
INTRODUCTION
Autophagy is a tightly regulated catabolic process in which cytoplasmic organelles and proteins are degraded in the lysosome [1]. This process enables cells to retain cellular environmental homeostasis, quality control and energy balance [2, 3]. Studies reveal that autophagy acts as housekeeping functions, can be induced by various stresses to adapt to the conditions of the environment change [4]. There are mainly three different autophagic pathways according to the substrates delivery mode to lysosome, including macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) [5]. CMA and macroautophagy are two well characterized pathways [6, 7]. Macroautophagy is a largely nonselective degradation system, which delivers cytosolic components into a double-membrane structure (autophagosome), and then fuses with lysosome. Whereas CMA is a selective process by which cytosolic substrate proteins bearing a KFERQ-like motif are transported into the lysosome for degradation. This selective degradation is mediated by binding to heat shock 70kDa protein 8 (HSC70) [7], and then substrates become unfolding before delivery into the lysosome by lysosome-associated membrane protein 2A (LAMP2A) [8]. Despite so many differences, the research has found the existence of connection between CMA and macroautophagy, with one compensating for another pathway if it is impaired [9, 10]. In general, microautophagy directly uptakes of cytosolic components into the lysosomal lumen by the invagination [11, 12]. However, the selective degradation system for substrates has also been found in an exceptive form of microautophagy, termed endosomal microautophagy (e-MI). Both CMA and e-MI require a pentapeptide motif related to KFERQ in substrate proteins for binding to HSC70. In contrast with CMA, selective e-MI does not need LAMP2A and proteins unfolding [13]. Consequently, CMA targets and degrades substrate proteins make it quite different from other two autophagic pathways.
In recent years, the researchers are keen on how CMA influences cancer pathophysiology. Kon et al. revealed an increase in the activity of CMA in a variety of cancer cells, with the up-regulated expression of LAMP2A. They also demonstrated that CMA is necessary for malignant cell growth and tumor metastasis [14]. Nevertheless, targeting the CMA in established cancer can inhibit the cancer cells is still being debated. Selective activation of CMA can eliminate cancer cells by inducing the aberrant mutant proteins degradation in the specific cancer cells [15]. Therefore, numerous researchers are particularly interested in the relationship between CMA substrate proteins and cancer biology. Through analysis of the CMA substrate proteins in cancer cells, CMA involves in glucose metabolism [14] and reducing the cellular stress [16]. In this review, we focus on substrate proteins which are degraded by CMA in cancer. We analyze the role of CMA substrate proteins in cancer and CMA substrate proteins are supposed to be developed into a curative approach for anticancer therapy.
MOLECULAR MECHANISM OF CMA
CMA is a kind of selective autophagy, which degrades the cytosolic proteins [17]. The process of CMA can be summarized as follows (Figure 1).
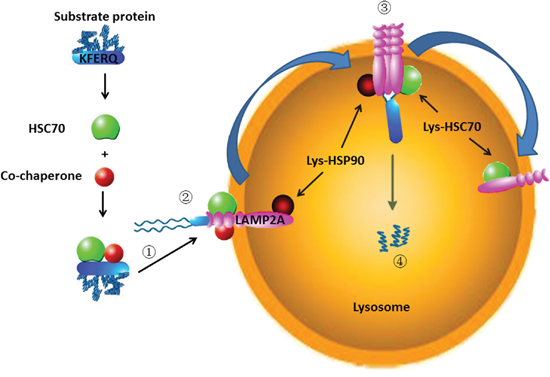
Figure 1: The process of CMA: (1) Recognizing substrate proteins and targeting them to lysosome; (2) Binding and unfolding substrate proteins; (3) Translocation into lysosomes; (4) Degradation by lysosome hydrolytic enzymes.
RECOGNIZING SUBSTRATE PROTEINS AND TARGETING THEM TO LYSOSOME
CMA substrate proteins must contain the amino acid sequence of the polypeptide motif KFERQ [18]. The amino acids consist of a glutamine (Q) residue preceded or followed by the four residues, a basic amino acid of the two positively charged residues, lysine (K) or arginine (R), an acidic amino acid of the two negatively charged residues, glutamic acid (E) or aspartic acid (D), and one or two of these hydrophobic residues, K, R, phenylalanine (F), valine (V), leucine (L) or isoleucine (I) [19]. Nevertheless, in some cases, substrates bearing more than one KFERQ-like motif, it is demonstrated experimentally that additional motif does not change the degradation of the substrates, and one motif is sufficient [20]. The KFERQ-like motif can be buried in both ends or the central domain of the substrate protein. The motif should be exposed or accessible for chaperone recognition. The following conditions can promote the exposure of the KFERQ-like motif: a partial unfolding of the protein; if the motif region is binding to the intracellular membranes, it should be released from the membranes for recognition; or disassemble from the protein-protein interaction [21]. Posttranslational modifications in proteins missing change the incomplete motif into a perfect KFERQ motif. Such as, by phosphorylating the tyrosine (Y), cysteine (C) or serine (S) residue can contribute a missing negative charge in incomplete motif, or acetylating the K residue can provide a missing Q [22]. Posttranslational modifications of CMA-targeting motifs provide a method for the regulation of CMA degradation. HSC70 can recognize the substrate proteins in the cytosol through interaction with the KFERQ-like motif [23]. During this process, many co-chaperones are involved in, such as Hsp40, Hsp90 and Hip [24, 25]. The chaperones/substrate protein is targeted to the surface of the lysosomal membrane [26].
BINDING AND UNFOLDING SUBSTRATE PROTEINS
Once recognized by the chaperone, the HSC70/substrate protein complex is delivered to the lysosomal membrane surface and interacted with the cytosolic tail of LAMP2A [26, 27]. LAMP2A, a single-span lysosomal membrane receptor protein, is a splice variant encoded by the lamp2 gene [26]. Through its 12-amino acid tail exposes in the cytoplasm, LAMP2A interacts with HSC70/substrate complex [26, 28]. Before translocation, substrate protein needs to be unfolded; this is mediated by HSC70 and its co-chaperones such as Hsp40, Hsp90 and Hip [29].
TRANSLOCATION INTO LYSOSOME
LAMP2A is located at the lysosome in monomeric form, since substrate proteins only interact with its monomers. This interaction induces the LAMP2A monomers aggregate into a 700 kDa protein complex. During the changing from monomeric to multimeric forms, lys-HSP90 (HSP90 in the lysosome) maintains the LAMP2A stability [24]. The multimeric forms of LAMP2A complexes assist CMA substrate proteins through the lysosome membrane. The substrate proteins translocation through the lysosome also needs the lys-hsc70 (HSC70 in the lysosome) normally resident in lysosomes [30].
DEGRADATION
After the substrate protein is pulled into the lysosome, it is degraded into amino acid by lysosome hydrolytic enzymes [31]. Lys-hsc70 (HSC70 in the lysosome) induces disassembly of LAMP2A from the multimeric form into the monomeric form, thus the next substrate protein can bind to LAMP2A in a new cycle [24].
PATHWAY OF CMA
CMA, acts as housekeeping functions, has crucial functions in cellular physiology and pathology. It is involved in cells at a low level under normal conditions. Regulation of CMA means a great deal to the steady state of a cell. However, there is little information on the signaling pathway of CMA. It can be induced by various stressors such as hypoxia [32], oxidative stress [33], DNA damage [34] and prolonged starvation [35]. The calcineurin/nuclear factor of activated T cells (NFAT) signaling pathway was the first CMA-activating signaling pathway identified [36]. Anguiano et al. showed that the activation of CMA depends on a functional retinoic acid receptor alpha (RARα) [37]. Studies have demonstrated that the targeting of rapamycin (mTOR) –protein kinase B (Akt) –pleckstrin homology domain and leucine-rich repeat protein phosphatase (PHLPP) to the surface of lysosomes can directly regulate CMA [38]. The critical mechanisms of CMA respond to mTORC2/PHLPP1/Akt signaling pathway still need further investigation. Many evidences show that there is a tight connection between the CMA and macroautophagy during the degradation of autophagic process. Through upregulating macroautophagy can block the activity of CMA [39]. Likewise, CMA can be induced by blocking macroautophagy [33]. Similarly, CMA pathway is also intimately connected with the ubiquitin–proteasome system [40]. Cross-talk between these pathways has been observed, with one compensating for others if one of them fails. The compensation among proteolytic pathways contributes to maintenance of protein homeostasis.
CMA SUBSTRATE PROTEIN IN CANCER
CMA is an alternative pathway of autophagy mediated substrate protein by HSC70 and LAMP2A; HSC70 recognizes and targets substrate protein bearing a KFERQ-like motif to lysosomal membrane. LAMP2A helps substrates to translocate into lysosome for degradation [18, 29, 30]. These features help us to identify the substrate proteins in the cancer cell. CMA substrate protein plays dual roles in the carcinogenesis and the progress of malignant tumor. It reveals the depth mechanism between the CAM and cancer. Following is the summary of CMA substrate proteins in cancer (Table 1 and Figure 2).
Table 1: CMA substrate proteins in cancer
Substrate protein |
Role in cancer |
Cancer type |
Ref. |
|---|---|---|---|
AF1Q |
promoter |
ML |
[55] |
Unphosphorylated PED |
suppressor |
NSCLC |
[78] |
Misfolded N-CoR |
promoter |
NSCLC |
[16] |
Vav1 |
promoter |
Pancreatic cancer |
[101] |
PKM2 |
promoter |
NSCLC |
[22] |
Eps8 |
promoter |
Pancreatic cancer |
[134] |
Rnd3 |
suppressor |
Gastric cancer |
[148] |
mutant p53 |
promoter |
Ovarian cancer |
[158] |
HK2 |
promoter |
Ovarian cancer |
[15] |
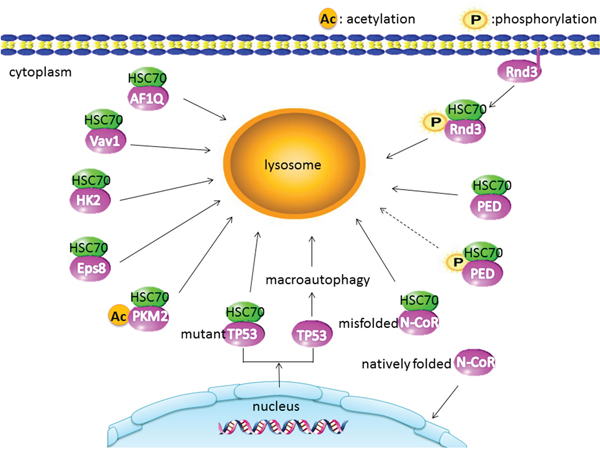
Figure 2: CMA substrate proteins in cancer: the acetylated PKM2 displays a stronger interaction with HSC70. Phosphorylation translocates Rnd3 from membrane to cytosol and promotes Rnd3 interaction with CMA. Unphosphorylated PED binds HSC70 and degradation by CMA. However, phosphorylated PED binds to HSC70 at a low level (dotted line). Misfolded N-CoR is associated with HSC70 and degraded through the CMA. The degradation of mutant TP53 is mediated by the CMA and the degradation of TP53 through macroautophagy.
AF1Q
AF1Q was first identified in acute myelomonocytic leukemia (AMMOL). It is described as a partner of mixed lineage leukemia gene fusion in AMMOL patients [41]. The upregulation expression of AF1Q has been observed in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [42–44]. High expression of AF1Q has been found in hematologic and solid malignancy patients with poor clinical outcomes [43–50]. Overexpression of AF1Q is significantly associated with a higher incidence of distant metastasis [49, 50]. AF1Q not only plays as an oncogenic factor but also has a vital function in apoptosis and chemotherapy drug resistance. Knockdown AF1Q protein in conjunction with the decreased apoptotic cell death is induced by doxorubicin or γ radiation [42, 51]. By upregulation of NF-κB p65, AF1Q can enhance the radiation-induced apoptosis, which may explain the oncogenic mechanism of AF1Q [52–54]. Bioinformatics analysis showed that AF1Q has six amino acids sequence correlated with KFERQ-like motifs. The researches support that AF1Q clearance is involved in CMA pathway, and CMA abnormal may lead to AF1Q related malignant tumors [55]. The molecular mechanisms by which AF1Q influences tumor suppressor gene loss and interacts with oncogene are not fully understood, however, the degradation of AF1Q via CMA pathway offers a promising new treatment option for cancer.
UNPHOSPHORYLATED PED
Phosphoprotein enriched in diabetes (PED) was first described by Helena over 20 years ago [56]. PED is a 15 kDa molecule consists of a NH2-terminal death effector domain (DED) and a COOH-terminus tail with the extracellular-regulated kinase (ERK) binding site and phosphorylation sites (Ser-104 and Ser-116) [57, 58]. PED is a highly conserved gene, which is located on human chromosome 1q21-22, and involves in regulating cellular functions, including survival and metabolism. There are three forms of PED in the cells: unphosphorylated, monophosphorylated and bisphosphorylated PED [59]. Combine unphosphorylated PED with ERK1/2 plays an important role in preventing PED translocation into the cell nucleus, which leads to inhibit the cell proliferation [60, 61]. As an endogenous substrate, PED can be phosphorylated by protein kinase C (PKC) at Ser-104 [62] and protein kinase B (PKB) or Ca2+/calmodulin-dependent protein kinase II (CaM kinase II) at Ser-116 [63, 64]. Phosphorylation of PED will result in the inhibition of apoptosis by preventing ERK1/2-binding and enhancing the binding to MORT1 and caspase 8 [62, 63]. Since the role of PED in apoptosis and ERK signal pathway, changes in PED expression may influence oncogenesis, cancer progression and chemotherapeutic sensitivity. PED has now been described as both the tumor suppressor and promoter. Unphosphorylated PED can inhibit proliferation and invasion of cells and correlates with good prognosis [65–68]. PED is known to be upregulated in several cancers and involved in resistance to TRAIL-mediated apoptosis [69–77]. Quintavalle et al. identified PED as a substrate protein of CMA pathway. In cancer cells, CMA targeted degrades phosphorylated PED (tumor promoter) at a reduced extent, however, unphosphorylated PED (tumor suppressor) at normal extent. This phenomenon fills the cancer cell with fuel. In cancer cells, CMA degrades unphosphorylated PED contributes to the resistance of chemotherapy and radiotherapy [78].
MISFOLDED N-COR
Nuclear receptor co-repressor (N-CoR) was first cloned as the protein associated with unliganded T3R-RXR heterodimers in 1995 [79]. By mediating active repression through nuclear receptors, N-CoR involves in cellular biological processes, including tumor initiation, differentiation and progression [80, 81]. Because of inaccuracy dissociation from nuclear receptors, N-CoR gains the inappropriate function, which causes a variety of diseases, including the human cancer [82]. Numerous studies have mainly focused on the function of N-CoR in transcription regulation [79]. N-CoR has emerged as a regulator of tumor suppressors via transcriptional control [83, 84]. Many evidences support that N-CoR is an essential component of many tumor suppressor proteins [79, 85, 86]. Knockdown of N-CoR suppressed the motility and proliferation of tumor cells [87]. However, the misfolded form of N-CoR loses the tumor suppression role and contributes to the development of non-small cell lung cancer (NSCLC). Bin et al. find misfolded N-CoR is associated with HSC70 and degraded through CMA in NSCLC. Degradation misfolded N-CoR by CMA can suppress the survival and growth in NSCLC cells [16].
VAV1
VAV family is one of the best-known proteins of Rho/Rac activators [88]. Vav guanine nucleotide exchange factor 1 (Vav1) is a 95 kDa protein of the Vav family (Vav1, Vav2 and Vav3). It is predominantly expressed in haematopoietic cells and consists of several functional domains including CH, DH, PH, SH2, and SH3 domains [89, 90]. Vav1 works as a signal transducer in maturation and immune response [91] and an adapter molecule, promoting interaction between the protein [89, 92]. Vav1 is a key driver of the dynamic regulation of actin cytoskeleton and numerous physical cellular processes of mature hematopoietic cells [93–95]. It is located on chromosome 19p12-p13.2, the domain of karyotypic abnormalities in human solid tumors or hematopoietic malignancies, therefore Vav1 has an essential function in human cancer [94]. Vav1 is specifically expressed in human cancer and plays a major role in carcinogenesis and progression [96–99], it has been defined as an oncogene [100]. Vav1 are regulated by its degradation through an HSC70-chaperone-mediated targeting to the lysosome [101]. Vav1 overexpression increases tumor cell survival, proliferation, and metastasis, thus drugs that targeted degradation Vav1 may be potent inhibitors of tumor cell migration.
PKM2
Cell proliferation is a process that consumes large amounts of energy, especially in cancer cells. In oncology, cells provide energy at a high rate of glycolysis accompanied with an increasing extrusion of lactic acid in the presence of oxygen, and this is called the Warburg effect [102, 103]. The high aerobic glycolysis has clear metabolic benefits for carcinogenesis and tumor growth. Pyruvate kinase (PK) regulates the last rate-limiting step of the glycolytic pathway and catalyzes the transfer of phosphoenolpyruvate and ADP into pyruvate and ATP [104, 105]. In mammals, the PK family has four known isoforms: PKM1, PKM2, PKL, and PKR [106, 107]. The isoenzyme of PK that allows the upregulation of phosphormetabolite pools in multicellular organisms is PKM2. It is an ancient variant of the pyruvate kinase enzyme found in unicellular organisms such as yeast and E. coli [108]. PKM2 possesses the less active dimer and the active tetramer forms [109, 110]. Dimeric PKM2 mainly facilitates the glycolytic intermediates towards biosynthesis and tumor growth, whereas tetrameric PKM2 promotes the activity of glycolysis for ATP production [111]. PKM2 makes an enormous contribution to cancer metabolism. It expresses and actives in cancer cells, which is correlated with the prognosis of tumor [112–114]. Numerous evidences support PKM2 as a tumor marker [115–117]. Through reducing the oxidative metabolism of cells, PKM2 sustains cell growth in hypoxic environments and provides cells with a growth advantage in metabolites [118]. PKM2 is important for cancer cell growth, therefore the inhibitor of PKM2 is very meaningful to the tumor treatment. Shikonin and its analog alkannin can selectively inhibit PKM2 [119]. Peptide aptamers [120] and RNA interference targeting PKM2 [121] also induce a significant decrease in cancer cell proliferation through the inhibition of PKM2. However, PKM2 inhibitors have become disputed since posttranslational modifications of them could promote tumor growth [22, 122]. Lei et al. found the acetylation of PKM2 can enhance the interaction with chaperone. PKM2 acetylation by high glucose reduces the activity of PKM2 and stimulates the degradation of PKM2 via CMA [22]. We can promote the degradation of PKM2 by CMA to reduce the energy of cancer cells by a minimal rate of glycolysis.
EPS8
The epidermal growth factor receptor pathway substrate 8 (Eps8) was originally characterised as a kinase activity substrate of the epidermal growth factor receptor (EGFR) [123]. Eps8 maps to the human chromosome 12p13.2 and play an important role conserved in evolution [124]. Fazioli et al. demonstrated that Eps8 exists in two isoforms: p97Eps8 and p68Eps8 [123], and most studies referring mainly to the p97Eps8 isoform. Eps8 is universally expressed [124], and overexpression of Eps8 leads to increased mitogenic signaling and malignant transformation [125]. Growing evidence reveals that the expression of Eps8 is elevated in most human solid tumor types and hematologic malignancies, including oral, thyroid, pituitary, esophageal, lung, breast, colorectal, pancreatic, ovarian, cervical cancer and mixed lineage leukemia [126–129]. Furthermore, elevated expression of Eps8 has been variously linked to tumorigenesis, proliferation and migration, and represents a poor prognosis in patients with cancer [126, 130–132]. To improve the prognosis of cancer patients, researchers make great effort to downregulate the expression of Eps8 by chemotherapeutics agents. Yang et al. demonstrated that mithramycin A could suppress tumor cell formation and metastasis in several cancer cell lines through an inhibition of Eps8 [133]. Furthermore, as a new CMA substrate protein, Eps8 has two KFERQ-like motifs for recognizing by HSC70 [134]. Since Eps8 exposes the imperative function in cancer progression, future research may accelerate protein degradation of Eps8 by CMA in cancer cells.
RND3
Rnd3, also known as RhoE, is a small signaling G protein. It is an atypical member of the Rho GTPase family [135], which involves in diverse cellular functions such as apoptosis, cell polarity and cell-cycle progression [136]. Such functions contribute to cancer cell migration and metastasis [137]. There is some controversy over the function of Rnd3 in tumor biology. Rnd3 has been considered either as an anti-oncogene or an oncogene. Interestingly, Rnd3 differentially expresses in various types of cancer. For example, Rnd3 is overexpressed in pancreatic cancer [138] and NSCLC [139, 140] and under-expressed in prostate cancer [141] and gastric cancers [142]. However, evidences strongly support Rnd3 may act as an anti-oncogene, such as hepatocellular carcinoma [143], squamous cell carcinoma [144], breast cancer [145, 146], prostate cancer [141], colorectal carcinoma [147], and lung cancer [145]. Researchers demonstrated that Rnd3 is a novel substrate protein for CMA, and the degradation of Rnd3 by CMA pathway can maintain cell proliferation in gastric cancer [148]. Thus, finding the natural products to reduce the degradation of Rnd3 through CMA as a treatment for cancer may evolve over the next several years.
MUTANT TP53
Tumor protein p53 (TP53), also known as p53, was first identified in 1979 as an SV40-binding protein by Lionel Crawford [149]. The TP53 maps to the human chromosome 17p13.1 and encodes a 53 kDa phosphoprotein [150]. TP53, the ‘guardian of the genome’, acts as a checkpoint control for cell cycle, cell differentiation, programmed cell apoptosis or death, and DNA synthesis and repair [151, 152]. The TP53 protein serves as a major barrier against cancer development, and the inactivation of TP53 pathway is found in human tumors [153, 154]. The majority of these mutations in TP53 are single-base substitution and loss of alleles [155]. TP53 mutations result in loss of tumor suppressor activities and gain of oncogenic functions [156]. TP53 mutants are contributing to tumor survival, proliferation, genomic instability, disruption of tissue architecture, angiogenesis, invasion, migration, and metastasis [157]. The researchers found that degradation of mutant TP53 is specifically mediated by the CMA pathway [158]. Thus, reducing the level of mutant TP53 proteins via CMA represents an attractive anticancer strategy.
HK2
Hexokinases (HKs) catalyze the cardinal process in glycolysis [159]. There are four isoforms of HK in mammals: HK1, HK2, HK3, and HK4 [160, 161]. HK2 is associated with the mitochondrial membrane, so loss HK2 can inhibit glucose metabolism and destroy the mitochondria [162]. Among these isozymes, the high expression of HK2 has been observed in lung, breast, pancreatic, ovarian cancers and hepatocellular carcinoma, and this is usually associated with poor prognosis [163–167]. HK2 is regulated by the transcription factors such as p53, Myc, and HIF-1 [168]. Besides, the effective anticancer drug 3-bromopyruvate (3BP) is a structural analog of pyruvic acid, which plays the inhibitory effects on HK2 [169–171]. HK2 is characterized as an oncoprotein since its role in tumor onset [164]. The researchers found HK2 degrades via CMA [15]. This suggests CMA may manipulate cellular metabolism and it may be a means of anticancer therapeutics.
SUBSTRATE GENES TARGET NETWORKS SUGGEST CMA INVOLVES IN WARBURG EFFECT OF CANCER
To identify the relevance of the cancer-associated substrate proteins, we analyze the substrate proteins by the Database for Annotation, Visualization and Integrated Discovery (DAVID). DAVID is a publicly available tool designed by the Laboratory of Immunopathogenesis and Bioinformatics, which is able to introduce the gene on KEGG pathway [172]. It can get access at http://david.abcc.ncifcrf.gov. The cancer-associated substrates were inputted into the Functional Annotation tool of DAVID. Table 2 lists the KEGG pathways association with substrate genes. Among the five pathways, the molecular pathway entitled “hsa05230: central carbon metabolism in cancer” shows a great association with substrates in cancer. The schematic illustration of CMA substrate proteins involve in Warburg effect is illustrated in Figure 3.
Table 2: List of five KEGG pathways and relative genes
KEGG pathway |
Genes |
P-Value |
Benjamini |
|---|---|---|---|
hsa05230:Central carbon metabolism in cancer |
HK2, PKM, TP53 |
5.0E-4 |
3.1E-2 |
hsa04930:Type II diabetes mellitus |
HK2, PKM |
2.8E-2 |
5.8E-1 |
hsa00010:Glycolysis/Gluconeogenesis |
HK2, PKM |
3.8E-2 |
5.5E-1 |
hsa01200:Carbon metabolism |
HK2, PKM |
6.4E-2 |
6.4E-1 |
hsa04919:Thyroid hormone signaling pathway |
N-CoR, TP53 |
6.4E-2 |
5.6E-1 |
hsa05202:Transcriptional misregulation in cancer |
N-CoR, TP53 |
9.4E-2 |
6.4E-1 |
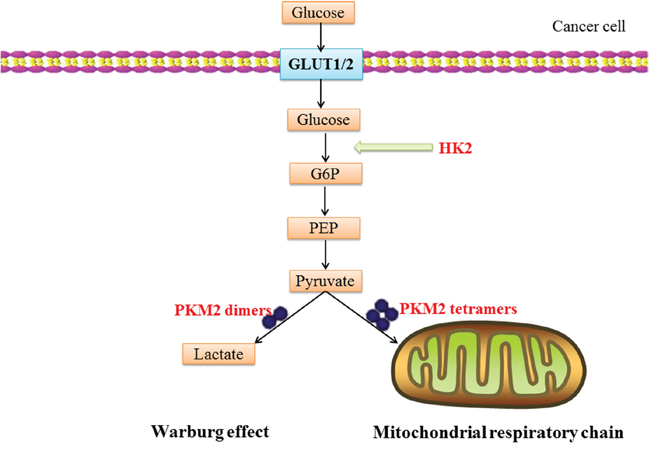
Figure 3: A schematic illustration of CMA substrate proteins involve in Warburg effect: glucose translocation through the plasma-membrane by glucose transporters (GLUT1/2) is rapidly phosphorylated to glucose-6-phosphate (G6P) by HK2. PKM2 dimers and tetramers possess low and high levels of pyruvate kinase activity, respectively. PKM2 dimer redirects the conversion of pyruvate to lactate; the PKM2 tetramer promotes the oxidative phosphorylation through the mitochondria respiratory chain. The Warburg effect describes the enhanced conversion of glucose to lactate by tumor cells, even in the presence of adequate oxygen that would ordinarily be used for oxidative phosphorylation.
In this pathway, HK2 and PKM2 are involved in Warburg effect by catalyzing glycolysis. In the beginning of the 20th century, Otto Warburg noticed that cancer cells show a high level of glycolysis accompanied with lactic acid fermentation even in the presence of oxygen [173]. This process is known as the Warburg effect [109]. This indicates that cancer cells prefer aerobic glycolysis for energy, rather than mitochondrial oxidative phosphorylation. The Warburg effect contributes to the survival and proliferation of tumor cell [174]. Previous studies reveal a critical role of PKM2 in tumorigenesis by promoting the Warburg effect. In cancer cells, knockdown of PKM2 increased oxygen consumption, reduced glucose uptake and lactate production [175]. HK2, a CMA substrate, catalyzes the first crucial step of glucose metabolism by phosphorylation of glucose to glucose-6-phosphate (G6P) [176]. The KFERQ-like motif of HK2 also binds to glucose molecules. The CMA motif of HK2 is hid in the protein when there is a glucose molecule. In other conditions, this motif is exposed for the recognizing by HSC70 [177]. CMA influences the glycolysis of malignant cell via degrading the enzymes of glycolytic and TCA cycle. Thus the maintenance of the Warburg effect requires functional CMA in cancer cells [14]. CMA substrate proteins may develop into a new anticancer therapeutic approach through decreasing the glycolysis of the cancer cell.
CONCLUSIONS AND FUTURE PERSPECTIVES
CMA is one of the autophagy-lysosome pathway which targets substrate proteins to the lysosomal membrane one by one for their degradation [7, 19]. CMA substrate protein plays a crucial role in cancer. However, substrate proteins found in CMA pathway may also degrade through other proteolytic systems, such as proteasomes or macroautophagy. For example, AF1Q and Rnd3 are degraded through the CMA and the proteasome-based system [55, 148]. This phenomenon suggests that CMA and proteasome system can co-regulate the substrate proteins. Prolonged glucose starvation can induce the degradation of mutant TP53 via macroautophagy [178]. Likewise, the cross-talk is existing in CMA and macroautophagy [179]. Mutant TP53 maybe select the mode of degradation between macroautophagy and CMA under different conditions. Different states of substrate proteins also influence the degradation through CMA, such as posttranslational modifications and different mutant alleles. In fact, acetylation contributes to the degradation of PKM2 by CMA [22].
By assessing the role of CMA substrate protein in cancer, we find PKM2 and HK2 are the key enzymes in glycolysis which contribute to the Warburg effect of the malignant cell. Abundant data indicate that blockade of CMA decrease the levels of glycolytic enzymes [180]. This result is somewhat counterintuitive because we insist that inhibiting CMA would lead to an accumulation of CMA-dependent glycolytic enzyme. Since blockade of CMA pathway is still debated, targeting to the CMA substrate proteins provides a new direction of cancer therapy. The fact that the CMA substrate protein motif is not in strict conformance with a five-amino acid residue sequence [20, 181], this makes it possible to create a motif out of an imperfect motif acquiring more effective recognition through posttranslational modifications [78, 182, 183]. Such as, acetylation the K residue can provide a missing Q, which explains that acetylation can increase the targeting of some glycolytic enzymes interaction with HSC70 [22]. HSC70 targets unphosphorylated PED, changing phosphorylated PED into unphosphorylated PED may suppress tumorigenesis. Selective modulation of cancer-associated CMA substrate proteins can also aid study the molecular mechanism of tumorigenesis. Such as, PKM2 is a crucial downstream protein of mammalian target of rapamycin (mTOR) [184]. Disruption of mTOR may suppress oncogenic PKM2-mediated tumorigenesis. Research shows a cross-talk between macroautophagy and CMA [185], mTOR also plays a crucial role in regulating macroautophagy [186]. PKM2 may as a key protein between macroautophagy and CMA for further cancer-associated study.
In principle, approximately 30% of proteins in the cytoplasm contain the HSC70-targeting sequence [187, 188], more CMA substrate proteins are pending further verification. CMA substrate proteins assist us to comprehend the relationship between CMA and cancer cells. Selective modulation of cancer-associated CMA proteins by posttranslational modifications shows the potential for cancer therapy.
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (81173161 to Dai-Han Zhou).
CONFLICTS OF INTEREST
The authors have declared no potential conflicts of interest.
REFERENCES
1. Kliosnky D. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd edition) (vol 12, pg 1, 2015). Autophagy. 2016; 12: 443.
2. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008; 451:1069–75.
3. Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005; 12 Suppl 2:1535-41.
4. Bejarano E, Cuervo AM. Chaperone-mediated autophagy. Proc Am Thorac Soc. 2010; 7:29–39.
5. Du P, Cao H, Wu HR, Zhu BS, Wang HW, Gu CW, Xing CG, Chen W. Blocking Bcl-2 leads to autophagy activation and cell death of the HEPG2 liver cancer cell line. Asian Pac J Cancer Prev. 2013; 14:5849–54.
6. Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010; 22:124–31.
7. Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010; 21:142–50.
8. Agarraberes FA, Terlecky SR, Dice JF. An intralysosomal hsp70 is required for a selective pathway of lysosomal protein degradation. J Cell Biol. 1997; 137:825–34.
9. Massey AC, Follenzi A, Kiffin R, Zhang C, Cuervo AM. Early cellular changes after blockage of chaperone-mediated autophagy. Autophagy. 2008; 4:442–56.
10. Rodríguez-Muela N, Koga H, García-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell. 2013; 12:478–88.
11. Ahlberg J, Glaumann H. Uptake—microautophagy—and degradation of exogenous proteins by isolated rat liver lysosomes. Effects of pH, ATP, and inhibitors of proteolysis. Exp Mol Pathol. 1985; 42:78–88.
12. Mortimore GE, Lardeux BR, Adams CE. Regulation of microautophagy and basal protein turnover in rat liver. Effects of short-term starvation. J Biol Chem. 1988; 263:2506–12.
13. Sahu R, Kaushik S, Clement CC, Cannizzo ES, Scharf B, Follenzi A, Potolicchio I, Nieves E, Cuervo AM, Santambrogio L. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011; 20:131–39.
14. Kon M, Kiffin R, Koga H, Chapochnick J, Macian F, Varticovski L, Cuervo AM. Chaperone-mediated autophagy is required for tumor growth. Sci Transl Med. 2011; 3:109ra117.
15. Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, Pan L, Liu J, Coloff JL, et al. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. J Cell Biol. 2015; 210:705–16.
16. Ali AB, Nin DS, Tam J, Khan M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PloS one. 2011; 6:e25268.
17. Wing SS, Chiang HL, Goldberg AL, Dice JF. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem J. 1991; 275:165–69.
18. Chiang HL, Terlecky SR, Plant CP, Dice JF. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science. 1989; 246:382–85.
19. Dice JF. Chaperone-mediated autophagy. Autophagy. 2007; 3:295–99.
20. Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990; 15:305–09.
21. Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012; 22:407–17.
22. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011; 42:719–30.
23. Arndt V, Dick N, Tawo R, Dreiseidler M, Wenzel D, Hesse M, Fürst DO, Saftig P, Saint R, Fleischmann BK, Hoch M, Höhfeld J. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010; 20:143–48.
24. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008; 28:5747–63.
25. Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005; 280:23727–34.
26. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996; 273:501–03.
27. Yang R, Gao G, Mao Z, Yang Q. Chaperone-Mediated Autophagy and Mitochondrial Homeostasis in Parkinson's Disease. Parkinsons Dis. 2016; 2016:2613401.
28. Cuervo AM, Dice JF. Unique properties of lamp2a compared to other lamp2 isoforms. J Cell Sci. 2000; 113:4441–50.
29. Salvador N, Aguado C, Horst M, Knecht E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J Biol Chem. 2000; 275:27447–56.
30. Agarraberes FA, Dice JF. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J Cell Sci. 2001; 114:2491–99.
31. Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014; 24:92–104.
32. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J Biol Chem. 2013; 288:10703–14.
33. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004; 15:4829–40.
34. Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015; 6:6823.
35. Finn PF, Dice JF. Ketone bodies stimulate chaperone-mediated autophagy. J Biol Chem. 2005; 280:25864–70.
36. Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014; 15:1046–54.
37. Anguiano J, Garner TP, Mahalingam M, Das BC, Gavathiotis E, Cuervo AM. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol. 2013; 9:374-82.
38. Arias E. Lysosomal mTORC2/PHLPP1/Akt axis: a new point of control of chaperone-mediated autophagy. Oncotarget. 2015; 6:35147–48. doi: 10.18632/oncotarget.5903.
39. Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci USA. 2006; 103:5805–10.
40. Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, Cuervo AM. Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell. 2015; 14:249–64.
41. Tse W, Zhu W, Chen HS, Cohen A. A novel gene, AF1q, fused to MLL in t(1;11) (q21;q23), is specifically expressed in leukemic and immature hematopoietic cells. Blood. 1995; 85:650–56.
42. Co NN, Tsang WP, Tsang TY, Yeung CL, Yau PL, Kong SK, Kwok TT. AF1q enhancement of gamma irradiation-induced apoptosis by up-regulation of BAD expression via NF-kappaB in human squamous carcinoma A431 cells. Oncol Rep. 2010; 24:547–54.
43. Tse W, Deeg HJ, Stirewalt D, Appelbaum FR, Radich J, Gooley T. Increased AF1q gene expression in high-risk myelodysplastic syndrome. Br J Haematol. 2005; 128:218–20.
44. Jacques C, Baris O, Prunier-Mirebeau D, Savagner F, Rodien P, Rohmer V, Franc B, Guyetant S, Malthiery Y, Reynier P. Two-step differential expression analysis reveals a new set of genes involved in thyroid oncocytic tumors. J Clin Endocrinol Metab. 2005; 90:2314–20.
45. Tse W, Meshinchi S, Alonzo TA, Stirewalt DL, Gerbing RB, Woods WG, Appelbaum FR, Radich JP. Elevated expression of the AF1q gene, an MLL fusion partner, is an independent adverse prognostic factor in pediatric acute myeloid leukemia. Blood. 2004; 104:3058–63.
46. Strunk CJ, Platzbecker U, Thiede C, Schaich M, Illmer T, Kang Z, Leahy P, Li C, Xie X, Laughlin MJ, Lazarus HM, Gerson SL, Bunting KD, et al. Elevated AF1q expression is a poor prognostic marker for adult acute myeloid leukemia patients with normal cytogenetics. Am J Hematol. 2009; 84:308–09.
47. Skotheim RI, Autio R, Lind GE, Kraggerud SM, Andrews PW, Monni O, Kallioniemi O, Lothe RA. Novel genomic aberrations in testicular germ cell tumors by array-CGH, and associated gene expression changes. Cell Oncol. 2006; 28:315–26.
48. Keshava C, Whipkey D, Weston A. Transcriptional signatures of environmentally relevant exposures in normal human mammary epithelial cells: benzo[a]pyrene. Cancer Lett. 2005; 221:201–11.
49. Li DQ, Hou YF, Wu J, Chen Y, Lu JS, Di GH, Ou ZL, Shen ZZ, Ding J, Shao ZM. Gene expression profile analysis of an isogenic tumour metastasis model reveals a functional role for oncogene AF1Q in breast cancer metastasis. Eur J Cancer. 2006; 42:3274–86.
50. Chang XZ, Li DQ, Hou YF, Wu J, Lu JS, Di GH, Jin W, Ou ZL, Shen ZZ, Shao ZM. Identification of the functional role of AF1Q in the progression of breast cancer. Breast Cancer Res Treat. 2008; 111:65–78.
51. Co NN, Tsang WP, Wong TW, Cheung HH, Tsang TY, Kong SK, Kwok TT. Oncogene AF1q enhances doxorubicin-induced apoptosis through BAD-mediated mitochondrial apoptotic pathway. Mol Cancer Ther. 2008; 7:3160–68.
52. Visconti R, Cerutti J, Battista S, Fedele M, Trapasso F, Zeki K, Miano MP, de Nigris F, Casalino L, Curcio F, Santoro M, Fusco A. Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NFkappaB p65 protein expression. Oncogene. 1997; 15:1987–94.
53. Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997; 100:2952–60.
54. Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, Scheidereit C. Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells. Leukemia. 2000; 14:399–402.
55. Li P, Ji M, Lu F, Zhang J, Li H, Cui T, Li Wang X, Tang D, Ji C. Degradation of AF1Q by chaperone-mediated autophagy. Exp Cell Res. 2014; 327:48–56.
56. Araujo H, Danziger N, Cordier J, Glowinski J, Chneiweiss H. Characterization of PEA-15, a major substrate for protein kinase C in astrocytes. J Biol Chem. 1993; 268:5911–20.
57. Formisano P, Ragno P, Pesapane A, Alfano D, Alberobello AT, Rea VE, Giusto R, Rossi FW, Beguinot F, Rossi G, Montuori N. PED/PEA-15 interacts with the 67 kD laminin receptor and regulates cell adhesion, migration, proliferation and apoptosis. J Cell Mol Med. 2012; 16:1435–46.
58. Kitsberg D, Formstecher E, Fauquet M, Kubes M, Cordier J, Canton B, Pan G, Rolli M, Glowinski J, Chneiweiss H. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFalpha-induced apoptosis. J Neurosci. 1999; 19:8244–51.
59. Danziger N, Yokoyama M, Jay T, Cordier J, Glowinski J, Chneiweiss H. Cellular expression, developmental regulation, and phylogenic conservation of PEA-15, the astrocytic major phosphoprotein and protein kinase C substrate. J Neurochem. 1995; 64:1016–25.
60. Callaway K, Abramczyk O, Martin L, Dalby KN. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry. 2007; 46:9187–98.
61. Renganathan H, Vaidyanathan H, Knapinska A, Ramos JW. Phosphorylation of PEA-15 switches its binding specificity from ERK/MAPK to FADD. Biochem J. 2005; 390:729–35.
62. Kubes M, Cordier J, Glowinski J, Girault JA, Chneiweiss H. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes: identification of Ser104 and Ser116 phosphorylated, respectively, by protein kinase C and calcium/calmodulin kinase II in vitro. J Neurochem. 1998; 71:1307–14.
63. Trencia A, Perfetti A, Cassese A, Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F, Condorelli G, Formisano P, Beguinot F. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol Cell Biol. 2003; 23:4511–21.
64. Formisano P, Perruolo G, Libertini S, Santopietro S, Troncone G, Raciti GA, Oriente F, Portella G, Miele C, Beguinot F. Raised expression of the antiapoptotic protein ped/pea-15 increases susceptibility to chemically induced skin tumor development. Oncogene. 2005; 24:7012–21.
65. Yu D, Wolf JK, Scanlon M, Price JE, Hung MC. Enhanced c-erbB-2/neu expression in human ovarian cancer cells correlates with more severe malignancy that can be suppressed by E1A. Cancer Res. 1993; 53:891–98.
66. Bartholomeusz C, Itamochi H, Nitta M, Saya H, Ginsberg MH, Ueno NT. Antitumor effect of E1A in ovarian cancer by cytoplasmic sequestration of activated ERK by PEA15. Oncogene. 2006; 25:79–90.
67. Glading A, Koziol JA, Krueger J, Ginsberg MH. PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Cancer Res. 2007; 67:1536–44.
68. Renault-Mihara F, Beuvon F, Iturrioz X, Canton B, De Bouard S, Léonard N, Mouhamad S, Sharif A, Ramos JW, Junier MP, Chneiweiss H. Phosphoprotein enriched in astrocytes-15 kDa expression inhibits astrocyte migration by a protein kinase C delta-dependent mechanism. Mol Biol Cell. 2006; 17:5141–52.
69. Condorelli G, Vigliotta G, Cafieri A, Trencia A, Andalò P, Oriente F, Miele C, Caruso M, Formisano P, Beguinot F. PED/PEA-15: an anti-apoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999; 18:4409–15.
70. Stassi G, Garofalo M, Zerilli M, Ricci-Vitiani L, Zanca C, Todaro M, Aragona F, Limite G, Petrella G, Condorelli G. PED mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 2005; 65:6668–75.
71. Zanca C, Garofalo M, Quintavalle C, Romano G, Acunzo M, Ragno P, Montuori N, Incoronato M, Tornillo L, Baumhoer D, Briguori C, Terracciano L, Condorelli G. PED is overexpressed and mediates TRAIL resistance in human non-small cell lung cancer. J Cell Mol Med. 2008; 12:2416–26.
72. Eramo A, Pallini R, Lotti F, Sette G, Patti M, Bartucci M, Ricci-Vitiani L, Signore M, Stassi G, Larocca LM, Crinò L, Peschle C, De Maria R. Inhibition of DNA methylation sensitizes glioblastoma for tumor necrosis factor-related apoptosis-inducing ligand-mediated destruction. Cancer Res. 2005; 65:11469–77.
73. Heikaus S, Kempf T, Mahotka C, Gabbert HE, Ramp U. Caspase-8 and its inhibitors in RCCs in vivo: the prominent role of ARC. Apoptosis. 2008; 13:938–49.
74. Incoronato M, Garofalo M, Urso L, Romano G, Quintavalle C, Zanca C, Iaboni M, Nuovo G, Croce CM, Condorelli G. miR-212 increases tumor necrosis factor-related apoptosis-inducing ligand sensitivity in non-small cell lung cancer by targeting the antiapoptotic protein PED. Cancer Res. 2010; 70:3638–46.
75. Garofalo M, Romano G, Quintavalle C, Romano MF, Chiurazzi F, Zanca C, Condorelli G. Selective inhibition of PED protein expression sensitizes B-cell chronic lymphocytic leukaemia cells to TRAIL-induced apoptosis. Int J Cancer. 2007; 120:1215–22.
76. Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, Yong VW, Parney IF, Roa WH, Petruk KC. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001; 61:1162–70.
77. Bellail AC, Tse MC, Song JH, Phuphanich S, Olson JJ, Sun SY, Hao C. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J Cell Mol Med. 2010; 14:1303–17.
78. Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, Mirabelli P, Monti M, Ballabio A, Pucci P, Cuervo AM, Condorelli G. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol. 2014; 229:1359–68.
79. Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Söderström M, Glass CK, Rosenfeld MG. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995; 377:397–404.
80. Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001; 1:181–93.
81. Baniahmad A. Nuclear hormone receptor co-repressors. J Steroid Biochem Mol Biol. 2005; 93:89–97.
82. Jepsen K, Rosenfeld MG. Biological roles and mechanistic actions of co-repressor complexes. J Cell Sci. 2002; 115:689–98.
83. Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Zhong S, Pandolfi PP, Ishii S. PML-RARalpha alleviates the transcriptional repression mediated by tumor suppressor Rb. J Biol Chem. 2001; 276:43491–94.
84. Khan MM, Nomura T, Kim H, Kaul SC, Wadhwa R, Shinagawa T, Ichikawa-Iwata E, Zhong S, Pandolfi PP, Ishii S. Role of PML and PML-RARalpha in Mad-mediated transcriptional repression. Mol Cell. 2001; 7:1233–43.
85. Heinzel T, Lavinsky RM, Mullen TM, Söderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, Seto E, Eisenman RN, Rose DW, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997; 387:43–48.
86. Laherty CD, Billin AN, Lavinsky RM, Yochum GS, Bush AC, Sun JM, Mullen TM, Davie JR, Rose DW, Glass CK, Rosenfeld MG, Ayer DE, Eisenman RN. SAP30, a component of the mSin3 corepressor complex involved in N-CoR-mediated repression by specific transcription factors. Mol Cell. 1998; 2:33–42.
87. Furuya F, Guigon CJ, Zhao L, Lu C, Hanover JA, Cheng SY. Nuclear receptor corepressor is a novel regulator of phosphatidylinositol 3-kinase signaling. Mol Cell Biol. 2007; 27:6116–26.
88. Movilla N, Bustelo XR. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol Cell Biol. 1999; 19:7870–85.
89. Tybulewicz VL. Vav-family proteins in T-cell signalling. Curr Opin Immunol. 2005; 17:267–74.
90. Oberley MJ, Wang DS, Yang DT. Vav1 in hematologic neoplasms, a mini review. Am J Blood Res. 2012; 2:1–8.
91. Katzav S. Vav1: a hematopoietic signal transduction molecule involved in human malignancies. Int J Biochem Cell Biol. 2009; 41:1245–48.
92. Bustelo XR. Vav proteins, adaptors and cell signaling. Oncogene. 2001; 20:6372–81.
93. Sanchez-Aguilera A, Lee YJ, Lo Celso C, Brumme K, Lin CP, Scadden DT, Williams DA. Regulation of Rho GTPases by the Hematopoietic-Specific Guanine Nucleotide Exchange Factor Vav1 Is Critical for Hematopoietic Stem Cell Retention in the Endosteal Niche and Engraftment. Blood. 2009; 114:37–38.
94. Katzav S. Flesh and blood: the story of Vav1, a gene that signals in hematopoietic cells but can be transforming in human malignancies. Cancer Lett. 2007; 255:241–54.
95. Fischer KD, Tedford K, Penninger JM. Vav links antigen-receptor signaling to the actin cytoskeleton. Semin Immunol. 1998; 10:317–27.
96. Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, Smyrk TC, Chari ST, Urrutia R, Billadeau DD. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005; 7:39–49.
97. Denicola G, Tuveson DA. VAV1: a new target in pancreatic cancer? Cancer Biol Ther. 2005; 4:509–11.
98. Lazer G, Idelchuk Y, Schapira V, Pikarsky E, Katzav S. The haematopoietic specific signal transducer Vav1 is aberrantly expressed in lung cancer and plays a role in tumourigenesis. J Pathol. 2009; 219:25–34.
99. Bartolomé RA, Molina-Ortiz I, Samaniego R, Sánchez-Mateos P, Bustelo XR, Teixidó J. Activation of Vav/Rho GTPase signaling by CXCL12 controls membrane-type matrix metalloproteinase-dependent melanoma cell invasion. Cancer Res. 2006; 66:248–58.
100. Katzav S, Martin-Zanca D, Barbacid M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. 1989; 8:2283–90.
101. Razidlo GL, Wang Y, Chen J, Krueger EW, Billadeau DD, McNiven MA. Dynamin 2 potentiates invasive migration of pancreatic tumor cells through stabilization of the Rac1 GEF Vav1. Dev Cell. 2013; 24:573–85.
102. Braun AC. On the origin of the cancer cells. Am Sci. 1970; 58:307–20.
103. Alfarouk KO, Verduzco D, Rauch C, Muddathir AK, Adil HH, Elhassan GO, Ibrahim ME, David Polo Orozco J, Cardone RA, Reshkin SJ, Harguindey S. Glycolysis, tumor metabolism, cancer growth and dissemination. A new pH-based etiopathogenic perspective and therapeutic approach to an old cancer question. Oncoscience. 2014; 1:777–802. doi: 10.18632/oncoscience.109.
104. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004; 84:1014–20.
105. Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, Loda M, Lane HA, Sellers WR. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004; 10:594–601.
106. Noguchi T, Inoue H, Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J Biol Chem. 1986; 261:13807–12.
107. Noguchi T, Yamada K, Inoue H, Matsuda T, Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. J Biol Chem. 1987; 262:14366–71.
108. Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E. Pyruvate kinase type M2: a crossroad in the tumor metabolome. Br J Nutr. 2002; 87 Suppl 1:S23-9.
109. Warburg O. On the origin of cancer cells. Science. 1956; 123:309–14.
110. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74.
111. Yang W, Lu Z. Regulation and function of pyruvate kinase M2 in cancer. Cancer Lett. 2013; 339:153–58.
112. Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol. 2005; 15:300–08.
113. Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005; 44:9417–29.
114. Koss K, Harrison RF, Gregory J, Darnton SJ, Anderson MR, Jankowski JA. The metabolic marker tumour pyruvate kinase type M2 (tumour M2-PK) shows increased expression along the metaplasia-dysplasia-adenocarcinoma sequence in Barrett’s oesophagus. J Clin Pathol. 2004; 57:1156–59.
115. Ahmed AS, Dew T, Lawton FG, Papadopoulos AJ, Devaja O, Raju KS, Sherwood RA. M2-PK as a novel marker in ovarian cancer. A prospective cohort study. Eur J Gynaecol Oncol. 2007; 28:83–88.
116. Elia S, Massoud R, Guggino G, Cristino B, Cortese C, De Massimi AR, Zenobi R. Tumor type M2-pyruvate-kinase levels in pleural fluid versus plasma in cancer patients: a further tool to define the need for invasive procedures. Eur J Cardiothorac Surg. 2008; 33:723–27.
117. Ugurel S, Bell N, Sucker A, Zimpfer A, Rittgen W, Schadendorf D. Tumor type M2 pyruvate kinase (TuM2-PK) as a novel plasma tumor marker in melanoma. Int J Cancer. 2005; 117:825–30.
118. Ferguson EC, Rathmell JC. New roles for pyruvate kinase M2: working out the Warburg effect. Trends Biochem Sci. 2008; 33:359–62.
119. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene. 2011; 30:4297–306.
120. Spoden GA, Rostek U, Lechner S, Mitterberger M, Mazurek S, Zwerschke W. Pyruvate kinase isoenzyme M2 is a glycolytic sensor differentially regulating cell proliferation, cell size and apoptotic cell death dependent on glucose supply. Exp Cell Res. 2009; 315:2765–74.
121. Goldberg MS, Sharp PA. Pyruvate kinase M2-specific siRNA induces apoptosis and tumor regression. J Exp Med. 2012; 209:217–24.
122. Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal. 2009; 2:ra73.
123. Fazioli F, Minichiello L, Matoska V, Castagnino P, Miki T, Wong WT, Di Fiore PP. Eps8, a substrate for the epidermal growth factor receptor kinase, enhances EGF-dependent mitogenic signals. EMBO J. 1993; 12:3799–808.
124. Ion A, Crosby AH, Kremer H, Kenmochi N, Van Reen M, Fenske C, Van Der Burgt I, Brunner HG, Montgomery K, Kucherlapati RS, Patton MA, Page C, Mariman E, Jeffery S. Detailed mapping, mutation analysis, and intragenic polymorphism identification in candidate Noonan syndrome genes MYL2, DCN, EPS8, and RPL6. J Med Genet. 2000; 37:884–86.
125. Ding X, Zhou F, Wang F, Yang Z, Zhou C, Zhou J, Zhang B, Yang J, Wang G, Wei Z, Hu X, Xiang S, Zhang J. Eps8 promotes cellular growth of human malignant gliomas. Oncol Rep. 2013; 29:697–703.
126. Chen YJ, Shen MR, Chen YJ, Maa MC, Leu TH. Eps8 decreases chemosensitivity and affects survival of cervical cancer patients. Mol Cancer Ther. 2008; 7:1376–85.
127. Griffith OL, Melck A, Jones SJ, Wiseman SM. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J Clin Oncol. 2006; 24:5043–51.
128. Wang H, Patel V, Miyazaki H, Gutkind JS, Yeudall WA. Role for EPS8 in squamous carcinogenesis. Carcinogenesis. 2009; 30:165–74.
129. Yao J, Weremowicz S, Feng B, Gentleman RC, Marks JR, Gelman R, Brennan C, Polyak K. Combined cDNA array comparative genomic hybridization and serial analysis of gene expression analysis of breast tumor progression. Cancer Res. 2006; 66:4065–78.
130. Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC, Wang ST, Huang CC, Chow NH, Leu TH. Eps8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J Biol Chem. 2007; 282:19399–409.
131. Yap LF, Jenei V, Robinson CM, Moutasim K, Benn TM, Threadgold SP, Lopes V, Wei W, Thomas GJ, Paterson IC. Upregulation of Eps8 in oral squamous cell carcinoma promotes cell migration and invasion through integrin-dependent Rac1 activation. Oncogene. 2009; 28:2524–34.
132. Welsch T, Endlich K, Giese T, Büchler MW, Schmidt J. Eps8 is increased in pancreatic cancer and required for dynamic actin-based cell protrusions and intercellular cytoskeletal organization. Cancer Lett. 2007; 255:205–18.
133. Yang TP, Chiou HL, Maa MC, Wang CJ. Mithramycin inhibits human epithelial carcinoma cell proliferation and migration involving downregulation of Eps8 expression. Chem Biol Interact. 2010; 183:181–86.
134. Welsch T, Younsi A, Disanza A, Rodriguez JA, Cuervo AM, Scita G, Schmidt J. Eps8 is recruited to lysosomes and subjected to chaperone-mediated autophagy in cancer cells. Exp Cell Res. 2010; 316:1914–24.
135. Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006; 16:522–29.
136. Riou P, Villalonga P, Ridley AJ. Rnd proteins: multifunctional regulators of the cytoskeleton and cell cycle progression. BioEssays. 2010; 32:986–92.
137. Trojan L, Steidler A, Haak M, Knoll T, Gretz N, Alken P, Michel MS. Identification of metastasis-associated genes in prostate cancer by genetic profiling of human prostate cancer cell lines. Eur Urol Suppl. 2005; 4:22.
138. Gress TM, Müller-Pillasch F, Geng M, Zimmerhackl F, Zehetner G, Friess H, Büchler M, Adler G, Lehrach H. A pancreatic cancer-specific expression profile. Oncogene. 1996; 13:1819–30.
139. Zhang C, Zhou F, Li N, Shi S, Feng X, Chen Z, Hang J, Qiu B, Li B, Chang S, Wan J, Shao K, Xing X, et al. Overexpression of RhoE has a prognostic value in non-small cell lung cancer. Ann Surg Oncol. 2007; 14:2628–35.
140. Cuiyan Z, Jie H, Fang Z, Kezhi Z, Junting W, Susheng S, Xiaoli F, Ning L, Xinhua M, Zhaoli C, Kang S, Bin Q, Baozhong L, et al. Overexpression of RhoE in Non-small Cell Lung Cancer (NSCLC) is associated with smoking and correlates with DNA copy number changes. Cancer Biol Ther. 2007; 6:335–42.
141. Bektic J, Pfeil K, Berger AP, Ramoner R, Pelzer A, Schäfer G, Kofler K, Bartsch G, Klocker H. Small G-protein RhoE is underexpressed in prostate cancer and induces cell cycle arrest and apoptosis. Prostate. 2005; 64:332–40.
142. Chen J, Zhou H, Li Q, Qiu M, Li Z, Tang Q, Liu M, Zhu Y, Huang J, Lang N, Liu Z, Deng Y, Zhang S, Bi F. Epigenetic modification of RhoE expression in gastric cancer cells. Oncol Rep. 2011; 25:173–80.
143. Luo H, Dong Z, Zou J, Zeng Q, Wu D, Liu L. Down-regulation of RhoE is associated with progression and poor prognosis in hepatocellular carcinoma. J Surg Oncol. 2012; 105:699–704.
144. Zhao H, Yang J, Fan T, Li S, Ren X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour Biol. 2012; 33:1363–74.
145. Zhu Y, Xu F, Zhou J, Liu S, Huang J, Luo D, Qiu M, Jin B, Bi F. [Expression of RhoE in lung and breast cancer and its clinical significance]. [Article in Chinese]. Zhongguo Fei Ai Za Zhi. 2008; 11:85-9.
146. Xia H, Li M, Chen L, Leng W, Yuan D, Pang X, Chen L, Li R, Tang Q, Bi F. Suppression of RND3 activity by AES downregulation promotes cancer cell proliferation and invasion. Int J Mol Med. 2013; 31:1081–86.
147. Luo H, Zou J, Dong Z, Zeng Q, Wu D, Liu L. Up-regulated miR-17 promotes cell proliferation, tumour growth and cell cycle progression by targeting the RND3 tumour suppressor gene in colorectal carcinoma. Biochem J. 2012; 442:311–21.
148. Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong J, Zhang S, Shang Y, Jiang X, Guo H, Chen N, Xiao X, Sheng J, et al. Chaperone-mediated autophagy regulates proliferation by targeting RND3 in gastric cancer. Autophagy. 2016; 12:515–28.
149. Chang C, Simmons DT, Martin MA, Mora PT. Identification and partial characterization of new antigens from simian virus 40-transformed mouse cells. J Virol. 1979; 31:463–71.
150. Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991; 351:453–56.
151. Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem. 1999; 274:36031–34.
152. Bálint E E, Vousden KH. Activation and activities of the p53 tumour suppressor protein. Br J Cancer. 2001; 85:1813–23.
153. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307–10.
154. Hussain SP, Harris CC. Molecular epidemiology of human cancer: contribution of mutation spectra studies of tumor suppressor genes. Cancer Res. 1998; 58:4023–37.
155. Rutz HP, Mirimanoff RO. Clinical implications of the p53 tumor-suppressor gene. N Engl J Med. 1994; 330:864–65.
156. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007; 26:2202–11.
157. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012; 26:1268–86.
158. Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, Yuan J. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013; 27:1718–30.
159. Smith TA. Mammalian hexokinases and their abnormal expression in cancer. Br J Biomed Sci. 2000; 57:170–78.
160. Wilson JE. HEXOKINASES. Reviews of Physiology Biochemistry and Pharmacology, Vol 126. 1995; 126: 65-198.
161. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003; 206:2049–57.
162. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008; 13:472–82.
163. Suh DH, Kim MA, Kim H, Kim MK, Kim HS, Chung HH, Kim YB, Song YS. Association of overexpression of hexokinase II with chemoresistance in epithelial ovarian cancer. Clin Exp Med. 2014; 14:345–53.
164. Patra KC, Wang Q, Bhaskar PT, Miller L, Wang Z, Wheaton W, Chandel N, Laakso M, Muller WJ, Allen EL, Jha AK, Smolen GA, Clasquin MF, et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013; 24:213–28.
165. Zhang Z, Huang S, Wang H, Wu J, Chen D, Peng B, Zhou Q. High expression of hexokinase domain containing 1 is associated with poor prognosis and aggressive phenotype in hepatocarcinoma. Biochem Biophys Res Commun. 2016; 474:673–79.
166. Sato-Tadano A, Suzuki T, Amari M, Takagi K, Miki Y, Tamaki K, Watanabe M, Ishida T, Sasano H, Ohuchi N. Hexokinase II in breast carcinoma: a potent prognostic factor associated with hypoxia-inducible factor-1α and Ki-67. Cancer Sci. 2013; 104:1380–88.
167. Palmieri D, Fitzgerald D, Shreeve SM, Hua E, Bronder JL, Weil RJ, Davis S, Stark AM, Merino MJ, Kurek R, Mehdorn HM, Davis G, Steinberg SM, et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Mol Cancer Res. 2009; 7:1438–45.
168. Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, Mueller-Klieser W. Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2001; 51:349–53.
169. Ko YH, McFadden BA. Alkylation of isocitrate lyase from Escherichia coli by 3-bromopyruvate. Arch Biochem Biophys. 1990; 278:373–80.
170. Azevedo-Silva J, Queirós O, Baltazar F, Ułaszewski S, Goffeau A, Ko YH, Pedersen PL, Preto A, Casal M. The anticancer agent 3-bromopyruvate: a simple but powerful molecule taken from the lab to the bedside. J Bioenerg Biomembr. 2016; 48:349–62.
171. Gandham SK, Talekar M, Singh A, Amiji MM. Inhibition of hexokinase-2 with targeted liposomal 3-bromopyruvate in an ovarian tumor spheroid model of aerobic glycolysis. Int J Nanomedicine. 2015; 10:4405–23.
172. Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
173. Warburg O, Wind F, Negelein E. The Metabolism of Tumors In the Body. J Gen Physiol. 1927; 8:519–30.
174. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004; 4:891–99.
175. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008; 452:230–33.
176. Ahn KJ, Kim J, Yun M, Park JH, Lee JD. Enzymatic properties of the N- and C-terminal halves of human hexokinase II. BMB Rep. 2009; 42:350–55.
177. Galan-Acosta L, Xia H, Yuan J, Vakifahmetoglu-Norberg H. Activation of chaperone-mediated autophagy as a potential anticancer therapy. Autophagy. 2015; 11:2370–71.
178. Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, Reynoso K, Bargonetti J, Wellstein A, Albanese C, Avantaggiati ML. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012; 11:4436–46.
179. Kaushik S, Massey AC, Mizushima N, Cuervo AM. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol Biol Cell. 2008; 19:2179–92.
180. Thorburn A, Debnath J. Targeting chaperone-mediated autophagy in cancer. Sci Transl Med. 2011; 3:109ps45.
181. Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011; 68:749–63.
182. Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, O’Rourke JG, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009; 187:1083–99.
183. Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004; 36:2435–44.
184. Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci USA. 2011; 108:4129–34.
185. Li CL, Wei HL, Chen J, Wang B, Xie B, Fan LL, Li LJ. Ebb-and-flow of macroautophagy and chaperone-mediated autophagy in Raji cells induced by starvation and arsenic trioxide. Asian Pac J Cancer Prev. 2014; 15:5715–19.
186. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Arozena AA, Adachi H, Adams CM, Adams PD, Adeli K, Adhihetty PJ, Adler SG, Agam G, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016; 12:1-222.
187. Arias E, Cuervo AM. Chaperone-mediated autophagy in protein quality control. Curr Opin Cell Biol. 2011; 23:184–89.
188. Kaushik S, Bandyopadhyay U, Sridhar S, Kiffin R, Martinez-Vicente M, Kon M, Orenstein SJ, Wong E, Cuervo AM. Chaperone-mediated autophagy at a glance. J Cell Sci. 2011; 124:495–99.