
INTRODUCTION
The somatic activating mutations in PIK3CA encoding the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA, also known as p100α) cause heterogeneous mosaic or segmental overgrowth disorders including fibroadipose overgrowth (FAO) [1], hemihyperplasia multiple lipomatosis (HHML) [2], congenital lipomatous overgrowth, vascular malformations, epidermal nevi, scoliosis/skeletal and spinal (CLOVES) syndrome [3–5], macrodactyly and muscle hemihypertrophy [6], megalencephaly syndromes such as megalencephaly-capillary malformation (MCAP) [7] and hemimegalencephaly [8], skin disorders such as benign lichenoid keratosis (BLK) [9] and seborrheic keratosis (SK) [10], and fibroadipose infiltrating lipomatosis [11]. Progressive segmental overgrowth in various regions of the body including visceral, subcutaneous, muscular, fibroadipose, and skeletal tissues is the common feature of these disorders [12]. The term, PIK3CA-related overgrowth spectrum (PROS), has been proposed to represent the broad range of clinical manifestations caused by PIK3CA mutations [13]. Similar and related disorders are caused by somatic mutations in the genes of the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway, which include PTEN [14], PIK3R2 (MCAP) [7], AKT1 (Proteus syndrome) [15], AKT2 (asymmetric overgrowth and hypoglycemia) [16], and AKT3 (hemimegalencephaly) [17].
In this study, we identified the most common somatic mosaic mutation of PIK3CA (c.3140A>G, [p.H1047R]) in a Japanese female patient with PROS. We analyzed the correlation between the severity of overgrowth and the mutant allele frequency from affected tissues by quantitative PCR and direct sequencing. Further, we established dermal fibroblast cell lines harboring high and low frequencies of the mosaic mutation from the patient’s affected tissues and assessed the therapeutic effects of four compounds (rapamycin, NVP-BEZ235, aspirin, and metformin) on PI3K/AKT/mTOR signaling inhibition by determining the phosphorylation status of associated proteins and measuring cell proliferation.
RESULTS
Patient
The patient was a 3-year-old female born to non-consanguineous parents, a 34-year-old father and a 38-year-old mother (Figure 1). She was born at 39 weeks and 4 days gestation by normal vaginal delivery following an uneventful pregnancy. Her birth weight was 2988 g (−0.3 SD), and she measured 50.5 cm (0.4 SD) in height, with an occipitofrontal circumference (OFC) of 32 cm (−0.7 SD). Deformity of the feet was noted at birth. The left second, third, and fourth toes showed cutaneous syndactyly, and the right second and third toes showed syndactyly. An increased volume of subcutaneous adipose tissue in the left chest, the perineum, and the inside of the right thigh was obvious at one week after birth. The entire left leg and the right foot were severely enlarged, and the toes of both feet showed dactylomegaly (Figure 1C and Figure 1D). Brain magnetic resonance imaging at one month showed normal findings. The patient was diagnosed with bilateral dactylomegaly at 9 months and subcutaneous adipose tissue at the left knee and the distal phalanx of the first, second, and third left toes were surgically removed. At 2 years and 9 months, subcutaneous adipose tissue at the left knee, lower abdomen, perineum, and left lower leg was surgically removed. She showed normal milestones of head control at 3 months, sitting up at 5 months, and walking at 1 year. At 2 years, her weight, height, and OFC were 12.5 kg (0 SD), 85 to 89.5 cm (−1.4 SD~0 SD, because of different sizes of the legs), and 46.5 cm (−1.1 SD), respectively. It was noted that she had sparse hair and that her skin was thin with a small volume of adipose tissue on parts of the body other than the regions of overgrowth. Intellectual development was normal as per age equivalency and development quotient at 2 years and 4 months was 111. The presented patient was diagnosed with PROS taking into account the following findings: 1) presence of somatic PIK3CA mutation (described below), 2) congenital onset, 3) overgrowth being sporadic and mosaic, 4) overgrowth in adipose and skeletal tissues, 5) isolated macrodactyly, overgrown splayed feet, and overgrown limb, and 6) truncal adipose overgrowth [13]. According to Martinez-Lopez et al. [18], this patient is categorized as CLOVES syndrome of PROS.
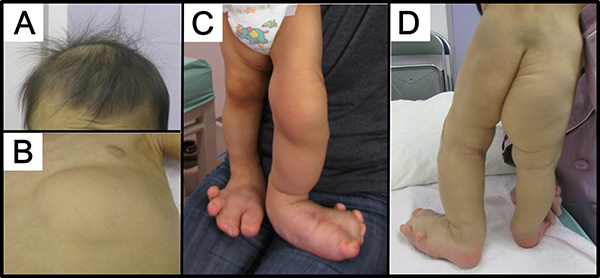
Figure 1: Patient. (A) The patient had sparse hair at 2 years of age. (B–D) Note the asymmetric distribution of truncal lipomatous masses and bilateral lower extremity involvement. Lipomatous tissue was thin in other areas. Regional lipohypoplasia was seen in the lower extremities and chest.
Identification and characterization of the PIK3CA H1047R mutation
Since mutations in PIK3CA contribute to unilateral and/or segmental overgrowth phenotypes, we determined nucleotide sequences of PIK3CA from the patient’s affected adipose tissues and skin by direct sequencing and identified a heterozygous mosaic mutation (c.3140A>G [p.H1047R]) in exon 20 (Figure 2A). The mutant allele was detected in all the affected tissues with the peak height ratio of A/(G+A) at nucleotide position 3140 determined by direct sequencing. Direct sequencing of PCR products and sequencing of 24 subcloned PCR products containing c.3140 confirmed that the patient’s blood cells did not contain the mutant allele. To determine the mutant allele frequency at nucleotide position 3140 of PIK3CA, we performed a quantitative multiplex PCR assay (Figure 2B). The calculated mutant allele frequencies of PIK3CA in the patient’s affected tissues and blood lymphocytes as determined by quantitative multiplex PCR and direct sequence are shown in Figure 2C. The frequencies of the PIK3CA mutant allele in the adipose tissue from the patient’s perineum, lower limb 1, lower limb 2, and chest determined by quantitative multiplex PCR were 6.9%, 9.9%, 11.1%, and 13.7%, respectively, and those in the skin of the perineum and lower limb were 3.4% and 8.7%, respectively. Of note, mutant allele frequency ratios determined by direct nucleotide sequence analysis were approximately double the frequency ratios determined by quantitative PCR analysis. This is possibly because of the large cycle number (36) in the PCR protocol for amplifying the region of genomic DNA that included the mutation. The mutant allele frequencies were higher in subcutaneous adipose tissues than in skin tissue. Among the affected regions, the mutation frequencies were higher in the lower limbs and chest than in the perineum. Thus, there was a higher mutant allele frequency in samples from regions with a larger mass of subcutaneous adipose tissue.
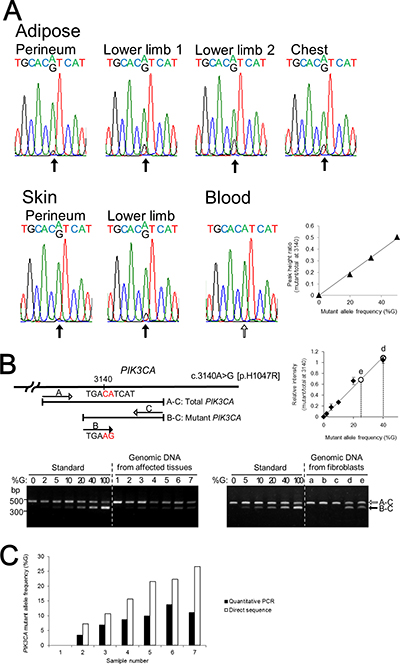
Figure 2: The identification and quantification of the mutation in PIK3CA. (A) Direct nucleotide sequence analysis of PIK3CA was performed using isolated genomic DNA from blood and six different regions including subcutaneous adipose tissue and skin from the lower limbs. A mosaic mutation (c.3140A>G [p.H1047R]) in exon 20 was identified from affected adipose tissues and skin. The ratios of the mutant allele are different in the patient’s affected tissues. Direct nucleotide sequence analysis was performed with plasmids containing normal or mutant PIK3CA fragment as standard reference materials. The right panel shows a standard curve for calculating PIK3CA mutant frequency by direct sequencing. (B) Multiplex PCR products of genomic DNA isolated from affected tissues, blood cells, and fibroblast cells were run through a 1.5% agarose gel and stained with ethidium bromide. Plasmids containing PCR products of wild-type and mutant PIK3CA exon 20 were used as standard reference materials. A 370-bp DNA fragment was generated from the mutant allele of PIK3CA (black arrow) and a 480-bp fragment was generated from both the wild-type and mutant PIK3CA alleles, as the internal control (white arrow). The sizes of the DNA markers are indicated on the left side. The upper right panel shows the standard curve for calculating PIK3CA mutant frequency by multiplex PCR. Lane: 1, blood; 2, skin from the perineum; 3, adipose tissue from the perineum; 4, skin from the lower limb; 5, adipose tissue from lower limb–1; 6, adipose tissue from the chest; 7, adipose tissue from lower limb–2; a, control fibroblast C2; b, control fibroblast C3; c, NHDF-c; d, PROS fibroblast from the skin of the lower limb; e, PROS fibroblast from the skin of the perineum. (C) PIK3CA mutant allele frequencies at nucleotide position 3140 in the patient’s affected tissues and blood lymphocytes were calculated by quantitative multiplex PCR and direct sequencing. The x-axis labels are the same as in (B).
Effect of direct or indirect inhibition of PI3K/AKT/mTOR signaling in fibroblasts
Previous studies demonstrated that mutations in PIK3CA cause gain-of-function of PI3K, which leads to activation of downstream pathways and enhances cellular growth in cancer and a spectrum of overgrowth disorders [5, 19–21]. We hypothesized that inhibitors of the PI3K/AKT/mTOR signaling pathway could be used for effective therapeutic treatment for PROS. To evaluate the therapeutic effect of such compounds, we established dermal fibroblast cell lines from skins of the lower limb and perineum of the presented patient, as an experimental model. It is noted that, the cell lines from the lower limb and perineum have 40.9% and 25.8% mutant allele frequency ratios, respectively, as determined by quantitative multiplex PCR (Figure 2B). Immunoblot analysis revealed that the levels of phosphorylated AKT and ribosomal protein S6 (S6) were increased in PROS fibroblasts as compared to those in normal fibroblasts (Figure 4A). To assess the therapeutic effects on segmental overgrowth of PROS, we treated the two fibroblast lines derived from the patient’s affected skin (PROS fibroblasts) and three normal controls (control fibroblasts) with rapamycin (mTOR inhibitor), NVP-BEZ235 (dual PI3K/mTOR inhibitor), aspirin, and metformin (Figure 3). We used metformin at the concentration used previously for cultured cancer cells [22–26]. First, we analyzed the changes in phosphorylation of the PI3K/AKT/mTOR signaling molecules S6 and AKT following treatment with these agents. Western blotting showed that rapamycin, NVP-BEZ235, and aspirin significantly inhibited phosphorylation of S6 in both PROS and control fibroblasts (Figure 4B and Figure 4C). Compare with rapamycin, metformin mildly reduced the level of phosphorylated S6 only in two of the PROS fibroblast lines (Figure 4C). In addition, NVP-BEZ235 decreased phosphorylation of AKT at Ser473 in PROS fibroblasts (Figure 4B). However, rapamycin induced phosphorylation of AKT at Ser473 in both control and PROS cells (Figure 4B and Figure 4C). Next, cell growth of PROS and control fibroblasts after treatment with each agent was assayed (Figure 5). Rapamycin, NVP-BEZ235, and aspirin showed remarkable growth inhibition of both PROS and control fibroblasts. Metformin also effectively suppressed the growth of both PROS fibroblast lines, which had different frequency of mosaic mutations. However, the effect on the growth of control cells was mild.
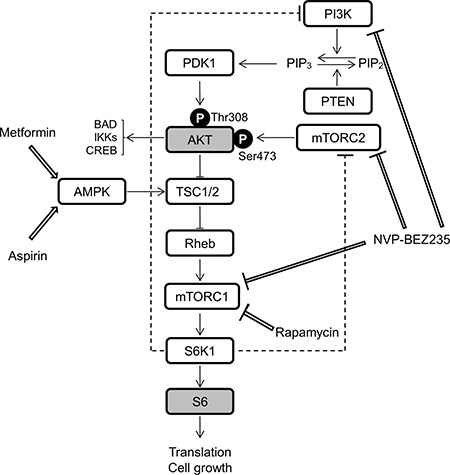
Figure 3: A schematic representation of simplified PI3K/AKT/mTOR signaling and the effects of rapamycin, NVP-BEZ235, aspirin, and metformin. The following abbreviations are used: PTEN, phosphatase and tensin homolog; PDK1, 3-phosphoinositide dependent kinase 1; TSC1/2, tuberous sclerosis complex proteins 1 and 2; Pheb, Ras homolog enriched in brain; S6K1, ribosomal protein S6 kinase 1; BAD, BCL-2-associated death promoter; IKKs, IκB kinases; CREB, cyclic AMP response element binding protein.
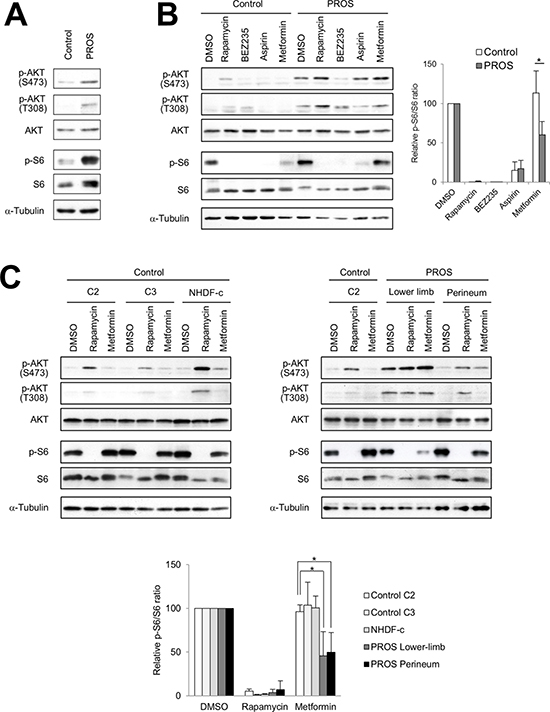
Figure 4: Effect of inhibitors of PI3K/AKT/mTOR signaling in fibroblasts. (A) Immunoblot analysis of phospho-AKT (Ser473), phospho-AKT (Thr308), pan-AKT, phospho-S6 (Ser235/236), and pan-S6 in the PROS fibroblasts from the lower limb of the patient compared with control fibroblasts (C2). (B) Fibroblast cells were seeded 35-mm dished. After 24 hours, the control fibroblasts (C2) and PROS fibroblasts from the lower limb were treated with 1 nM rapamycin, 100 nM NVP-BEZ235, 5 mM aspirin, or 10 mM metformin for 2 days. (C) Control fibroblast lines C2 and C3, NHDF-c, and PROS fibroblast lines from the lower limb and perineum were treated with 1 nM rapamycin or 10 mM metformin for 2 days. In the right panel, extracts from control fibroblast line C2 were loaded as controls. Effects of these agents on the PI3K/AKT/mTOR pathway in fibroblast cells were analyzed by western blotting. α-Tubulin served as a loading control. Representative blots from three independent experiments are shown. All figures show the results of independent experiments. Quantification is presented as the relative ratio of phosphorylated S6 to total S6 in fibroblast cells. The quantification of the band intensity is normalized to the corresponding DMSO control treatment. Data are expressed as mean ± S.D. (n = 3). *p < 0.05, Student’s t-test.
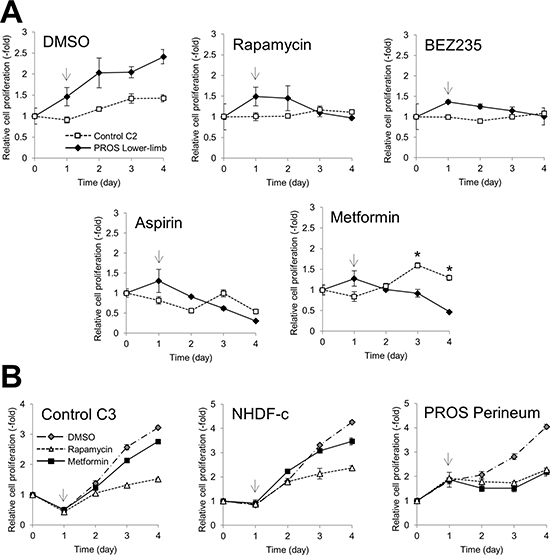
Figure 5: Effect of inhibitors of cell growth in fibroblasts. Cell growth of PROS and control fibroblasts was assessed using the MTT assay. (A) Control fibroblasts C2 and PROS fibroblasts from the lower limb were treated with 1 nM rapamycin, 100 nM NVP-BEZ235, 5 mM aspirin, and 10 mM metformin. (B) Control fibroblast line C3, NHDF-c, and PROS fibroblasts from the perineum were treated with 1 nM rapamycin and 10 mM metformin. Arrows indicate the time point when the inhibitor was administrated. The data are normalized to Day 0. Data are expressed as mean ± S.D. (n = 3). *p < 0.05, Student’s t-test.
To examine the effect of these compounds on PI3K/AKT/mTOR signaling in detail, dose-response studies were carried out on control (C2) and PROS (lower limb) fibroblasts (Figure 6). Western blots from fibroblasts treated with rapamycin show that there was an inverse relationship between the phosphorylation of S6 and AKT. NVP-BEZ235 diminished phosphorylated S6 and AKT at Ser473 in PROS fibroblasts, but inhibition of AKT phosphorylation at Ser473 required higher concentrations of NVP-BEZ235 than that of S6 phosphorylation. Although NVP-BEZ235 also inhibited PI3K activity, phosphorylation of AKT at Thr308, which is phosphorylated by PDK1, was induced by treatment of 100 nM NVP-BEZ235 in PROS fibroblasts. High concentrations of aspirin and metformin (5 mM and 10 mM, respectively) were required for inhibition of S6 phosphorylation on PROS cells. Since it is known that metformin and, in some cases, aspirin can activate AMP-activated protein kinase (AMPK) [27, 28], the phosphorylation state of AMPK in the fibroblasts was investigated. Metformin at concentrations of 1 mM and higher induced phosphorylation of AMPK of PROS and control fibroblasts, while aspirin did not affect AMPK phosphorylation in PROS and control fibroblasts at the concentrations tested.
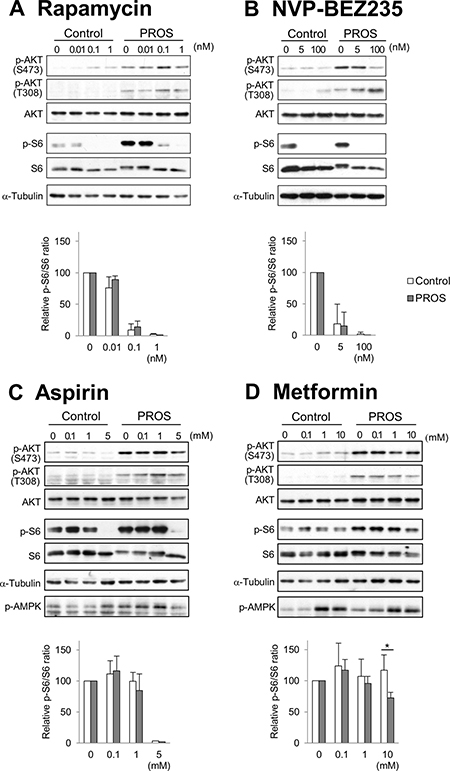
Figure 6: Dose-response study of the effects of inhibitors on PI3K/AKT/mTOR signaling. After seeding the control fibroblasts (C2) and PROS fibroblasts from the lower limb, the cells were treated with different concentrations of rapamycin (A) NVP-BEZ235 (B) aspirin (C) and metformin (D) for 2 days. Activity of PI3K/AKT/mTOR was monitored by examining phosphorylation of AKT-S473, AKT-T308, and S6 by western blotting. AMPK activation was monitored by phosphorylation of threonine 172 with a phosphor-specific antibody. α-Tubulin served as a loading control. Representative blots from three independent experiments are shown. Quantification is presented as the relative ratio of phosphorylated S6 to total S6 in the control (white boxes) and the PROS (gray boxes) fibroblast cells. The quantification of the band intensity is normalized to the corresponding DMSO control treatment. Data are expressed as mean ± S.D. (n = 3). *p < 0.05, Student’s t-tests.
DISCUSSION
We report the characterization of a PROS patient harboring a mosaic PIK3CA (c.3140A>G [p.H1047R]) mutation in the affected tissues. H1047R is the most common amino acid substitution, occurring in a helix domain at the end of the activation loop of the p110α/p85α complex. A recent study demonstrated that the H1047R mutation in PIK3CA causes extensive remodeling of gene signatures, thus this single base mutation has an unexpectedly deep and broad impact on the phenotypic properties of the cell [29]. Therefore, it is important to investigate whether the patient’s tissues contain this mutation. We demonstrate the correlation between mutant allele frequency and the size of adipose tissues in the presented patient. Moreover, we also show that direct sequencing is a useful diagnostic genetic test to identify a mosaic mutation in affected adipose tissue or fibroblast cells, even though these are small overgrown regions.
A major manifestation of PROS is progressive segmental overgrowth in various regions of the body present congenitally or starting in early childhood [13]. Since surgical debulking and orthopedic procedures are currently the only available treatments for patients with segmental overgrowth syndromes [30, 31], it is imperative to develop new therapeutic approaches and effective treatments to combat these syndromes. Recently, the therapeutic effects of small compounds on the inhibition of the PI3K/AKT/mTOR signaling pathway for venous malformation, lymphatic malformations (LMs), and PROS with PIK3CA mutants have been reported [32–34]. These studies have reported the following findings: 1) Treatment with rapamycin and BYL719 (a PI3K p110α specific inhibitor) diminished AKT phosphorylation in human umbilical vein endothelial cells (HUVEC) expressing PIK3CA mutants cultured in a medium containing 10% fetal calf serum (FCS). However, the effect on cell proliferation was not described. 2) Rapamycin and GDC-0941 (a potent inhibitor of PI3K p110α) effectively inhibited the proliferation of both LM-mutant and control lymphatic endothelial cells (LECs). Rapamycin had a stronger effect on proliferation of LM-LECs than that of control LEC, but the effect of rapamycin and GDC-0941 on the phosphorylation of AKT in LM-LECs was not described. 3) Wortmannin and LY294002 (PI3K inhibitors) abrogated the over-activation of AKT and p70S6K in fibroblasts derived from two PROS patients in the absence of serum. Thus, wortmannin and LY294002 decrease growth factor-independent proliferation and AKT phosphorylation. However, the effect of these inhibitors on the growth of normal cells was not described.
Constitutive activation of PI3K is a characteristic feature in PROS patients with PIK3CA mutations and overgrowth of cells in patients’ organs usually occurs with the onset of expression of certain growth factors during development. Previous studies have shown PI3K activation of lymphoblastoid cells harboring PIK3CA mutations cultured in media with 15% FCS [7, 35]. Therefore, we established PROS patient-derived fibroblasts harboring a high frequency (40.9%) and a low frequency (25.8%) of the mutation to study the therapeutic effects of small compounds on control and PROS fibroblasts in medium containing 10% FCS. First, we analyzed the effects of the direct inhibitors of PI3K/AKT/mTOR signaling pathway, rapamycin and NVP-BEZ235, on PROS fibroblasts. Rapamycin acts as a specific inhibitor of mTORC1 (Figure 3) and has synergistic anti-cancer effects [36]. NVP-BEZ235 is a dual PI3K and mTOR inhibitor [37] (Figure 3) and is currently in phase I/II clinical trials for treating advanced solid tumors [38]. In this study, we show that 1 nM rapamycin completely suppressed the phosphorylation of S6 and the cell growth of both control and PROS fibroblasts (Figures 4, 5, and 6). Thus, the PROS fibroblasts are sensitive to a low concentration of rapamycin, which is a similar dose as is used to treat rapamycin-sensitive cancer cells [39]. In this model, rapamycin increased phosphorylated AKT, likely as a result of rapamycin-induced feedback activation of AKT signaling [40] (Figures 3, 4C, and 6A). Thus, PI3K/AKT signaling upstream of mTOR is more highly activated in PROS fibroblasts than in control cells, leading to the activation of the collateral pathways upstream of mTOR. Similarly, 5 nM NVP-BEZ235 inhibited mTOR activity of control and PROS fibroblasts by completely suppressing phosphorylation of S6 while slightly increasing the phosphorylation of AKT at Thr308 in PROS fibroblasts (Figures 3, 4B, and 6B). This phenomenon could be explained by induction of feedback activation of AKT signaling by mTOR inhibition. Importantly, the findings of this study are similar to those of a previously reported study on cancer cells with a PI3K active mutation [41]. Thus, rapamycin and NVP-BEZ235 are therapeutically effective to treat PROS, but these inhibitors suppress cell growth of both PROS cells and control cells. Moreover, side effects such as mucositis, elevation of ALT/AST, hypercholesterolemia, headache, and neutropenia were reported in the treatment of complicated vascular anomalies in children with rapamycin [42]. These findings indicate that administration of both compounds should be carefully performed to avoid serious side effects in growing children.
Next, we analyzed the effect of indirect inhibitors of the PI3K/AKT/mTOR signaling pathway aspirin and metformin on control and PROS fibroblasts. Aspirin is an anti-inflammatory drug and has been shown to increase the five-year survival rate of patients with several types of cancer [43], including colorectal cancer harboring PIK3CA mutations in exons 9 and 20 [44, 45]. Aspirin inhibits cyclooxygenase (COX), leading to reduction of prostaglandin production [46]. Recent studies have shown that aspirin inhibits mTOR signaling by activating AMPK in cancer cells [28, 47] (Figure 3). We demonstrated that 5 mM aspirin completely blocked the phosphorylation of S6 and inhibited cell growth of control and PROS fibroblasts, but we did not observe phosphorylation of AMPK in PROS cells (Figures 4B, 5A, and 6C). Importantly, the aspirin concentration (5 mM) used in this study is similar to the effective aspirin concentration (1 to 3 mM) in the blood of patients taking high-dose aspirin (100 mg/kg/day) for type 2 diabetes and Kawasaki disease [48, 49]. However, the use of high-dose aspirin may increase the risk of gastrointestinal hemorrhage [50], sensorineural hearing loss [51], and Reye syndrome [52] in children. Metformin (1,1-dimethylbiguanide hydrochloride), a biguanide derivate, is the drug most commonly used in treating type 2 diabetes [53]. Previous epidemiological studies have demonstrated that metformin can reduce the risk of cancer in diabetic patients [54, 55]. Recently, metformin has been shown to decrease the risk of several cancers, including colon, breast, prostate, and pancreatic cancers [56]. One of the molecular mechanisms underlying the anti-cancer effects of metformin is the activation of AMPK (Figure 3), which results in suppression of mTOR signaling [56]. In this study, we found that 10 mM metformin treatment activates AMPK and reduces S6 phosphorylation without affecting AKT phosphorylation in both control and PROS fibroblast cells (Figure 6D). Importantly, metformin strongly inhibited cell growth of the PROS fibroblasts containing either high or low frequency of the mosaic H1047R mutation, while the effect on the three control cell lines was mild (Figure 5). Compared with the other three compounds, metformin had a milder effect of suppressing phosphorylation of S6 in PROS cells (Figure 4 and Figure 6). Therefore, the suppression of the phosphorylation of S6 is difficult to explain in light of the selective cell growth inhibition of PROS cells by metformin, and so, other molecular mechanisms may be involved in the selective growth inhibition. A high concentration of metformin (~10 mM) was necessary to suppress the growth of PROS fibroblasts in this study likely because of the fact that metformin does not passively diffuse through cell membranes because of its low lipophilicity and so it must be transported into cells by the transmembrane protein organic cation transporters 1-3 [57–59]. Similarly, a concentration of at least 1 mM metformin was required to suppress the growth of two types of endometrial cancer-derived cell lines, one of which has a mutation in PTEN in the PI3K/AKT/mTOR signaling pathway [60] (Figure 3). On the other hand, a recent clinical study on patients with endometrial cancer has also shown that oral administration of 1500–2250 mg/day of metformin for 4 weeks resulted in a significantly reduced expression of the cell proliferation marker Ki-67 and decreased phosphorylated S6 in endometrial cancer tissues [60]. The metformin concentrations in the plasma and endometrial cancer tissues of these patients were 6.8 to 18.1 μM and 1.2 to 5.1 μmol/kg wet weight, respectively [60]. Although we do not have sufficient evidence to explain the difference in effective metformin concentrations, low concentrations having been reported in patients’ blood and tissue compared with high concentrations required for efficacy in cell culture medium for PROS fibroblasts in this study, it is possible that metformin acts on PROS fibroblasts with different frequencies of mosaic mutations in the same manner as it does in cancer cells [60]. In a randomized clinical trial of metformin to treat obese insulin-resistant children with dosages ranging from 500 to 2000 mg/day, side effects such as nausea, loose stool, and fatigue were reported, but serious or life-threatening events were not noted [61]. Therefore, administration of regular daily doses of metformin is one of the candidate treatments for PROS patients.
In summary, we studied the effect of four small compounds, rapamycin, NVP-BEZ235, aspirin, and metformin on control and PROS fibroblasts and demonstrated that all four compounds could suppress the activated PIK3/AKT/mTOR signaling pathway and the growth of PROS patient-derived fibroblasts by suppressing phosphorylation of S6. However, only metformin showed mild inhibition of the growth of control fibroblasts. Since PROS is a developmental disorder in children, metformin is a candidate drug for treating PROS. However, further investigation is necessary to elucidate the molecular mechanism governing the higher sensitivity of cell growth of PROS fibroblasts to metformin than that of control cells and to determine the optimal concentration of metformin required to effectively suppress the growth of mutant cells in PROS patients.
MATERIALS AND METHODS
Bioethics approval
Written informed consent was obtained from the family members who participated in this study. The study was conducted after approval from the institutional review board at the Institute for Developmental Research, Aichi Human Service Center and in accordance with the principles embodied in the Declaration of Helsinki.
Identification of somatic mutation
Genomic DNA was isolated from seven different regions, including subcutaneous adipose tissue from the chest, lower limb, and perineum, skin from the lower limb and perineum, and white blood cells by phenol/chloroform extraction. To identify the mutations in PIK3CA, DNA fragments including all exons and exon-intron junctions of PIK3CA were amplified from genomic DNA by 36 cycles of polymerase chain reaction (PCR) using specific primer sets. The resulting PCR products were separated by agarose gel electrophoresis, purified with QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany), and then directly sequenced using the GenomeLab GeXP Genetic Analysis System (AB SCIEX, Framingham, MA, USA) and the GenomeLab Dye Terminator Cycle Sequencing with Quick Start Kit (Beckman Coulter, Brea, CA, USA) with 50 to 100 fmol of template DNA. For quantification, 380 bp of wild-type and mutant PCR products of the PIK3CA gene including exon 20 were amplified from genomic DNA isolated from lower limb adipose tissue by PCR using a specific primer set (sense S1: 5′-caatgatgcttggctctgga-3′; antisense A1: 5′-tcaaaccctgtttgcgtttac-3′) and subcloned into pGEM-T Easy vector (Promega, Madison, WI, USA).
Determination of H1047R frequency in tissues
To determine frequencies of the missense mutation of PIK3CA gene (c.3140A>G) in patient tissues, we established a quantification method for determining the dosage of genomic DNA using multiplex PCR (Figure 2B). Specific primer pairs (mismatch sense S2: 5′-atgaaacaa atgaatgatgcaag-3′ and antisense A2: 5′-acaaacaatcttcaaagt ttacct-3′; --cacg was changed to --caag) were designed to specifically amplify a 370 bp DNA fragment containing only the mutant PIK3CA gene (c.3140A>G). Another pair of primers (S1 and A2) were also designed to amplify a 480 bp genomic DNA fragment containing wild-type and mutant PIK3CA. Aliquots (0.05 μg) of genomic DNA from each affected tissue and a normal blood sample were amplified by PCR in a total volume of 20 μL, containing 0.45 μM of total primers, 30 mM of each dNTP, 2.5 mM MgCl2, 10 mM Tris–HCl (pH 8.3), 50 mM KCl, and 1 U of AmpliTaq-Gold (Applied Biosystems). The ratio of the concentrations of the three primers was 1 : 4 : 5 (S1 : S2 : A2). PCR samples were preheated at 94°C for 10 min and 32 PCR cycles were performed. Each cycle consisted of 30 s denaturation at 94°C, 30 s annealing at 56°C, and 30 s extension at 72°C. The PCR products were electrophoresed on 1.5% agarose gels and band intensities were measured using Image Quant TL 7.0 software (GE healthcare).
Treatment of PROS fibroblast cells with inhibitors of the PI3K/AKT/mTOR signaling pathway
Two PROS fibroblast cell lines were established from the patient’s lower limb and perineum. Two control fibroblast cell lines (C2 and C3), which were previously established from the skins of two unrelated healthy volunteers of a 36-year-old male and a 27-year-old female, respectively, [62] were used. Additionally, a normal human dermal fibroblast cell line established from the skin of a 14-year-old male (NHDF-c, c-12300) was purchased from PromoCell GmbH (Heidelberg, Germany). Fibroblast cells were cultured as previously described [62]. Briefly, fibroblasts were maintained in Eagle’s minimal essential medium (EMEM; Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% FCS, penicillin (100 U/mL), streptomycin (0.1 mg/mL), L-glutamine (1.75 mM), HEPES (10 mM), and NaHCO3 (0.13%). All supplemental reagents, except for FCS, were purchased from Nacalai Tesque (Kyoto, Japan). The cells were plated on 35-mm collagen-coated dishes at a density of 1.1 × 104 cells/cm2 for biochemical experiments. Fibroblast cells were treated with rapamycin (LKT Laboratories, St. Paul, MN, USA), NVP-BEZ235 (Chemscene, Monmouth Junction, NJ, USA), aspirin (acetylsalicylic acid; Wako, Osaka, Japan) and metformin hydrochloride (Sigma-Aldrich) for 2 days, washed with PBS, and then harvested and subjected to SDS-PAGE and western blotting.
Western blotting
Samples were electrophoresed on 11% SDS polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Merck Millipore, Darmstadt, Germany). The membranes were incubated in 5% bovine serum albumin in Tris-buffered saline containing Tween-20 (50 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.03% Tween-20) and then incubated with primary antibodies anti-pan-AKT, anti-phospho-AKT (Ser473), anti-phospho-AKT (Thr308), anti-pan-S6, anti-phospho-S6 (Ser235/236) (Cell Signaling Technologies, Danvers, MA, USA), and anti-α-tubulin (Sigma-Aldrich), followed by incubation with a horseradish peroxidase-conjugated secondary antibody. Immunoreactive proteins were visualized with Western Lightning Plus-ECL (PerkinElmer, MA, USA). Densitometric measurement of bands on western blotting was performed using ImageJ software (US National Institutes of Health, Bethesda, MD, USA).
MTT assay
Cell growth was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, fibroblast cells were seeded at a density of 3.1 × 104 cells/cm2 onto collagen-coated 96-well culture plates. Each day, 10 μL of MTT solution (5 mg/mL in PBS) was added to each well. After incubation for 2 h at 37°C, the purple-blue MTT formazan precipitate was dissolved in 200 μL of MTT solvent (40 mM HCl in isopropanol) and the absorbance was measured with a multiplate reader at 570 nm.
Statistical analysis
Data are expressed as means ± SD. Statistical analysis was performed by Student’s t-test. A p value of < 0.05 was considered to be statistically significant.
Abbreviations
AMPK, AMP-activated protein kinase; COX, cyclooxygenase; mTOR, mammalian target of rapamycin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PI3K, phosphatidylinositol 3-kinase; PIK3CA, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PROS, PIK3CA-related overgrowth spectrum; S6, ribosomal protein S6.
Authors’ contributions
YS contributed to the acquisition, analysis, and interpretation of data and the drafting of the manuscript. YE established the patient-derived fibroblasts cell lines. KY provided technical expertise with genetic analysis and drafting of the manuscript. MI, KK, NH, TM, and SM provided clinical data and biopsy samples obtained by therapeutic surgery from the patient. NW is accountable for design of the work, interpretation of data, and revision for intellectual content.
ACKNOWLEDGMENTS
The authors are grateful to the patient and her family for agreeing to participate in this study. We also wish to acknowledge Noriko Nomura and Arisa Yamano for helpful assistance.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported by Takeda Science Foundation a grant for specific research No.10 in 2009 (to NW) and JSPS KAKENHI Grant Numbers JP15K06764 (to YS) and JP15K09610 (to KY).
REFERENCES
1. Lindhurst MJ, Parker VE, Payne F, Sapp JC, Rudge S, Harris J, Witkowski AM, Zhang Q, Groeneveld MP, Scott CE, Daly A, Huson SM, Tosi LL, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012; 44:928–933.
2. Biesecker LG, Peters KF, Darling TN, Choyke P, Hill S, Schimke N, Cunningham M, Meltzer P, Cohen MM Jr. Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am J Med Genet. 1998; 79:311–318.
3. Sapp JC, Turner JT, van de Kamp JM, van Dijk FS, Lowry RB, Biesecker LG. Newly delineated syndrome of congenital lipomatous overgrowth, vascular malformations, and epidermal nevi (CLOVE syndrome) in seven patients. Am J Med Genet A Paet. 2007; 143A:2944–2958.
4. Alomari AI. Characterization of a distinct syndrome that associates complex truncal overgrowth, vascular, and acral anomalies: a descriptive study of 18 cases of CLOVES syndrome. Clin Dysmorphol. 2009; 18:1–7.
5. Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, Warman ML. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012; 90:1108–1115.
6. Rios JJ, Paria N, Burns DK, Israel BA, Cornelia R, Wise CA, Ezaki M. Somatic gain-of-function mutations in PIK3CA in patients with macrodactyly. Hum Mol Genet. 2013; 22:444–451.
7. Rivière JB, Mirzaa GM, O’Roak BJ, Beddaoui M, Alcantara D, Conway RL, St-Onge J, Schwartzentruber JA, Gripp KW, Nikkel SM, Worthylake T, Sullivan CT, Ward TR, et al. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012; 44:934–940.
8. Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012; 44:941–945.
9. Groesser L, Herschberger E, Landthaler M, Hafner C. FGFR3, PIK3CA and RAS mutations in benign lichenoid keratosis. Br J Dermatol. 2012; 166:784–788.
10. Hafner C, Lopez-Knowles E, Luis NM, Toll A, Baselga E, Fernandez-Casado A, Hernandez S, Ribe A, Mentzel T, Stoehr R, Hofstaedter F, Landthaler M, Vogt T, et al. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc Natl Acad Sci U S A. 2007; 104:13450–13454.
11. Maclellan RA, Luks VL, Vivero MP, Mulliken JB, Zurakowski D, Padwa BL, Warman ML, Greene AK, Kurek KC. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast Reconstr Surg. 2014; 133:12e–9e.
12. Keppler-Noreuil KM, Sapp JC, Lindhurst MJ, Parker VE, Blumhorst C, Darling T, Tosi LL, Huson SM, Whitehouse RW, Jakkula E, Grant I, Balasubramanian M, Chandler KE, et al. Clinical delineation and natural history of the PIK3CA-related overgrowth spectrum. Am J Med Genet A. 2014; 164:1713–1733.
13. Keppler-Noreuil KM, Rios JJ, Parker VE, Semple RK, Lindhurst MJ, Sapp JC, Alomari A, Ezaki M, Dobyns W, Biesecker LG. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. Am J Med Genet A. 2015; 167:287–295.
14. Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, Milas K, Pederson H, Remzi B, Orloff MS, Eng C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011; 88:42–56.
15. Lindhurst MJ, Sapp JC, Teer JK, Johnston JJ, Finn EM, Peters K, Turner J, Cannons JL, Bick D, Blakemore L, Blumhorst C, Brockmann K, Calder P, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011; 365:611–619.
16. Hussain K, Challis B, Rocha N, Payne F, Minic M, Thompson A, Daly A, Scott C, Harris J, Smillie BJ, Savage DB, Ramaswami U, De Lonlay P, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011; 334:474.
17. Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012; 74:41–48.
18. Martinez-Lopez A, Blasco-Morente G, Perez-Lopez I, Herrera-Garcia JD, Luque-Valenzuela M, Sanchez-Cano D, Lopez-Gutierrez JC, Ruiz-Villaverde R, Tercedor-Sanchez J. CLOVES syndrome: review of a PIK3CA-related overgrowth spectrum (PROS). Clin Genet. 2017; 91:14–21.
19. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004; 304:554.
20. Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA. 2005; 102:802–807.
21. Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci USA. 2006; 103:1475–1479.
22. Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66:10269–10273.
23. Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9:1057–1064.
24. Sikka A, Kaur M, Agarwal C, Deep G, Agarwal R. Metformin suppresses growth of human head and neck squamous cell carcinoma via global inhibition of protein translation. Cell Cycle. 2012; 11:1374–82.
25. Nangia-Makker P, Yu Y, Vasudevan A, Farhana L, Rajendra SG, Levi E, Majumdar AP. Metformin: a potential therapeutic agent for recurrent colon cancer. PLoS One. 2014; 9:e84369.
26. Huang D, He X, Zou J, Guo P, Jiang S, Lv N, Alekseyev Y, Luo L, Luo Z. Negative regulation of Bmi-1 by AMPK and implication in cancer progression. Oncotarget. 2016; 7:6188–6200. doi: 10.18632/oncotarget.6748.
27. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001; 108:1167–1174.
28. Din FV, Valanciute A, Houde VP, Zibrova D, Green KA, Sakamoto K, Alessi DR, Dunlop MG. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology. 2012; 142:1504–1515.e3.
29. Hart JR, Zhang Y, Liao L, Ueno L, Du L, Jonkers M, Yates JR 3rd, Vogt PK. The butterfly effect in cancer: a single base mutation can remodel the cell. Proc Natl Acad Sci USA. 2015; 112:1131–1136.
30. Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006; 14:1151–1157.
31. Tosi LL, Sapp JC, Allen ES, O’Keefe RJ, Biesecker LG. Assessment and management of the orthopedic and other complications of Proteus syndrome. J Child Orthop. 2011; 5:319–327.
32. Limaye N, Kangas J, Mendola A, Godfraind C, Schlögel MJ, Helaers R, Eklund L, Boon LM, Vikkula M. Somatic Activating PIK3CA Mutations Cause Venous Malformation. Am J Hum Genet. 2015; 97:914–921.
33. Osborn AJ, Dickie P, Neilson DE, Glaser K, Lynch KA, Gupta A, Dickie BH. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Hum Mol Genet. 2015; 24:926–938.
34. Loconte DC, Grossi V, Bozzao C, Forte G, Bagnulo R, Stella A, Lastella P, Cutrone M, Benedicenti F, Susca FC, Patruno M, Varvara D, Germani A, et al. Molecular and Functional Characterization of Three Different Postzygotic Mutations in PIK3CA-Related Overgrowth Spectrum (PROS) Patients: Effects on PI3K/AKT/mTOR Signaling and Sensitivity to PIK3 Inhibitors. PLoS One. 2015; 10:e0123092.
35. Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, Eng C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. 2013; 92:76–80.
36. Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000; 19:6680–6686.
37. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008; 7:1851–1863.
38. Bellozi PM, Lima IV, Dória JG, Vieira ÉL, Campos AC, Candelario-Jalil E, Reis HJ, Teixeira AL, Ribeiro FM, de Oliveira AC. Neuroprotective effects of the anticancer drug NVP-BEZ235 (dactolisib) on amyloid-β 1-42 induced neurotoxicity and memory impairment. Sci Rep. 2016; 6:25226.
39. Sawyers CL. Will mTOR inhibitors make it as cancer drugs? Cancer Cell. 2003; 4:343–348.
40. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007; 26:1932–1940.
41. Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008; 68:8022–8030.
42. Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, Dasgupta R, Azizkhan RG, Adams DM. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011; 57:1018–1024.
43. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011; 377:31–41.
44. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K, Sun R, Nosho K, Meyerhardt JA, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012; 367:1596–1606.
45. Domingo E, Church DN, Sieber O, Ramamoorthy R, Yanagisawa Y, Johnstone E, Davidson B, Kerr DJ, Tomlinson IP, Midgley R. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J Clin Oncol. 2013; 31:4297–305.
46. Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol. 1971; 231:232–235.
47. Gao M, Kong Q, Hua H, Yin Y, Wang J, Luo T, Jiang Y. AMPK-mediated up-regulation of mTORC2 and MCL-1 compromises the anti-cancer effects of aspirin. Oncotarget. 2016; 7:16349–16361. doi: 10.18632/oncotarget.7648.
48. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J Clin Invest. 2002; 109:1321–1326.
49. Palazzi DL, McClain KL, Kaplan SL. Hemophagocytic syndrome after Kawasaki disease. Pediatr Infect Dis J. 2003; 22:663–666.
50. Matsubara T, Mason W, Kashani IA, Kligerman M, Burns JC. Gastrointestinal hemorrhage complicating aspirin therapy in acute Kawasaki disease. J Pediatr. 1996; 128:701–703.
51. Sundel RP, Newburger JW, McGill T, Cleveland SS, Miller WW, Berry B, Klein AM, Burns JC. Sensorineural hearing loss associated with Kawasaki disease. J Pediatr. 1990; 117:371–377.
52. Ninove L, Daniel L, Gallou J, Cougard PA, Charpentier A, Viard L, Roquelaure B, Paquis-Flucklinger V, de Lamballerie X, Zandotti C, Charrel RN. Fatal case of Reye’s syndrome associated with H3N2 influenza virus infection and salicylate intake in a 12-year-old patient. Clin Microbiol Infect. 2011; 17:95–97.
53. Stumvoll M, Nurjhan N, Perriello G, Dailey G, Gerich JE. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995; 333:550–554.
54. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005; 330:1304–1305.
55. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006; 29:254–258.
56. Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. 2012; 122:253–270.
57. Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002; 302:510–515.
58. Kimura N, Masuda S, Tanihara Y, Ueo H, Okuda M, Katsura T, Inui K. Metformin is a superior substrate for renal organic cation transporter OCT2 rather than hepatic OCT1. Drug Metab Pharmacokinet. 2005; 20:379–386.
59. Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R, Zanger UM, Keppler D, Schwab M, Schaeffeler E. Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology. 2009; 50:1227–1240.
60. Mitsuhashi A, Kiyokawa T, Sato Y, Shozu M. Effects of metformin on endometrial cancer cell growth in vivo: a preoperative prospective trial. Cancer. 2014; 120:2986–2995.
61. Yanovski JA, Krakoff J, Salaita CG, McDuffie JR, Kozlosky M, Sebring NG, Reynolds JC, Brady SM, Calis KA. Effects of metformin on body weight and body composition in obese insulin-resistant children: a randomized clinical trial. Diabetes. 2011; 60:477–485.
62. Yamada K, Aiba K, Kitaura Y, Kondo Y, Nomura N, Nakamura Y, Fukushi D, Murayama K, Shimomura Y, Pitt J, Yamaguchi S, Yokochi K, Wakamatsu N. Clinical, biochemical and metabolic characterisation of a mild form of human short-chain enoyl-CoA hydratase deficiency: significance of increased N-acetyl-S-(2-carboxypropyl)cysteine excretion. J Med Genet. 2015; 52:691–698.