, Stuart C, Yin D. β-arrestin2/miR-155/GSK3β regulates transition of 5›-azacytizine-induced Sca-1-positive cells to cardiomyocytes. J Cell Mol Med. 2014; 18:1562–1570.
18. McClure C, Brudecki L, Ferguson DA, Yao ZQ, Moorman JP, McCall CE, El Gazzar M. MicroRNA 21 (miR-21) and miR-181b couple with NFI-A to generate myeloid-derived suppressor cells and promote immunosuppression in late sepsis. Infect Immun. 2014; 82:3816–3825.
19. Gao M, Wang X, Zhang X, Ha T, Ma H, Liu L, Kalbfleisch JH, Gao X, Kao RL, Williams DL, Li C. Attenuation of Cardiac Dysfunction in Polymicrobial Sepsis by MicroRNA-146a Is Mediated via Targeting of IRAK1 and TRAF6 Expression. J Immunol. 2015; 195:672–682.
20. Alves-Filho JC, de Freitas A, Spiller F, Souto FO, Cunha FQ. The role of neutrophils in severe sepsis. Shock. 2008; 30:3–9.
21. Müller-Werdan U, Prondzinsky R, Werdan K. Effect of inflammatory mediators on cardiovascular function. Curr Opin Crit Care. 2016; 22:453–463.
22. Zhou R, O’Hara SP, Chen XM. MicroRNA regulation of innate immune responses in epithelial cells. Cell Mol Immunol. 2011; 8:371–379.
23. Brudecki L, Ferguson DA, Yin D, Lesage GD, McCall CE, El Gazzar M. Hematopoietic stem-progenitor cells restore immunoreactivity and improve survival in late sepsis. Infect Immun. 2012; 80:602–611.
24. Moore FA, Moore EE. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am. 1995; 75:257–277.
25. Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016; 126:23–31.
26. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, Bricker TL, Jarman SD 2nd, Kreisel D, Krupnick AS, Srivastava A, Swanson PE, Green JM, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011; 306:2594–2605.
27. Schorr CA, Dellinger RP. The Surviving Sepsis Campaign: past, present and future. Trends Mol Med. 2014; 20:192–194.
28. Gao M, Ha T, Zhang X, Wang X, Liu L, Kalbfleisch J, Singh K, Williams D, Li C. The Toll-like receptor 9 ligand, CpG oligodeoxynucleotide, attenuates cardiac dysfunction in polymicrobial sepsis, involving activation of both phosphoinositide 3 kinase/Akt and extracellular-signal-related kinase signaling. J Infect Dis. 2013; 207:1471–1479.
29. Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med. 2010; 38:1276–1283.
30. Qiu S, Feng Y, LeSage G, Zhang Y, Stuart C, He L, Li Y, Caudle Y, Peng Y, Yin D. Chronic morphine-induced microRNA-124 promotes microglial immunosuppression by modulating P65 and TRAF6. J Immunol. 2015; 194:1021–1030.
31. Sun HX, Zeng DY, Li RT, Pang RP, Yang H, Hu YL, Zhang Q, Jiang Y, Huang LY, Tang YB, Yan GJ, Zhou JG. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012; 60:1407–1414.
32. Koch M, Mollenkopf HJ, Klemm U, Meyer TF. Induction of microRNA-155 is TLR- and type IV secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc Natl Acad Sci USA. 2012; 109:E1153–1162.
33. Wang P, Hou J, Lin L, Wang C, Liu X, Li D, Ma F, Wang Z, Cao X. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010; 185:6226–6233.
34. Verschoor CP, Dorrington MG, Novakowski KE, Kaiser J, Radford K, Nair P, Anipindi V, Kaushic C, Surette MG, Bowdish DM. MicroRNA-155 is required for clearance of Streptococcus pneumoniae from the nasopharynx. Infect Immun. 2014; 82:4824–4833.
35. Bhela S, Mulik S, Reddy PB, Richardson RL, Gimenez F, Rajasagi NK, Veiga-Parga T, Osmand AP, Rouse BT. Critical role of microRNA-155 in herpes simplex encephalitis. J Immunol. 2014; 192:2734–2743.
36. Aird WC. The role of the endothelium in severe sepsis and multiple organ dysfunction syndrome. Blood. 2003; 101:3765–3777.
37. Henninger DD, Panés J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997; 158:1825–1832.
38. Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007; 116:1413–1423.
39. Petrich BG, Gong X, Lerner DL, Wang X, Brown JH, Saffitz JE, Wang Y. c-Jun N-terminal kinase activation mediates downregulation of connexin43 in cardiomyocytes. Circ Res. 2002; 91:640–647.
40. Rouquette-Jazdanian AK, Kortum RL, Li W, Merrill RK, Nguyen PH, Samelson LE, Sommers CL. miR-155 Controls Lymphoproliferation in LAT Mutant Mice by Restraining T-Cell Apoptosis via SHIP-1/mTOR and PAK1/FOXO3/BIM Pathways. PLoS One. 2015; 10:e0131823.
41. Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G. Association of beta-arrestin and TRAF6 negatively regulates Toll-like receptor-interleukin 1 receptor signaling. Nat Immunol. 2006; 7:139–147.
42. Yu MC, Su LL, Zou L, Liu Y, Wu N, Kong L, Zhuang ZH, Sun L, Liu HP, Hu JH, Li D, Strominger JL, Zang JW, et al. An essential function for beta-arrestin 2 in the inhibitory signaling of natural killer cells. Nat Immunol. 2008; 9:898–907.
43. Otto GP, Sossdorf M, Claus RA, Rödel J, Menge K, Reinhart K, Bauer M, Riedemann NC. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. 2011; 15:R183.
44. Wang H, Bei Y, Huang P, Zhou Q, Shi J, Sun Q, Zhong J, Li X, Kong X, Xiao J. Inhibition of miR-155 Protects Against LPS-induced Cardiac Dysfunction and Apoptosis in Mice. Mol Ther Nucleic Acids. 2016; 5:e374.
45. Chua CC, Gao J, Ho YS, Xu X, Kuo IC, Chua KY, Wang H, Hamdy RC, Reed JC, Chua BH. Over-expression of a modified bifunctional apoptosis regulator protects against cardiac injury and doxorubicin-induced cardiotoxicity in transgenic mice. Cardiovasc Res. 2009; 81:20–27.
46. Hu D, Wan L, Chen M, Caudle Y, LeSage G, Li Q, Yin D. Essential role of IL-10/STAT3 in chronic stress-induced immune suppression. Brain Behav Immun. 2014; 36:118–127.
47. Li H, Sun X, LeSage G, Zhang Y, Liang Z, Chen J, Hanley G, He L, Sun S, Yin D. β-arrestin 2 regulates Toll-like receptor 4-mediated apoptotic signalling through glycogen synthase kinase-3β. Immunology. 2010; 130:556–563.