
Background
Non-small cell lung cancer - standard therapy and outcomes
Lung cancer is the leading cause of cancer mortality worldwide accounting for 24% of global cancer related deaths. Each year, in North America over 250,000 patients are diagnosed with lung cancer and 180,000 die from the disease (American and Canadian Cancer Societies, 2016) [1]. Non-Small Cell Lung Cancer (NSCLC) represents 80% of all cases and includes three main histologies, adenocarcinomas, squamous cell carcinomas and large cell carcinomas. Surgery [2, 3] and modern stereotactic body radiotherapy (RT) (SBRT) [4] provide reasonable control for early stage tumors. However, locally advanced NSCLC is frequently in-operable and treated with combined Chemo-RT [5], with poor outcomes [6]. Current standard chest RT for locally advanced NSCLC (60-63Gy in 30 fractions) was developed more than 30 years ago [7] and is associated with 60-85% rates of recurrence [8]. Platinum-based concurrent Chemo-RT, established in late 1990s as standard therapy for locally advanced NSCLC [9, 10], shows a median survival of 24-28 months in studies with modern radiotherapy [11]. Unfortunately, escalation of RT dose (74Gy) failed to show benefit in phase III studies (RTOG-0617) [11]. There is an obvious need for well-tolerated therapies to enhance the response of NSCLC to Chemo-RT.
Molecular pathways of cancer cell survival and radio-resistance
The Epidermal Growth Factor Receptor (EGFR), the oncogene K-Ras, the tumor suppressors TP53 and Liver Kinase B 1 (LKB1) are the most frequently mutated genes in NSCLC [12]. EGFR is an established driver of growth and survival (Figure 1A). It is mutated in 7-19% of adenocarcinomas and over-expressed in up to 50% of lung cancers [12]. EGFR inhibitors have been investigated intensively in the last 15 years [13]. Pre-clinical studies showed radio-sensitization of NSCLC to such inhibitors [14], and phase II studies showed promising results (IDEAL 1-2). However, phase III trials (INTACT-1/2) did not show improved response to chemotherapy [13]. Although the chimerized monoclonal antibody against EGFR cetuximab showed promising results in a phase II study (RTOG 0324) [15], it did not demonstrate benefit with either standard (60Gy) or dose-escalated (74Gy) Chemo-RT in phase III trials (RTOG-0617) [6].
The oncogene Ras is activated by EGFR to attract and activate multiple effector pathways promoting growth, survival and resistance to cytotoxic therapy (Figure 1A). Of the three mammalian Ras genes (H, K and N), K-Ras has the highest frequency of mutations in NSCLC (6.5% in squamous cell and 26% of adenocarcinomas, in Western patients) [12]. Lung adenocarcinomas associated with smoking reveal mutations in codons 12 or 13 of this gene, involving typically G-T transitions. K-Ras activates Phosphatidylinositol 3-kinase (PI3k) and Akt, which regulate the mammalian Target of Rapamycin (mTOR) complex 1 (mTORC1) [16] (see Figure 1A). Preclinical studies with PI3k and mTOR inhibitors did show sensitization of NSCLC xenografts to radiation [17, 18]. Currently, there is no evidence of clinical benefit of such inhibitors in combination with RT or standard Chemo-RT in lung cancer. mTOR inhibitors are reported to have a significant risk for pneumonitis (relative risk of 31 for mild- and 8.8 for grade 3-4) [19], which questions their use in lung cancer, particularly in combination with RT. These agents are suspected to have failed to show benefit due to a feedback activation of Akt, leading to resistance to cancer therapies [20].
Radiotherapy: DNA damage-activated signaling pathways
RT kills cancer cells through lethal DNA double strand breaks (DSB). Sub-lethal doses permit DNA repair and survival initiated through detection of DNA breaks by the MRE11 complex and the kinase Ataxia Telengiectasia Mutated (ATM) [21-23] (Figure 1B). ATM and ATR (ATM and Rad3-related) regulate genotoxic stress-induced cell cycle checkpoints to facilitate DNA repair and preserve genomic stability [24], through phosphorylation and activation of p53 [25] and checkpoint kinases (Chk1 and Chk2) [26]. p53 and cyclin-dependent kinase (CDK) inhibitor p21cip1 mediate G1-S and G2-M checkpoints through, Cdk2-cyclin E [27] and Cdk1-cyclin B [28], respectively.
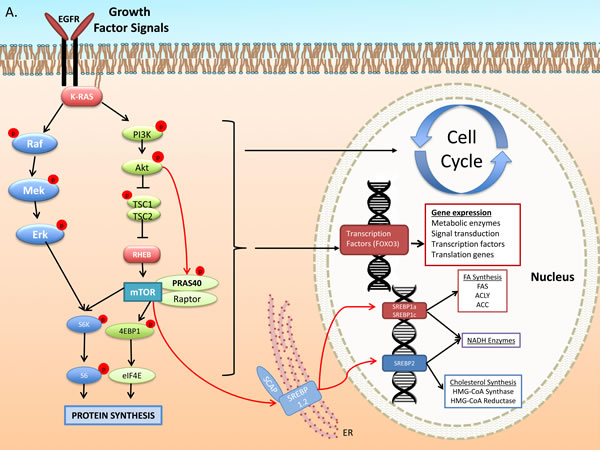
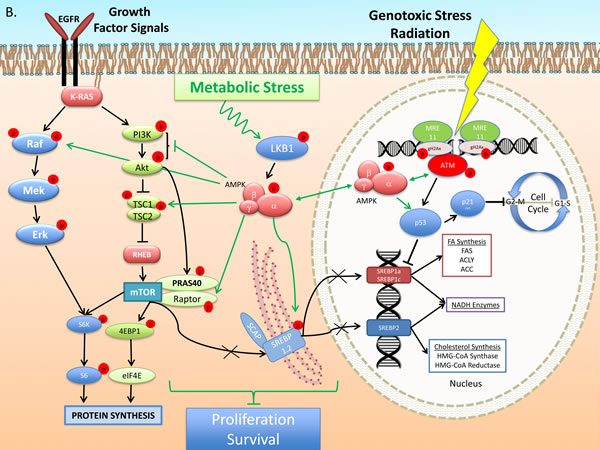
Figure 1: Growth Factor and DNA Damage Response (DDR) activated signal transduction: regulation of cell cycle, and metabolic gene expression. A. The Epidermal Growth Factor Receptor (EGFR) activates signaling pathways that mediate, gene expression, protein and lipid synthesis, growth of cellular biomass and survival after cytotoxic therapy [146]. EGFR transduces signals through the well-described kinase pathways initiated by Ras, including the Raf/Mek/Erk and Phosphatidyl-inositol 3-kinase (PI3k)/Akt/mammalian Target of Rapamycin (mTOR). Akt activates mTOR through, i) phosphorylation and inhibition of Tuberous Sclerosis Complex 2 (TSC2), which inactivates the mTOR activating GTP-binding protein Rheb and/or ii) phosphorylation of PRAS40 a member of mTOR complex 1 (mTORC1) (one of the two functional complexes of mTOR that includes mLST8/Gbl and the scaffold protein Raptor [147]). mTORC1 promotes Cap-dependent gene expression and translation through phosphorylation-mediated activation of p70S6-kinase (p70s6k) and phosphorylation-mediated inhibition of translation initiation inhibitor eIF4E binding protein 1 (4EBP1) [146, 147]. The PI3k-mTOR pathway stimulates glucose uptake and de novo lipogenesis (DNL). Regulation of the latter is mediated at the transcriptional, translational and post-translational level. PI3k-Akt facilitate cleavage of Sterol Regulatory Element-Binding Proteins (SREBP) 1a,c and 2 [148] which, translocate to nucleus to stimulate expression of lipogenic enzymes mediating both fatty acid and cholesterol synthesis. Transcription of ACLY, ACC1 and FASN is stimulated by the PI3k-Akt pathway through SREBPs [149, 150]. SREBP1a and 1c regulate genes involved in FA metabolism while SREBP2 regulates cholesterol synthesis genes [150]. B. Metabolic stress activates a metabolic stress response signals through mediators, such as Liver Kinase B 1 (LKB1), which phosphorylates and activates AMP-activated kinase (AMPK). AMPK suppresses mTOR activation through dual action to phosphorylate and enhance the activity of Tuberous Sclerosis Complex 1 and 2 (an inhibitor of mTOR) and through inhibitory phosphorylation of mTORC1 protein Raptor. Further, it suppresses lipogenic gene transcription through inhibitory SREBP phosphorylation that prevents cleavage and nuclear translocation to induce transcription. Genotoxic Stress induced by ionizing radiation activates the DNA Damage Response (DDR). Chromosomal damage is recognized by MRE11 complex and ATM, a key mediator of DDR, which phosphorylates histone H2Ax (γH2Ax), a step leading to recruitment of molecular DNA repair complexes at the sites of strand breaks. ATM induces p53 expression and phosphorylation and expression of the cyclin dependent kinase inhibitor p21cip1, which facilitates the G1-S and G2-M checkpoints. Further, p53 blocks lipogenic gene expression at the transcriptional level. In lung cancer cells ATM induction of p53-p21cip1 appears to be mediated through AMPK [89]. Genotoxic stress, such as radiation, induces in lung cancer cells and tumors sustained activation of the ATM-AMPK-p53-p21cip1 and suppression of the Akt-mTOR pathways [73, 89, 151].
Cancer Metabolism
Hypoxia of the tumor microenvironment stimulates anaerobic metabolism in tumors, but even in the presence of oxygen tumors maintain anaerobic glycolysis, a paradox that Otto Warburg described nine decades ago [29, 30]. Today, studies continue to clarify the degree of metabolic adaptation taking place in cancer cells to meet the demands of rapid proliferation. Cancer cells exhibit enhanced glucose, protein and lipid metabolism, stimulated by the EGFR-PI3k-Akt-mTOR pathway [30]. These events lead to the Warburg phenotype to support unrestrained cell division. The enhanced metabolic activity of tumors contributes to cancer diagnosis and staging. For example, the high levels of glucose uptake in solid tumors forms the basis of positron emission tomography (PET), using 18F-labeled 2-deoxy-D-glucose (F18-FDG) as a marker of glucose uptake and tumor metabolic activity.
Glucose transport and glycolysis
Epithelial tumor cells express increased number of plasma membrane facilitative glucose transporters (GLUTs) [31] and glycolytic enzymes to mediate enhanced glucose uptake and catabolism (Figure 2). Loss of p53 and activation of the Akt pathway is critical in promoting the glycolytic phenotype of cancer cells by stimulating, i) transcription and translation of GLUT1 glucose transporter in cancer models [32, 33] and ii) hexokinase activity and localization with the mitochondria. This localization commits glucose to glycolysis once it is phosphorylated by hexokinase to glucose-6-phosphate, and is involved in Akt-mediated prevention of early apoptotic events [34].
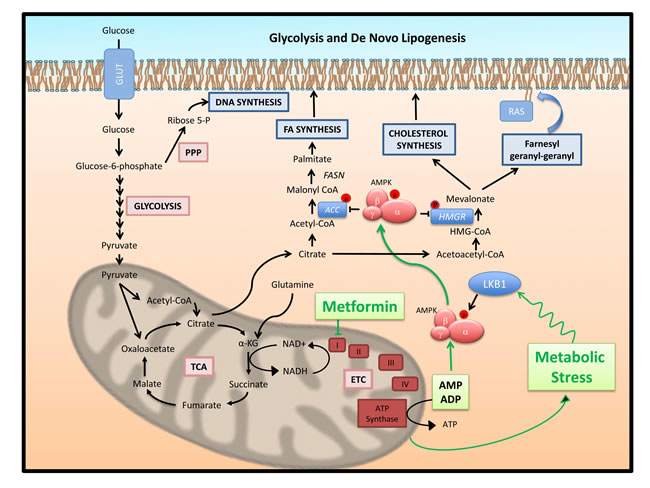
Figure 2: Glucose metabolism and lipogenesis, post-translational regulation by AMPK: Mechanism of action of metformin. Glucose provides cancer cells with substrates not only for energy production but also for biosynthesis of proteins, nucleic acids and fats. Anaerobic glycolysis generates, (i) ribose 5-phosphate to support nucleotide biosynthesis through the pentose phosphate pathway and (ii) pyruvate to generate Acetyl CoA to feed the Krebs (TCA) cycle. The TCA cycle supports the electron transport chain (ETC) to produce ATP from AMP and ADP. However, TCA produced citrate is also transported to the cytoplasm by the tricarboxylate transporter to be cleaved by ATP Citrate Lyase (ACLY) and generate cytoplasmic acetyl-CoA. This is a crucial common step for the initiation of fatty acid (FA) and cholesterol synthesis. A rate limiting step in FA synthesis is the generation of malonyl-CoA from acetyl-CoA, catalyzed by Acetyl-CoA Carboxylase (ACC). Malonyl-CoA is further converted to palmitic acid by fatty acid synthase (FASN) [36]. On the other hand, conversion of acetyl-CoA to acetoacetyl-CoA, HMG-CoA and finally mevalonate initiates the pathway of synthesis of cholesterol and isoprenoids. A rate limiting step of this pathway is the conversion of HMG-CoA to mevalonate catalyzed by HMG-CoA reductase (HMGR). Products of the mevalonate pathway are important for tumor cell growth. Cholesterol is required for structure and function of plasma membranes, while isoprenoids (farnesyl-PP and geranyl-PP diphosphates) mediate the post-translational modification of oncogenes such as Ras [38]. Metformin mediates in cells a state of mild metabolic stress through inhibition of aerobic energy production at the mitochondria. It blocks the oxidative phosphorylation (OxPhos) complex I leading to enhancement of cellular AMP/ADP levels and stimulation of AMPK activity through binding to the enzyme’s γ-subunit. The metabolic stress induced by metformin also induces LKB1 induced AMPK phosphorylation on AMPKα subunit T172 leading to greater activation. Through AMPK metformin blocks energy consuming biosynthetic pathways such as DNL. At the post-translational level, activated AMPK phosphorylates and inhibits both ACC and HMGR, leading to blockade of both fatty acid and cholesterol synthesis. Metformin blocks lipogenesis and this may be one of the key actions mediating its cytostatic activity.
De novo lipogenesis (DNL)
Tumor cells activate endogenous lipid bio-synthesis to maintain fatty acid and cholesterol levels required for elevated rates of cellular replication. Early cancer research suggested that this takes place despite adequate extracellular lipid supply [35, 36]. Energy storage is perhaps the least important function of DNL in cancer since this function supports, i) the supply of building blocks for membrane biogenesis, ii) second messenger function, and iii) post-translational modification of signaling proteins, all of which are vital for rapidly dividing cells. Catabolism of glucose (glycolysis), lipids (mitochondrial β-oxidation) and proteins (glutamine) converge at the Krebs cycle to facilitate not only the generation of energy through oxidative phosphorylation (OxPhos), but also the generation of citrate to support lipogenesis [37]. Citrate is converted to acetyl-CoA by ATP Citrate Lyase (ACLY) (Figure 2), which supports both fatty acid and cholesterol synthesis. Acetyl-CoA Carboxylase (ACC) and Fatty acid synthase (FASN) are rate limiting steps in fatty acid synthesis, while HMG-CoA Reductase (HMGR) has this role for the mevalonate - cholesterol synthesis pathway and is the target of the anti-cholesterol agents statins [38].
The significance of DNL in cancer is evident by the up-regulation of lipogenic genes in many aggressive tumors including lung, breast, prostate and colon cancer [39, 40]. Increased expression and activation of ACLY is a negative prognostic factor in lung and colon cancer, and is associated with poor survival [41, 42]. Molecular or biochemical blockade of ACLY suppresses DNL and inhibits NSCLC cell growth [41, 43, 44], and combined inhibition of ACLY and HMGR by lovastatin enhanced the anti-proliferative effects of ACLY inhibition. Interestingly, combining ACLY knockdown and statin treatment also blocked PI3k/Akt and Mitogen Activated Protein Kinase (MAPK) signaling in EGFR and K-Ras mutant NSCLC lines [45]. Biochemical inhibition of ACC was also found to be toxic for NSCLC cells [46, 47]. Recently, Svensson et al (2016) [47] showed inhibition of ACC affected NSCLC cell and tumor growth, enhanced cytotoxic effects of chemotherapy, and observed increased activity in K-Ras mutant p53-/- and K-Ras mutant - LKB1-/- mouse models. While FASN inhibition has been shown to inhibit growth of other cancer types [48, 49], no significant work has been done in NSCLC. There is strong evidence that growth factor receptor pathways mediate expression of FASN through PI3k and Akt [36]. This suggests that DNL is potential target for mitigating NSCLC growth. However, development of compounds that could target DNL pathways and simultaneously activate AMPK and p53, could have advantages in slowing malignant growth by targeting cell cycle progression and enhancing radiosensitivity.
LKB1 - AMPK pathway
The anabolic events at the cancer cell plasma membrane, cytoplasm and mitochondria are also under the tight regulation of the Liver Kinase 1 (LKB1) - AMP-activated (AMP-activated kinase) kinase pathway. LKB1 is a tumor suppressor that is mutated in Peutz-Jeghers syndrome and is associated with hamartomas, primary gut polyps, breast, colon, and lung cancer [50]. Studies show up to 30% rate of Lkb1 point mutations and deletions in NSCLC, ubiquitous expression of LKB1 in adult lung bronchial epithelium and a progressive loss of LKB1 as pre-malignant adenomatous hyperplasia progresses to frank invasive disease [50]. The highest rate of LKB1 defects is detected in lung adenocarcinomas (34%). LKB1 defects alone are early events in lung cancer carcinogenesis, but within K-Ras mutant tumors, they help develop an aggressive metastatic phenotype [51]. Low copy number of LKB1 gene was associated with higher risk for brain metastasis in patients with advanced NSCLC expressing mutant K-Ras [52].
LKB1 mediates many of its metabolic and anti-proliferative functions through phosphorylation and activation of the AMPK. This is an evolutionarily-conserved sensor of metabolic stress that at times of low energy levels, i) inhibits anabolic processes including energy storage, cellular growth and proliferation, and ii) increases nutrient uptake and catabolism to enhance energy availability [50]. Homologues of AMPK in yeast and C. Elegans were shown to regulate fundamental processes such as cellular growth, survival under environmental stress and longevity [53]. Human AMPK is a heterotrimer, consisting of a catalytic α-subunit, and regulatory β and γ subunits. In humans two α (α1 and α2), two β (β1 and β2) and three γ subunit (γ1, γ2 and γ3) isoforms exist [54]. AMPK is activated by direct LKB1 phosphorylation on the α-subunit at threonine 172 (T172), and by increases in the AMP/ATP ratio induced by natural metabolic stressors such as exercise, starvation, and hypoxia. At times of energy stress increased levels of AMP/ADP bind to four tandem cystathionine β synthase (CBS) repeats of the γ-subunit (now known as Bateman domains [55]) and induce an allosteric activation of AMPK leading to a 5-fold increase of the α-subunit catalytic activity [56]. However, phosphorylation of T172 within an activation loop of α-subunit, by LKB1, removes its auto-inhibition and stimulates the activity of the kinase domains by 100-fold [57]. The regulatory β-subunit of AMPK acts as a scaffold on which the α and γ-subunits can bind and form a functional AMPK heterotrimeric complex [56]. The β-subunit contains a carbohydrate (glycogen) binding domain (CBM), the role of which is still being elucidated. It likely aids in the localization of AMPK close to substrates that also bind glycogen, such as glycogen synthase [58]. The β1-subunit, specifically, undergoes post-translational modification by myristoylation and phosphorylation, which is required for AMPK localization and activity [59]. Phosphorylation of β1 subunit Ser108, in the CBM domain, leads to allosteric activation of the kinase which prevents the de-phosphorylation of T172. This takes place in response to direct AMPK β1 activators A-769662 and salicylate [60].
AMPK regulation of signaling and metabolic events
Early events in growth signal transduction
AMPK is described to regulate both the PI3k-Akt-mTOR and the Raf-Mek-Erk pathway that stimulate gene expression, cellular growth and survival (Figure 1B). It is suggested to inhibit early steps of insulin and Insulin-like growth factor I (IGF-1) receptor signal transduction through an inhibitory phosphorylation of Ser789 on Insulin Receptor Substrate (IRS)-1 leading to reduced activation of PI3k-Akt axis [61], an event observed in many tumor types. Further, AMPK attenuates the B-RAF-MEK-ERK pathway, through phosphorylation of BRAF on Ser729. This event promotes its association with the adaptor 14-3-3 and disrupts its interaction with the Kinase Suppressor of Ras (KSR) 1, which mediates activation of the B-RAF-MEK-ERK pathway [62].
Protein synthesis
AMPK blocks protein synthesis through inhibition of mTOR complexes, mTOR complex 1 (mTORC1), and its downstream action (Figure 1A). This is mediated through, i) phosphorylation of the tumor suppressor Tuberous Sclerosis 2 (TSC2, on Thr1227 and Ser1345 residues), which activates the TSC1:TSC2 complex, and induces the GTPase activity (inhibits) the small G-protein Rheb, an activator of mTORC1 and ii) through phosphorylation and inhibition of the mTORC1 complex protein Raptor, preventing mTORC1 from phosphorylating downstream targets [63]. These events block regulation of mTORC1 targets such as the ribosomal p70-S6 kinase (p70S6K), that stimulates ribosomal activity, and the eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4EBP1), which normally blocks initiation of translation. [64]
Lipogenesis
AMPK inhibits DNL through modulation of lipogenic gene expression as well as direct regulation of enzymatic activity. It inhibits transcriptional activity of sterol regulatory binding proteins (SREBP) 1c and 2 through phosphorylation on Ser372/374 [65] (Figure 1), a step that blocks proteolytic cleavage of SREBP and their ability to translocate to nucleus to activate lipogenic gene transcription. In this fashion, AMPK exerts global control over de novo fatty acid synthesis. However, AMPK is a key regulator of ACC that controls the synthesis of FA [66]. It inhibits ACC through phosphorylation of Ser79 of ACC1 and Ser212 on ACC2. G. Steinberg and colleagues (2013) examined the regulation of lipid homeostasis by ACC by inhibiting the ability of ACC to be regulated by AMPK. Generation of ACC1 and ACC2 knock-in animals, as well as double knock-in (DKI) ACC1/ACC2 animals, which carry substitutions of Ser 79 and 212, in the two genes with alanine [67], showed that such animals have elevated lipogenesis, lower fatty acid oxidation, insulin resistance, glucose intolerance and fatty livers [67]. ACC DKI animals become obese on high-fat diet and in that setting are refractory to the insulin-sensitizing and lipid lowering effects of metformin [67].
Cell cycle and apoptosis
A key effect of AMPK on proliferation is the mediation of a metabolic checkpoint and induction of apoptosis and autophagy [68]. AMPK blocks cell cycle by increasing total and phosphorylated levels of p53 and its downstream effector p21cip1. Jones et al (2005) [69] suggested in cancer cells that glucose deprivation stimulates AMPK that in-turn phosphorylates p53 on Ser15, leading to increase in p53 activity [70] and stabilization of the molecule [71]. Further, AMPK phosphorylates the CDK inhibitor (CDKI) p27kip1 on Thr198 to sequester it in the cytoplasm and promote survival in response to nutrient or growth-factor withdrawal [72]. Interestingly, we [73] and others [74, 75] described nuclear localization of AMPK. The potential role of AMPK in mitosis was discussed earlier (see Sanli et al (2014) [76]). Other mechanisms described to link AMPK and apoptosis include the inhibition of FASN and stimulation of the mitochondrial apoptotic pathway [77-79].
Transcription factors
Transcription factors, their co-activators, and histones are modulated downstream of AMPK to regulate gene expression and nuclear events leading to metabolic reprogramming and cell survival. AMPK is described to regulate transcription factors involved in hepatic gluconeogenesis, helping to regulate hepatic glucose output. In the context of cancer and NSCLC, several other transcription factors are affected by AMPK activation, including SREBP, p53, hypoxia induced factor 1α (HIF1α) (discussed below), and forkhead box 03a (FOXO3a). SREBP1c regulates fatty acid synthesis genes, and is inhibited by AMPK [80]. p53 which regulates expression of apoptotic proteins, including members of the Bcl-2 family and caspase 6, as described, is induced and stabilized by AMPK [81]. AMPK suppresses protein levels of HIF1α, which when induced by hypoxia, facilitates expression of several pro-survival genes, including vascular endothelial growth factor (VEGF), and glucose transporters (GLUT1) [82]. Finally, AMPK regulates forkhead box protein O3 (FOXO3a) by direct phosphorylation, which regulates glucose metabolism and apoptosis, during metabolic stress [83].
Sensor of genotoxic stress
AMPK is activated in response to both chemotherapy and RT. We demonstrated that therapeutic doses of RT (2-8Gy) induce potent time- and dose-dependent activation of AMPK in cancer cells [70]. Phosphorylated AMPK is detected in the nucleus followed by translocation of the activated complex to the cytoplasm. Interestingly, RT-induced activation of AMPK was not dependent on LKB1, as it was observed in LKB1-null A549 cells. This action of AMPK was blocked upon inhibition of ATM. We proposed that AMPK, i) transduces signals through a DNA damage response (DDR)-activated ATM-AMPK-p53-p21cip1 axis, ii) facilitates the DDR-induced G2-M checkpoint and iii) mediates RT-induced cytotoxicity in NSCLC [70, 76] (see models Figure 1B). AMPK subunit gene expression is acutely stimulated by RT in cancer cells [73]. Work with embryonic fibroblasts from genetically engineered mice (MEFs) lacking AMPK α-subunit expression, suggested that AMPK plays in stabilizing the basal activity of DDR and survival signaling pathways. We observed that lack of AMPK destabilizes both the ATM-p53 and the Akt-mTOR signals. Untreated AMPKα1,2-/-MEFs have enhanced ATM and p53 as well as Akt-mTOR signals but these pathways failed to respond to radiation. These cells lacked a G2-M checkpoint response and showed evidence of radio-resistance [73]. In human NSCLC xenograft models RT leads to a chronic sustained expression and activation of the entire ATM-AMPK-p53/p21cip1 pathway but inhibition of mTOR signals [84] that is associated with inhibition of tumor growth, expression of apoptosis markers and inhibition of angiogenesis [84]. RT-induced expression and activation of AMPK is mediated through sestrin 2, a member of a family of stress-induced genes that is activated by RT and mediates radio-sensitization of breast and NSCLC cells [85]. Overall, RT causes sustained activation and increased expression of AMPK and its effectors in lung cancer cells and tumors leading to inhibition of survival.
Targeting cellular metabolisms to enhance response to cytotoxic therapy
AMPK’s ability to regulate cellular growth and cell cycle revived the concept that targeting cellular metabolism may be able to control tumor growth. This triggered investigation of agents mediating metabolic stress in cells but the anti-diabetic agent metformin gained the greatest popularity due to its favorable toxicity profile, wide-spread use, low cost and excellent tolerability in non-diabetics also.
Metformin
Metformin (1-(diaminomethylidene)-3,3-dimethyl-guanidine) is a biguanide, a class of anti-diabetic drugs containing two linked guanidine rings that was derived from galegine, a guanidine found in French lilac (galega officinalis). Two other biguanides, phenoformin and buformin have potent activity and were used earlier but were withdrawn from clinical use due to increased risk of lactic acidosis. Metformin is an effective and well-tolerated anti-diabetic agent used by more than 120 million patients worldwide [86] and it is also indicated for the treatment of polycystic ovary syndrome and non-alcoholic liver disease.
Metformin and cancer
Pre-clinical studies - molecular mechanism of action
Metformin inhibits complex I of the mitochondrial OxPhos chain, leading to increased levels of ADP and AMP and activation of AMPK [56] (Figures 2, 3). Thereby, metformin induces all pathways, metabolic and cytostatic events attributed to AMPK [87]. Many studies observed anti-tumor activity of metformin in NSCLC cell lines but fewer examined the drug in combination with targeted or cytotoxic therapy (see Table 1). Although, most groups analyzed very similar or the same NSCLC cell lines, significant controversies exist in the reported mechanism of action of metformin and phenformin. Studies observed anti-tumor activity of metformin in most lung cancer lines and histologies, including small cell lung cancer, but responses were generally detected at high (mM) concentrations [88, 89].
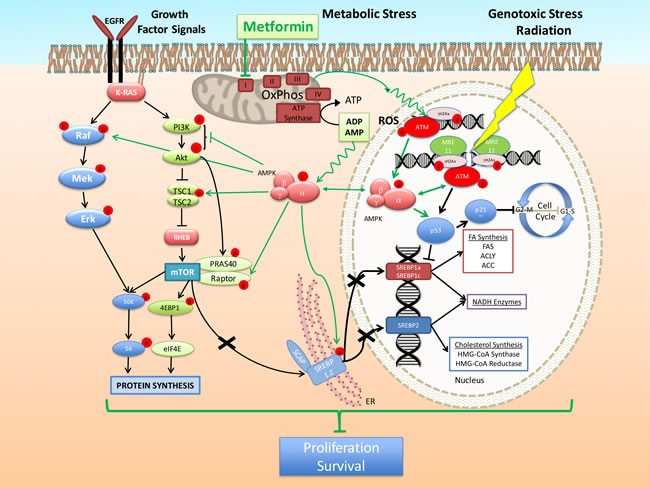
Figure 3: Model of the mechanism of action of metformin alone and in combination with cytotoxic therapy. Inhibition of mitochondria OxPhos complex I by metformin creates in cancer cells a metabolic stress characterized by increased levels of AMP/ADP and potential generation of Reactive Oxygen Species (ROS). The first leads to AMPK activation through binding on the AMPK γ subunit, while the second is likely responsible for the observed trigger of DDR, activation of ATM and induction of γH2Ax foci and activation of AMPK. These effects enhance further the activation of AMPK mediated by genotoxic stress leading to improved suppression of the downstream events such as the mTOR pathway, metabolic gene expression, cell growth, cell cycle progression and survival.
Role of LKB1
In mouse Lewis lung model, Algire et al (2011) [90], detected opposite roles of LKB1 on the response of LLC1 cells to metformin depending on glucose availability. In cells grown in standard high glucose media (25mM), LKB1 knockdown blocked AMPK and ACC phosphorylation and abolished metformin-induced growth inhibition. However, in low glucose media (2.5mM) lack of LKB1 enhanced significantly the cellular sensitivity to metformin. These findings are contrary to the notion that the LKB1-AMPK axis mediates tumor suppression and indicate that LKB1 may trigger a specific mechanism of resistance to tumor suppressive effects of metabolic stress.
K-Ras
Adenocarcinoma cells harboring K-Ras with or without loss of LKB-1 expression are reported to show greater sensitivity to biguanides [88, 91]. However, the mechanism behind this sensitivity is not defined. One would expect that enhanced signaling downstream of K-Ras (including PI3k-Akt-mTOR) would stimulate a glycolytic and lipogenic phenotype that would resemble the Warburg effect and mediate independence from OxPhos, the site of action of biguanides. Shackelford et al (2013) [92], studied mutant K-RasG12S-LKB1-/- adenocarcinoma lines and detected LKB1-dependent activation of AMPK and blockade of mTOR by both metformin and phenformin when wild type LKB1 was transfected, but detected apoptosis only in response to phenformin. Using the adenovirus Cre-induced lung specific model of K-RasG12D expression they studied the impact of floxed alleles of p53 or LKB1. p53 loss is well-described to increase significantly tumor burden, metastasis and radio-resistance in this model but they found that loss of LKB1, and not p53, mediated phenformin sensitivity.
AMPK
A number of groups verified the dependence of metformin action on AMPK activity. In the background of K-Ras mutation, Salani et al (2012) [93] suggested that AMPK activation is dependent on Caveolin-1. We detected LKB1-independent but AMPK-dependent inhibition of survival of adeno- and squamous cell carcinoma lines with microM concentrations of metformin [89]. Similar results were obtained by Guo et al (20156) [94], albeit with higher drug doses. Inhibition of proliferation and clonogenic survival was independent of p53 or LKB1 status. Metformin was equally active in A549 (K-RasG12S-LKB1 null) and in H1299 (N-RasQ12K-p53 null) cells and xenografts [89], and in squamous SK-MES (p53 null) cells.
Recent studies, using the same cell lines (H1299, A549) suggested that the response to metfromin is mediated solely by, i) suppression of mitochondria metabolism, which suppresses vital cellular functions, like lipogenesis [95] or ii) by mitochondria-mediated apoptosis [96].
ATM
Metformin induces expression of ATM and activation, detected as ATM phosphorylation and induction of γH2Ax foci, and this is required for metformin activation of AMPK [89]. The mechanisms by which metformin activates ATM and how this contributes to AMPK activation are not well understood. Other groups also described the concept of ATM-induced AMPK activation is response to hormone (IGF-I) [97] and cytotoxic therapy (etoposide) [98], metabolic stress [99] and in response to metformin in esophageal and epidermoid cancer cells [100, 101]. The site of metformin-induced ATM activation may not be limited to the nucleus. ROS were suggested to activate ATM in the cytoplasm in breast cancer cells, leading to activation of LKB1-AMPK-mTOR axis [99]. Other groups also detected nuclear activation of ATM. Vazquez-Martin et al (2011) [101] observed presence of γH2Ax foci and ATM phosphorylation in the absence of p53 binding protein foci (indicative of DNA strand breaks) or positive comet assays. They suggested that metformin may mediate changes in the chromatin structure that triggers activation of DDR and phosphorylation of ATM, leading to checkpoint activation. ATM is felt to function as a guardian of the genome in the face of genotoxic stress induced by cytotoxic and metabolic insults [101, 102].
Mitochondria - ROS
Whether metformin induces or reduces ROS levels in cells remains controversial. It was suggested that blockade of complex I leads to reduction of mitochondrial ROS production [103], but OxPhos blockade leads to acidosis and others did show enhanced ROS levels with prolonged metformin incubation in breast cancer cells [104]. It is possible that blockade of mitochondrial OxPhos by metformin induces a slow generation of ROS, which diffuse into the nucleus to induce activation of ATM in the absence of true DNA breaks (see model of Figure 3). Although, the exact mechanism of ATM activation by metformin remains unclear, the presence of γH2Ax foci indicate that byproducts of metabolic stress indeed enter the nucleus and mediate a state of genomic stress that activates DDR. Alternatively, metformin may trigger cytoplasmic activation of ATM that subsequently shuttles into the nucleus. Future studies need to elucidate the exact mechanism by which ATM regulates AMPK.
Hypoxia induced factor (HIF)1α
HIF1α has a well-described role in supporting cellular survival in the hypoxic tumor micro-environment, a mechanism well-linked to radio-resistance and tumor metastatic potential [105, 106]. Although one would expect that induction of energy stress by metformin, through complex I inhibition, would likely induce HIF1α, studies demonstrate suppression of HIF1α by metformin [107, 108], which is suggested to contribute to the anti-tumor action of the drug [109]. In the absence of LKB1, HIF1α mediates expression of the Warburg metabolic phenotype and LKB1 and AMPK are shown to suppress transcription and translation of HIF1α, albeit by different mechanisms [110]. Whether the metformin-induced suppression of HIF1α involves LKB1 or AMPK or neither, this mechanism is dependent on the blockade of mitochondrial complex I. Wheaton et al (2014) [111] showed that expression of the metformin-resistant Saccharomyces cerevisiae NADH dehydrogenase NDI1 blocks the suppression of HIF1a by metformin, indicating the importance of OxPhos inhibition in suppressing HIF1α.
Interaction with cytostatic therapies
In our hands, the anti-proliferative effects of µM doses of metformin were comparable to those of EGFR (Gefitinib) and mTOR (rapamycin) inhibitors (supplemental data in Storozhuk et al (2013)) [90]. NSCLC cell line and xenograft work indicated that metformin could enhance the activity of EGFR inhibitor Gefitinib [112] and suppress tumor growth after Gefitinib withdrawal [113] (Table 1). Further, we showed that in NSCLC cells metformin enhances the anti-tumor activity of other AMPK-activating agents such as salicylate in both p53 null and LKB1 null cells, through a mechanism that involves AMPK β1 subunit and DNL [114] (Table 1).
Table 1: Pre-clinical studies of metformin in lung cancer: Investigation of metformin alone and in combination with chemotherapy and radiation.
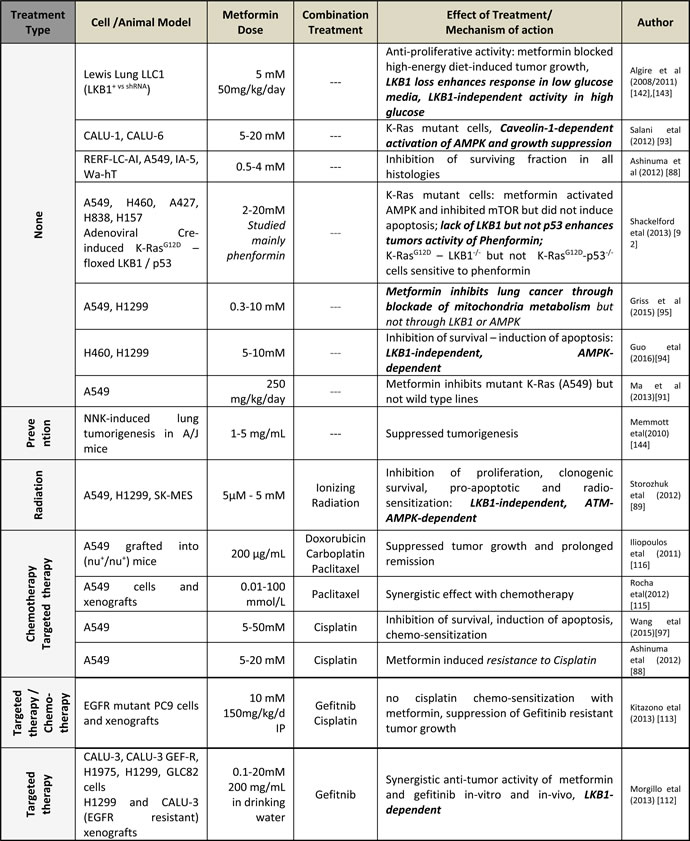
In combination with chemotherapy
Rocha et al (2011) [115] showed that in lung cancer models paclitaxel activates AMPK and inhibits mTOR, effects that are enhanced by metformin which enhances paclitaxel cytotoxicity (Table 1). While one group suggested that cisplatin may antagonize the anti-proliferative effects of metformin in some cell lines [88], other studies [116] suggested that metformin could function in combination with doxorubicin to reduce the chemotherapy dose needed to suppress tumor growth [116] and can mediate synergistic cytotoxicity with cisplatin [117, 118].
Radio-sensitizing activity
We observed that metformin sensitizes lung cancer cells and tumors to RT [89] (Table 1). Metformin enhances the RT-induced activation of the ATM-AMPK-p53/p21cip1 axis, while it mediates suppression of the Akt-mTOR-4EBP1 pathway [89]. Importantly, we detected these effects of metformin at low micro-molar (5-100µM) doses. Metformin enhanced the induction of γH2Ax and total ATM levels in irradiated lung cancer cells and tumors, indicating a sustained increased expression and activation of ATM. This increase was present in metformin treated tumors even 8 weeks after RT [89]. Inhibition of ATM activity with knockdown using specific ATM siRNA or with the inhibitor KU60019 blocked both metformin- and RT-induced histone phosphorylation (γH2Ax) and AMPK activation. Further, knockdown of AMPK α-subunit, i) blocked metformin- and RT induction of p21cip1, ii) allowed activation of Akt and mTOR and iii) blocked the inhibition of lung cancer cell proliferation mediated by metformin and RT [89]. In NSCLC xenografts metformin given orally (in drinking water) at a dose of 250mg/kg/day enhanced the activation of ATM - AMPK - p53/p21cip1 axis induced by RT, inhibited tumor growth and enhanced the cytotoxicity of RT, inhibited angiogenesis and enhanced the induction of apoptotic markers. This work suggested that metabolic stress with metformin can be combined with RT to enhance the tumor suppressive effects of RT (Figure 3).
Metformin bio-availability in tumors
Some of the work described above, suggests that metformin could have direct activity in lung tumors at doses that can be achieved in human circulation. However, doubt developed in the scientific community, as to whether metformin is able to mediate direct anti-tumor activity and activate AMPK in tumor cells, since many studies showed activity of the drug in cancer cell lines only at mM concentrations of metformin that cannot be achieved clinically [87, 119, 120]. Diabetic patients are typically treated with 1-2.5g of metformin per day, a dose that achieves typically plasma levels of 7-10 µM [121, 122] but peak plasma concentrations of 30-40 µM have been described [122]. In our studies such plasma levels could mediate direct anti-tumor activity in lung cancer. The discrepancy between achievable doses of metformin in humans and the ones needed to mediate anti-tumor action in other tumors led groups to suggest that most of the anti-tumor activity of metformin may be due to the indirect hepatic action of the drug where, due to portal circulation, metformin reaches greater concentration [123]. Metformin action in liver could lead to improved control of glycemia and reduced circulating levels of insulin and IGF-1, which can reduced trophic effects on tumors. Nevertheless, recent studies examined metformin levels in the circulation of animals used in xenograft studies and in tumor tissue, using liquid chromatography and mass spectroscopy. Dowling et al (2016) [124] observed that intraperitoneal injections of 125mg/kg caused average plasma and tumor levels of metformin (of 145 µM and 77 µM, respectively), while delivery of the drug in the drinking water (at 5mg/ml) provided a stable average level of 34 µM and similar tumor concentration of 32 µM. At those concentrations of metformin caused activation of AMPK in colon carcinoma xenografts (HCT116), while activation of AMPK in the same cells in vitro required 10-20 mM metformin. In our studies with A549 and H1299 lung adenocarcinoma xenografts we observed sustained activation of AMPK with oral delivery of metformin in drinking water at a concentration of 250mg/kg/day (about 1.25mg/ml) [89]. It is suggested that, being a cation, metformin could accumulate in the mitochondria at 100-500 times higher concentrations due to the mitochondria membrane potential, leading to an effective inhibition of complex I. Overall, metformin should be able to mediate AMPK activation and anti-tumor activity, in human tumors with serum levels that are clinically achievable with standard anti-diabetic doses of the drug, indicating that metformin doses used in current clinical trials are appropriate.
Clinical Investigation of Metformin in cancer
Retrospective clinical studies in cancer: Prevention vs improved response to cytotoxic therapy
Retrospective work has suggested that metformin may decrease the incidence of cancer in patients with type 2 diabetes [125, 126], and may be able to improve cancer treatment outcomes. Single center studies and a meta-analysis showed that metformin use was associated with a decrease in cancer mortality in all sites, including lung cancer (RR 0.66, p = 0.005) [127, 128]. Importantly, the beneficial effects of metformin were not shared with other hypoglycemic agents and potentially worse outcomes have been reported in patients treated with sulfonylureas and insulin [129-131].
Lung cancer prevention and outcomes
Reduced incidence of NSCLC was also reported with metformin in type II diabetic patient in some [128, 130, 132] but not all studies [133] (Table 2). A retrospective study from Cleveland Clinic observed that diabetic patients with lung cancer, who were previously exposed to metformin and/or another class of anti-diabetic drugs (thiazolidenediones: also known to activate AMPK), are less likely to present with metastatic disease, are more likely to present with an adenocarcinoma, and they may survive longer [132]. Tan and colleagues (2011) [134] suggested that locally advanced (LA-)NSCLC patients taking metformin had a median OS of 20 months compared to 13 months for those who took other hypoglycemic agents (insulin or other). This was also observed by retrospective analysis in other tumors such as esophageal, head and neck and prostate cancer [135-140].
Table 2: Retrospective clinical studies with metformin in lung cancer
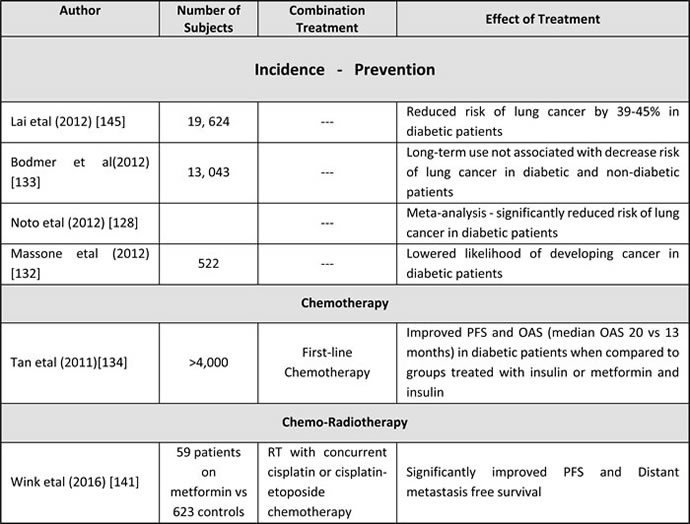
Retrospective results in LA-NSCLC treated with chemo-radiotherapy
Recently, Wink et al (2016) [141] suggested that metformin use is associated with improved Progression Free Survival (PFS) in patient with LA-SCLC that were treated with chemo-radiotherapy (Table 2). They reviewed patients treated in three radiation oncology departments in Netherlands, between 2008 and 2013, and identified 623 controls and 59 who received metformin at least during their radiotherapy course. Systemic therapy included either daily cisplatin or three-week cycle cisplatin-etoposide regimens, while chest RT was equivalent to 50 Gy or higher. Overall survival (OAS) and loco-regional recurrence free survival were not statistically significant between metformin users and non-users but PFS and distant metastasis free survival were found to be significantly increased in the metformin group (p < 0.01) [141].
We also pursued a retrospective analysis of patients with locally advanced NSCLC treated at the Juravinski Cancer Center (Hamilton, Ontario, Canada). Stage II-III patients were selected that received 50 Gy or more of chest RT with or without concurrent chemotherapy. Figure 4 illustrates a Kaplan-Meyer survival plot of the two groups. Median survival was 12.3 months for patients not treated with metformin vs 18.7 months for those who were treated with metformin. The benefit associated with metformin use appeared to diminish with prolonged survival. Results showed a trend for improved OAS in patients treated with metformin vs the controls, which approached but did not reach statistical significance (p = 0.06). These results are consistent with the observations of Wink et al (2016) [141] and other groups indicating that metformin may indeed be associated with improved outcomes in NSCLC. Recognizing the limited value of retrospective data, these and other results suggested the need to examine metformin in combination with standard cytotoxic therapy (Chemo-RT) in prospective controlled clinical trials.
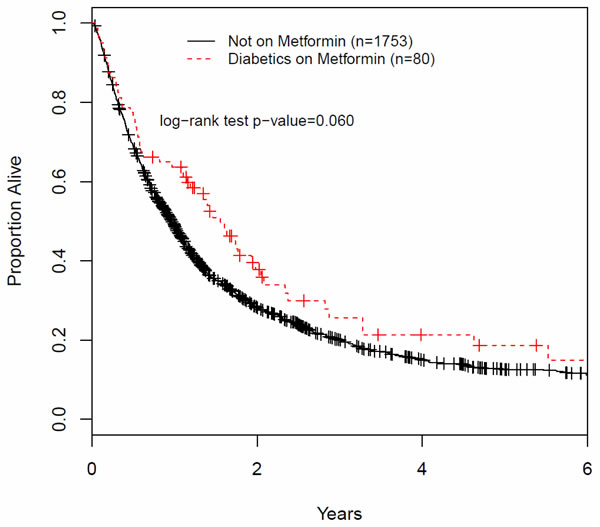
Figure 4: Overall survival of patients with LA-NSCLC treated with chemotherapy and radiotherapy, with or without metformin. A retrospective case review of patients treated at the Juravinski Cancer Center, Hamilton, Ontario, in the period of 1998 - 2013. Patients with stage II and III disease were selected that received 50Gy or more of chest RT with or without concurrent chemotherapy. We identified 80 patients who were treated with metformin, as therapy for type II diabetes, during the period of cytotoxic therapy and beyond vs 1753 patient not treated with metformin. Patients received variable metformin doses of 1000 - 2500mg daily and the length of treatment with metformin after cytotoxic therapy was not determined. Patients and treatment characteristics of the two groups (patients NOT on Metformin vs ON Metformin) were similar: i) age: 68.82 +/- 10.72 and 68.79 +/- 8.38; ii) female to male ratio was 0.8 in both groups; iii) % of stage II: 14.3 vs 15.3; iv) % of stage III: 85.6 vs 84.6; v) % treated with chemotherapy: 77.05 vs 62.16, respectively.
Active prospective cancer trials
Presently, there are over 130 clinical trials investigating metformin in cancer overall, while 12 studies focus on lung cancer (www.ClinicalTrials.govOctober 2016). Table 3 groups these studies in terms of their primary and additional intervention.
Table 3: Active clinical trials with metformin in lung cancer (www.ClinicalTrials.gov, Oct. 2016)
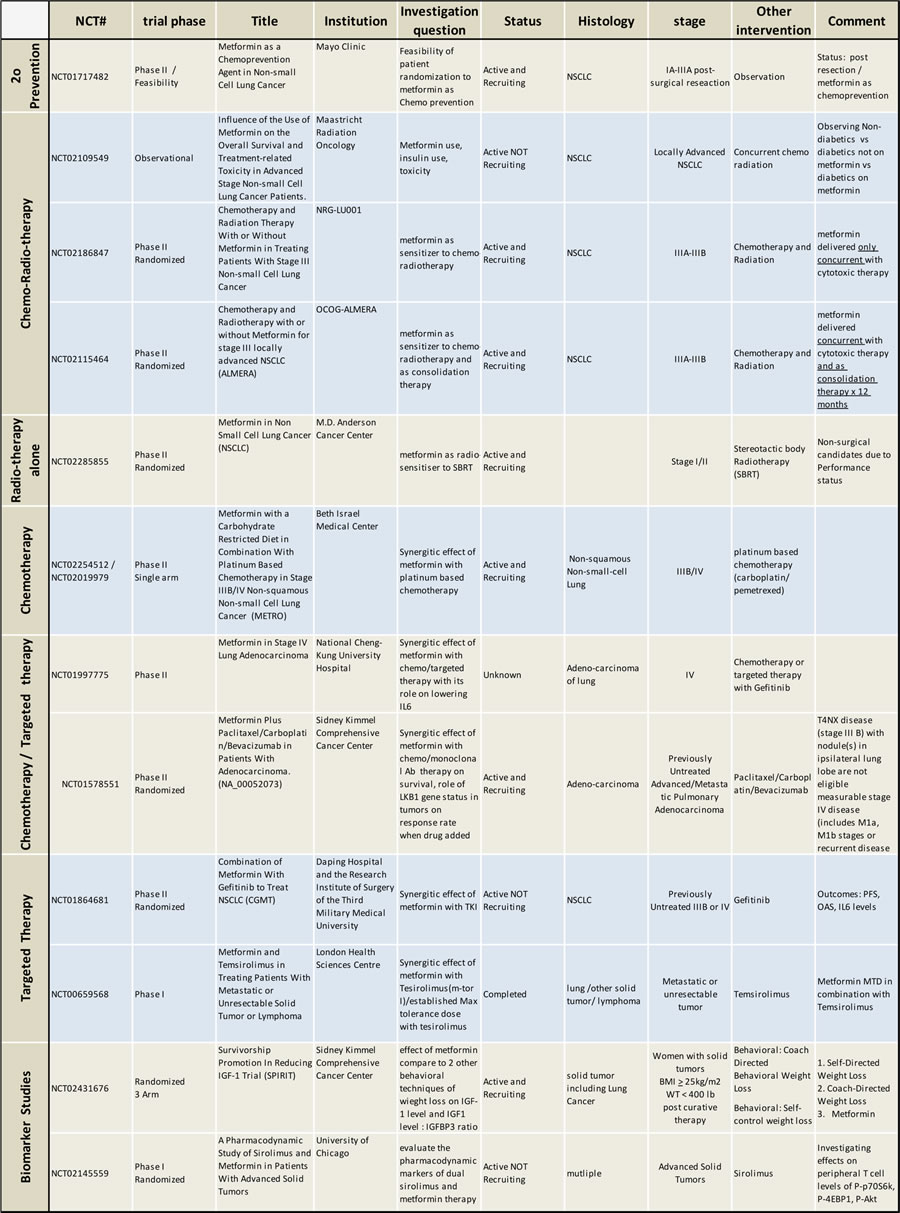
Prevention studies
A study from Mayo Clinic (NCT01717482) investigates the feasibility to pursue secondary prevention studies with metformin in Stage IA-IIIA resected NSCLC (Table 3). Patients randomized to metformin vs none, and rates of randomization, accrual and tissue collection are observed.
In combination with Chemo-RT
An observational study from Maastricht monitors use of metformin and insulin and induced toxicity in diabetic and control patients with locally advanced NSCLC treated with chemo-RT. Phase II clinical trials investigate metformin as a Chemo-RT-sensitizer and consolidation therapy in locally advanced NSCLC.
The NSABP-RTOG-GOG (NRG) LU001 trial (NCT02186847), is an US NCI CTEP funded study that investigates metformin as a sensitizer to chemotherapy and RT in stage III NSCLC through addition of metformin only during cytotoxic therapy. This includes concurrent Chemo-RT for 6 weeks followed by 6 weeks of consolidation chemotherapy treatment. Chemotherapy, in both the concurrent and the consolidation phase of this study, is the widely used carboplatin-paclitaxel doublet, while patients receive standard chest RT of 60Gy in 30 fractions. Metformin dose is escalated over 2 weeks from 1000mg to 2000mg daily, at which point, is combined with cytotoxic therapy and remains constant for the duration of cytotoxic therapy. This study completed accrual at the end of 2016. A total of 168 patients were accrued. Sample size was designed to detect a 15% improvement in PFS, with the addition of metformin, at 12 months. The first results of this trial may be available by the end of 2017.
An additional, multi-center phase II study that runs in Canada through the Ontario Clinical Oncology Group (OCOG), ALMERA (NCT02115464), examines the potential of metformin to offer both chemo-radio-sensitization as well as consolidation therapy as a single agent following cytotoxic therapy. A total of 98 patients will be randomized in this screening trial to standard Chemo-RT with or without treatment with 2000mg of metformin, which in this study is delivered during cytotoxic Chemo-RT (2 cycles of cisplatin-based chemotherapy, concurrent with chest RT of 63 Gy in 30 fractions) but continues also as consolidation therapy for a total of 12 months. Primary outcome is 12 month PFS. When completed NRG-LU001 and OCOG-ALMERA will provide the first randomized evidence on the potential of metformin to improve the outcomes of standard cytotoxic therapy in locally advanced NSCLC.
Metformin with RT alone
A randomized, placebo control, phase II study at MD Anderson Cancer Center investigates metformin in combination with RT alone in inoperable early stage NSCLC patients, treated with stereotactic RT. The primary objectives in this study are tumor response by RECIST criteria and by 18F-PET SUV response (NCT02285855).
In combination with chemotherapy
Three studies examine metformin in stage III-IV NSCLC in combination with chemotherapy: one in combination with carbohydrate restriction (NCT02254512), a second with anti-EGFR (Gefitinib: NCT01997775) and a third with anti-angiogenic (Bevacizumab: NCT01578551) targeted therapy, having disease control (PFS) or biomarker (IL6) outcomes.
In combination with targeted therapy
Two studies investigate metformin in combination with targeted therapy alone; a phase II study with anti-EGFR (gefitinib: NCT01864681) having PFS as primary outcome and a phase I with anti-mTOR (temsirolimus: NCT00659568) looking for the maximum tolerated dose (MTD) for metformin in this combination.
Biomarker studies
Finally, two additional studies examine biomarkers in response to metformin. A three arm study compares metformin to self-directed or coach-directed weight loss and examines effects on IGF-I and IGF binding protein 3 (IGFBP3) levels (NCT02431676) and a second combines metformin with sirolimus and examines effects on the phosphorylation levels of p70S6k, 4EBP1 and Akt in peripheral blood T-cells (NCT02145559).
Biomarkers for patient selection
To date, there are no established predictive biomarkers of metformin sensitivity in any tumor site. p53, LKB1 and K-Ras status have been proposed as markers of metformin action but, as discussed, pre-clinical studies remain divided on this issue with evidence suggesting that metformin has wide anti-tumor activity independent of the expression levels or mutation status of these signal mediators. It is essential that prospective studies with metformin in lung cancer pursue systematic biospecimen collection to allow investigation of predictive biomarkers of metformin response.
Investigating metformin in diabetics with lung cancer
A large number of lung cancer patients are diabetic. Some of these patients are already treated with metformin but many are treated with insulin or other hypoglycemic agents alone. For methodological reasons, it has not been possible to include diabetics in the majority of on-going lung cancer clinical trials with metformin. Since, retrospective evidence suggests potential improvement of lung cancer outcomes with metformin treatment for diabetics, efforts should be made to include such patients in future phase III trials. Although, it is very challenging to introduce new anti-diabetic agents in diabetics being prepared for cytotoxic therapy, this may be possible with appropriate supervision by diabetes care teams and introduction of proper stratification criteria in the phase III trial design. Alternatively, prospective studies in diabetics alone could be considered.
Conclusions
NSCLC requires urgently effective and well-tolerated chemo-/radio-sensitization therapy. The work discussed here shows that targeting tumor metabolism to enhance the response of NSCLC to cytotoxic therapy is a promising concept. Metformin’s ability to activate AMPK and inhibit survival and resistance to chemotherapy and radiation indicates a capacity to compete well with other novel therapeutics. The molecular profile of metformin sensitivity is yet to be determined. K-Ras mutations and loss of LKB1 or p53 need to be investigated further as potential predictive biomarkers. The evidence of chemo- and radio-sensitizing activity and its excellent tolerability allowed metformin to progress rapidly into clinical trials in NSCLC. The on-going randomized studies will begin reporting results in the next 1-2 years. If positive, these studies will assist in rational design of biomarker work and phase III trials.
Conflicts of interests
The authors declare that they have no competing interests.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30. doi: 10.3322/caac.21332.
2. Chang MY, Sugarbaker DJ. Surgery for early stage non-small cell lung cancer. Semin Surg Oncol. 2003; 21: 74-84. doi: 10.1002/ssu.10024.
3. Ginsberg RJ, Rubinstein LV. Randomized trial of lobectomy versus limited resection for T1 N0 non-small cell lung cancer. Lung Cancer Study Group. Ann Thorac Surg. 1995; 60: 615-22; discussion 22-3.
4. Senan S. Surgery versus stereotactic radiotherapy for patients with early-stage non-small cell lung cancer: more data from observational studies and growing clinical equipoise. Cancer. 2013; 119: 2668-70. doi: 10.1002/cncr.28101.
5. Bezjak A, Temin S, Franklin G, Giaccone G, Govindan R, Johnson ML, Rimner A, Schneider BJ, Strawn J, Azzoli CG. Definitive and Adjuvant Radiotherapy in Locally Advanced Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Evidence-Based Clinical Practice Guideline. J Clin Oncol. 2015; 33: 2100-5. doi: 10.1200/JCO.2014.59.2360.
6. Bradley JD, Paulus R, Komaki R, Masters G, Blumenschein G, Schild S, Bogart J, Hu C, Forster K, Magliocco A, Kavadi V, Garces YI, Narayan S, et al. Standard-dose versus high-dose conformal radiotherapy with concurrent and consolidation carboplatin plus paclitaxel with or without cetuximab for patients with stage IIIA or IIIB non-small-cell lung cancer (RTOG 0617): a randomised, two-by-two factorial phase 3 study. Lancet Oncol. 2015; 16: 187-99. doi: 10.1016/S1470-2045(14)71207-0.
7. Perez CA, Stanley K, Rubin P, Kramer S, Brady L, Perez-Tamayo R, Brown GS, Concannon J, Rotman M, Seydel HG. A prospective randomized study of various irradiation doses and fractionation schedules in the treatment of inoperable non-oat-cell carcinoma of the lung. Preliminary report by the Radiation Therapy Oncology Group. Cancer. 1980; 45: 2744-53.
8. Le Chevalier T, Arriagada R, Tarayre M, Lacombe-Terrier MJ, Laplanche A, Quoix E, Ruffie P, Martin M, Douillard JY. Significant effect of adjuvant chemotherapy on survival in locally advanced non-small-cell lung carcinoma. J Natl Cancer Inst. 1992; 84: 58.
9. Furuse K, Fukuoka M, Kawahara M, Nishikawa H, Takada Y, Kudoh S, Katagami N, Ariyoshi Y. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with mitomycin, vindesine, and cisplatin in unresectable stage III non-small-cell lung cancer. J Clin Oncol. 1999; 17: 2692-9.
10. Curran WJ Jr, Paulus R, Langer CJ, Komaki R, Lee JS, Hauser S, Movsas B, Wasserman T, Rosenthal SA, Gore E, Machtay M, Sause W, Cox JD. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst. 2011; 103: 1452–60. Erratum in: J Natl Cancer Inst. 2012; 104: 79.
11. Cox JD. Are the results of RTOG 0617 mysterious? Int J Radiat Oncol Biol Phys. 2012; 82: 1042-4.
12. Dearden S, Stevens J, Wu YL, Blowers D. Mutation incidence and coincidence in non small-cell lung cancer: meta-analyses by ethnicity and histology (mutMap). Ann Oncol. 2013; 24: 2371-6. doi: 10.1093/annonc/mdt205.
13. Kim DW, Choy H. Potential role for epidermal growth factor receptor inhibitors in combined-modality therapy for non-small-cell lung cancer. Int J Radiat Oncol Biol Phys. 2004; 59: 11-20.
14. Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE. Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res. 2008; 14: 1266-73. doi: 10.1158/1078-0432.CCR-07-1606.
15. Blumenschein GR Jr, Paulus R, Curran WJ, Robert F, Fossella F, Werner-Wasik M, Herbst RS, Doescher PO, Choy H, Komaki R. Phase II study of cetuximab in combination with chemoradiation in patients with stage IIIA/B non-small-cell lung cancer: RTOG 0324. J Clin Oncol. 2011; 29: 2312-8. doi: 10.1200/JCO.2010.31.7875.
16. Bussink J, van der Kogel AJ, Kaanders JH. Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol. 2008; 9: 288-96. doi: 10.1016/S1470-2045(08)70073-1
17. Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA, Scaglioni PP. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009; 69: 7644-52.
18. Mauceri HJ, Sutton HG, Darga TE, Kocherginsky M, Kochanski J, Weichselbaum RR, Vokes EE. Everolimus exhibits efficacy as a radiosensitizer in a model of non-small cell lung cancer. Oncol Rep. 2012; 27: 1625-9.
19. Iacovelli R, Palazzo A, Mezi S, Morano F, Naso G, Cortesi E. Incidence and risk of pulmonary toxicity in patients treated with mTOR inhibitors for malignancy. A meta-analysis of published trials. Acta Oncol. 2012.
20. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. If Mammalian Target of Metformin Indirectly Is Mammalian Target of Rapamycin, Then the Insulin-Like Growth Factor-1 Receptor Axis Will Audit the Efficacy of Metformin in Cancer Clinical Trials. J Clin Oncol. 2009.
21. Jackson SP. Detecting, signalling and repairing DNA double-strand breaks. Biochem Soc Trans. 2001; 29: 655-61.
22. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003; 3: 155-68.
23. Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003; 13: 458-62.
24. Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci. 2008; 13: 5016-29.
25. Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998; 281: 1677-9.
26. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003; 3: 421-9.
27. Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009; 9: 400-14.
28. Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006; 21: 3-9.
29. Upadhyay M, Samal J, Kandpal M, Singh OV, Vivekanandan P. The Warburg effect: insights from the past decade. Pharmacol Ther. 2013; 137: 318-30. doi: 10.1016/j.pharmthera.2012.11.003.
30. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324: 1029-33. doi: 10.1126/science.1160809.
31. Barron CC, Bilan PJ, Tsakiridis T, Tsiani E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism. 2016; 65: 124-39. doi: 10.1016/j.metabol.2015.10.007.
32. Bao YY, Zhou SH, Lu ZJ, Fan J, Huang YP. Inhibiting GLUT-1 expression and PI3K/Akt signaling using apigenin improves the radiosensitivity of laryngeal carcinoma in vivo. Oncol Rep. 2015; 34: 1805-14. doi: 10.3892/or.2015.4158.
33. Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999; 274: 20281-6.
34. Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, Hay N. Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 2001; 15: 1406-18. doi: 10.1101/gad.889901.
35. Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953; 13: 27-9.
36. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007; 7: 763-77. doi: 10.1038/nrc2222.
37. Swinnen JV, Brusselmans K, Verhoeven G. Increased lipogenesis in cancer cells: new players, novel targets. Curr Opin Clin Nutr Metab Care. 2006; 9: 358-65. doi: 10.1097/01.mco.0000232894.28674.30.
38. Osmak M. Statins and cancer: current and future prospects. Cancer Lett. 2012; 324: 1-12. doi: 10.1016/j.canlet.2012.04.011.
39. Furuta E, Okuda H, Kobayashi A, Watabe K. Metabolic genes in cancer: their roles in tumor progression and clinical implications. Biochim Biophys Acta. 2010; 1805: 141-52. doi: 10.1016/j.bbcan.2010.01.005.
40. Menendez JA. Fine-tuning the lipogenic/lipolytic balance to optimize the metabolic requirements of cancer cell growth: molecular mechanisms and therapeutic perspectives. Biochim Biophys Acta. 2010; 1801: 381-91. doi: 10.1016/j.bbalip.2009.09.005.
41. Migita T, Narita T, Nomura K, Miyagi E, Inazuka F, Matsuura M, Ushijima M, Mashima T, Seimiya H, Satoh Y, Okumura S, Nakagawa K, Ishikawa Y. ATP citrate lyase: activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008; 68: 8547-54. doi: 10.1158/0008-5472.CAN-08-1235.
42. Zhou Y, Bollu LR, Tozzi F, Ye X, Bhattacharya R, Gao G, Dupre E, Xia L, Lu J, Fan F, Bellister S, Ellis LM, Weihua Z. ATP citrate lyase mediates resistance of colorectal cancer cells to SN38. Mol Cancer Ther. 2013; 12: 2782-91. doi: 10.1158/1535-7163.MCT-13-0098.
43. Hatzivassiliou G, Zhao F, Bauer DE, Andreadis C, Shaw AN, Dhanak D, Hingorani SR, Tuveson DA, Thompson CB. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005; 8: 311-21. doi: 10.1016/j.ccr.2005.09.008.
44. Zaidi N, Swinnen JV, Smans K. ATP-citrate lyase: a key player in cancer metabolism. Cancer Res. 2012; 72: 3709-14. doi: 10.1158/0008-5472.CAN-11-4112.
45. Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, Sukhatme VP. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J Cell Physiol. 2012; 227: 1709-20. doi: 10.1002/jcp.22895.
46. Wang C, Rajput S, Watabe K, Liao DF, Cao D. Acetyl-CoA carboxylase-a as a novel target for cancer therapy. Front Biosci (Schol Ed). 2010; 2: 515-26.
47. Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, Gerken L, Greenwood J, Bhat S, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016; 22: 1108-19. doi: 10.1038/nm.4181.
48. Agostini M, Almeida LY, Bastos DC, Ortega RM, Moreira FS, Seguin F, Zecchin KG, Raposo HF, Oliveira HC, Amoedo ND, Salo T, Coletta RD, Graner E. The fatty acid synthase inhibitor orlistat reduces the growth and metastasis of orthotopic tongue oral squamous cell carcinomas. Mol Cancer Ther. 2014; 13: 585-95. doi: 10.1158/1535-7163.MCT-12-1136.
49. Bandyopadhyay S, Zhan R, Wang Y, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Furuta E, Iiizumi M, Mohinta S, Watabe M, Chalfant C, et al. Mechanism of apoptosis induced by the inhibition of fatty acid synthase in breast cancer cells. Cancer Res. 2006; 66: 5934-40. doi: 10.1158/0008-5472.CAN-05-3197.
50. Makowski L, Hayes DN. Role of LKB1 in lung cancer development. Br J Cancer. 2008; 99: 683-8.
51. Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008; 359: 1367-80.
52. Zhao N, Wilkerson MD, Shah U, Yin X, Wang A, Hayward MC, Roberts P, Lee CB, Parsons AM, Thorne LB, Haithcock BE, Grilley-Olson JE, Stinchcombe TE, et al. Alterations of LKB1 and KRAS and risk of brain metastasis: comprehensive characterization by mutation analysis, copy number, and gene expression in non-small-cell lung carcinoma. Lung Cancer. 2014; 86: 255-61. doi: 10.1016/j.lungcan.2014.08.013.
53. Hedbacker K, Carlson M. SNF1/AMPK pathways in yeast. Front Biosci. 2008; 13: 2408-20.
54. Riek U, Scholz R, Konarev P, Rufer A, Suter M, Nazabal A, Ringler P, Chami M, Muller SA, Neumann D, Forstner M, Hennig M, Zenobi R, et al. Structural properties of AMP-activated protein kinase: dimerization, molecular shape, and changes upon ligand binding. J Biol Chem. 2008; 283: 18331-43.
55. Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997; 22: 12-3.
56. Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009; 89: 1025-78.
57. Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001; 23: 1112-9.
58. Jorgensen SB, Nielsen JN, Birk JB, Olsen GS, Viollet B, Andreelli F, Schjerling P, Vaulont S, Hardie DG, Hansen BF, Richter EA, Wojtaszewski JF. The alpha2-5’AMP-activated protein kinase is a site 2 glycogen synthase kinase in skeletal muscle and is responsive to glucose loading. Diabetes. 2004; 53: 3074-81.
59. Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. beta-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci U S A. 2010; 107: 19237-41.
60. Hawley SA, Fullerton MD, Ross FA, Schertzer JD, Chevtzoff C, Walker KJ, Peggie MW, Zibrova D, Green KA, Mustard KJ, Kemp BE, Sakamoto K, Steinberg GR, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2013; 336: 918-22.
61. Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010; 123: 271-9.
62. Shen CH, Yuan P, Perez-Lorenzo R, Zhang Y, Lee SX, Ou Y, Asara JM, Cantley LC, Zheng B. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell. 2013; 52: 161-72. doi: 10.1016/j.molcel.2013.08.044.
63. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008; 30: 214-26.
64. van Veelen W, Korsse SE, van de Laar L, Peppelenbosch MP. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene. 2011; 30: 2289-303.
65. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, et al. AMPK Phosphorylates and Inhibits SREBP Activity to Attenuate Hepatic Steatosis and Atherosclerosis in Diet-Induced Insulin-Resistant Mice. Cell Metab. 2011; 13: 376-88.
66. Carlson CA, Kim KH. Regulation of hepatic acetyl coenzyme A carboxylase by phosphorylation and dephosphorylation. J Biol Chem. 1973; 248: 378-80.
67. Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen ZP, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, Hardie DG, Macaulay SL, Schertzer JD, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013.
68. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: A novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2013; 15.
69. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005; 18: 283-93.
70. Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T. Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys. 2010; 78: 221-9.
71. Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. MolCell. 2005; 18: 283-93.
72. Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007; 9: 218-24.
73. Sanli T, Storozhuk Y, Linher-Melville K, Bristow RG, Laderout K, Viollet B, Wright J, Singh G, Tsakiridis T. Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: Modulation of cellular signals regulating cell cycle and survival. Radiother Oncol. 2012; 102: 459-65.
74. Vazquez-Martin A, Lopez-Bonet E, Oliveras-Ferraros C, Perez-Martinez MC, Bernado L, Menendez JA. Mitotic kinase dynamics of the active form of AMPK (phospho-AMPKalphaThr172) in human cancer cells. Cell Cycle. 2009; 8: 788-91.
75. Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. The active form of the metabolic sensor: AMP-activated protein kinase (AMPK) directly binds the mitotic apparatus and travels from centrosomes to the spindle midzone during mitosis and cytokinesis. Cell Cycle. 2009; 8: 2385-98.
76. Sanli T, Steinberg GR, Singh G, Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol Ther. 2014; 15: 156-69. doi: 10.4161/cbt.26726.
77. Baumann P, Mandl-Weber S, Emmerich B, Straka C, Schmidmaier R. Inhibition of adenosine monophosphate-activated protein kinase induces apoptosis in multiple myeloma cells. Anticancer Drugs. 2007; 18: 405-10. doi: 10.1097/CAD.0b013e32801416b6.
78. Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol. 2004; 67: 2005-11. doi: 10.1016/j.bcp.2004.01.020.
79. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004; 101: 3329-35.
80. Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B, Park O, Luo Z, Lefai E, Shyy JY, Gao B, Wierzbicki M, Verbeuren TJ, et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011; 13:376-88. doi: 10.1016/j.cmet.2011.03.009.
81. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003; 22: 9030-40. doi: 10.1038/sj.onc.1207116.
82. Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, Kim SS, Ha J. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003; 278: 39653-61. doi: 10.1074/jbc.M306104200.
83. Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007; 282: 30107-19.
84. Storozhuk Y, Sanli T, Hopmans SN, Schultz C, Farrell T, Cutz JC, Steinberg G, Wright J, Singh G, Tsakiridis T. Chronic modulation of AMP-Kinase, Akt and mTOR pathways by ionizing radiation in human lung cancer xenografts. Radiat Oncol. 2012; 7: 71.
85. Sanli T, Linher-Melville K, Tsakiridis T, Singh G. Sestrin2 Modulates AMPK Subunit Expression and Its Response to Ionizing Radiation in Breast Cancer Cells. PLoS One. 2012; 7: e32035.
86. Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. Bmj. 2003; 327: 951-3.
87. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007; 67: 10804-12.
88. Ashinuma H, Takiguchi Y, Kitazono S, Kitazono-Saitoh M, Kitamura A, Chiba T, Tada Y, Kurosu K, Sakaida E, Sekine I, Tanabe N, Iwama A, Yokosuka O, et al. Antiproliferative action of metformin in human lung cancer cell lines. Oncol Rep. 2012; 28: 8-14. doi: 10.3892/or.2012.1763.
89. Storozhuk Y, Hopmans SN, Sanli T, Barron C, Tsiani E, Cutz JC, Pond G, Wright J, Singh G, Tsakiridis T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013; 108: 2021-32. doi: 10.1038/bjc.2013.187.
90. Algire C, Amrein L, Bazile M, David S, Zakikhani M, Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011; 30:1174-82. doi: 10.1038/onc.2010.483
91. Ma Y, Guo FC, Wang W, Shi HS, Li D, Wang YS. Kras gene mutation as a predictor of cancer cell responsiveness to metformin. Mol Med Rep. 2013; 8: 763-8. doi: 10.3892/mmr.2013.1596.
92. Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013; 23: 143-58. doi: 10.1016/j.ccr.2012.12.008.
93. Salani B, Maffioli S, Hamoudane M, Parodi A, Ravera S, Passalacqua M, Alama A, Nhiri M, Cordera R, Maggi D. Caveolin-1 is essential for metformin inhibitory effect on IGF1 action in non-small-cell lung cancer cells. FASEB J. 2012; 26: 788-98. doi: 10.1096/fj.11-192088.
94. Guo Q, Liu Z, Jiang L, Liu M, Ma J, Yang C, Han L, Nan K, Liang X. Metformin inhibits growth of human non-small cell lung cancer cells via liver kinase B-1-independent activation of adenosine monophosphate-activated protein kinase. Mol Med Rep. 2016; 13: 2590-6. doi: 10.3892/mmr.2016.4830.
95. Griss T, Vincent EE, Egnatchik R, Chen J, Ma EH, Faubert B, Viollet B, DeBerardinis RJ, Jones RG. Metformin Antagonizes Cancer Cell Proliferation by Suppressing Mitochondrial-Dependent Biosynthesis. PLoS Biol. 2015; 13: e1002309. doi: 10.1371/journal.pbio.1002309.
96. Wang J, Gao Q, Wang D, Wang Z, Hu C. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol Lett. 2015; 10: 1343-9. doi: 10.3892/ol.2015.3450.
97. Suzuki A, Kusakai G, Kishimoto A, Shimojo Y, Ogura T, Lavin MF, Esumi H. IGF-1 phosphorylates AMPK-alpha subunit in ATM-dependent and LKB1-independent manner. Biochem Biophys Res Commun. 2004; 324: 986-92.
98. Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE. 2008; 3: e2009.
99. Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010; 107: 4153-8.
100. Feng T, Li L, Ling S, Fan N, Fang M, Zhang H, Fang X, Lan W, Hou Z, Meng Q, Jin D, Xu F, Li Y. Metformin enhances radiation response of ECa109 cells through activation of ATM and AMPK. Biomed Pharmacother. 2015; 69: 260-6. doi: 10.1016/j.biopha.2014.11.021.
101. Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Martin-Castillo B, Menendez JA. Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle. 2011; 10: 1499-501.
102. Menendez JA, Cufi S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, Vazquez-Martin A. Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging (Albany NY). 2011; 3: 1063-77. doi: 10.18632/aging.100407.
103. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab. 2014; 20: 953-66. doi: 10.1016/j.cmet.2014.09.018.
104. Queiroz EA, Puukila S, Eichler R, Sampaio SC, Forsyth HL, Lees SJ, Barbosa AM, Dekker RF, Fortes ZB, Khaper N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS One. 2014; 9: e98207. doi: 10.1371/journal.pone.0098207.
105. Graves EE, Maity A, Le QT. The tumor microenvironment in non-small-cell lung cancer. Semin Radiat Oncol. 2010; 20: 156-63. doi: 10.1016/j.semradonc.2010.01.003.
106. Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012; 18: 5585-94. doi: 10.1158/1078-0432.CCR-12-0858.
107. Wang J, Li G, Wang Y, Tang S, Sun X, Feng X, Li Y, Bao G, Li P, Mao X, Wang M, Liu P. Suppression of tumor angiogenesis by metformin treatment via a mechanism linked to targeting of HER2/HIF-1alpha/VEGF secretion axis. Oncotarget. 2015; 6: 44579-92. doi: 10.18632/oncotarget.6373.
108. Zhou X, Chen J, Yi G, Deng M, Liu H, Liang M, Shi B, Fu X, Chen Y, Chen L, He Z, Wang J, Liu J. Metformin suppresses hypoxia-induced stabilization of HIF-1alpha through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget. 2016; 7: 873-84. doi: 10.18632/oncotarget.6418.
109. Ranasinghe WK, Sengupta S, Williams S, Chang M, Shulkes A, Bolton DM, Baldwin G, Patel O. The effects of nonspecific HIF1alpha inhibitors on development of castrate resistance and metastases in prostate cancer. Cancer Med. 2014; 3: 245-51. doi: 10.1002/cam4.189.
110. Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, Jones RG. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A. 2014; 111: 2554-9. doi: 10.1073/pnas.1312570111.
111. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014; 3: e02242. doi: 10.7554/eLife.02242.
112. Morgillo F, Sasso FC, Della Corte CM, Vitagliano D, D’Aiuto E, Troiani T, Martinelli E, De Vita F, Orditura M, De Palma R, Ciardiello F. Synergistic effects of metformin treatment in combination with gefitinib, a selective EGFR tyrosine kinase inhibitor, in LKB1 wild-type NSCLC cell lines. Clin Cancer Res. 2013; 19: 3508-19. doi: 10.1158/1078-0432.CCR-12-2777.
113. Kitazono S, Takiguchi Y, Ashinuma H, Saito-Kitazono M, Kitamura A, Chiba T, Sakaida E, Sekine I, Tada Y, Kurosu K, Sakao S, Tanabe N, Iwama A, et al. Effect of metformin on residual cells after chemotherapy in a human lung adenocarcinoma cell line. Int J Oncol. 2013; 43: 1846-54. doi: 10.3892/ijo.2013.2120.
114. O’Brien AJ, Villani LA, Broadfield LA, Houde VP, Galic S, Blandino G, Kemp BE, Tsakiridis T, Muti P, Steinberg GR. Salicylate activates AMPK and synergizes with metformin to reduce the survival of prostate and lung cancer cells ex vivo through inhibition of de novo lipogenesis. Biochem J. 2015; 469:177-87. doi: 10.1042/BJ20150122.
115. Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB. Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res. 2011; 17: 3993-4005.
116. Iliopoulos D, Hirsch HA, Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011; 71: 3196-201.
117. Chen YQ, Chen G. Combined therapeutic effect and molecular mechanisms of metformin and cisplatin in human lung cancer xenografts in nude mice. J Cancer Res Ther. 2015; 11: 324-30. doi: 10.4103/0973-1482.151444.
118. Mitrakas AG, Kalamida D, Koukourakis MI. Effect of mitochondrial metabolism-interfering agents on cancer cell mitochondrial function and radio/chemosensitivity. Anticancer Drugs. 2014; 25: 1182-91. doi: 10.1097/CAD.0000000000000152.
119. Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27: 3576-86.
120. Iglesias DA, Yates MS, van der Hoeven D, Rodkey TL, Zhang Q, Co NN, Burzawa J, Chigurupati S, Celestino J, Bowser J, Broaddus R, Hancock JF, Schmandt R, et al. Another surprise from Metformin: novel mechanism of action via K-Ras influences endometrial cancer response to therapy. Mol Cancer Ther. 2013; 12: 2847-56. doi: 10.1158/1535-7163.MCT-13-0439.
121. Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, Furlong TJ, Greenfield JR, Greenup LC, Kirkpatrick CM, Ray JE, Timmins P, Williams KM. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 2011; 50: 81-98. doi: 10.2165/11534750-000000000-00000.
122. Lipska KJ, Bailey CJ, Inzucchi SE. Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care. 2011; 34: 1431-7.
123. Chandel NS, Avizonis D, Reczek CR, Weinberg SE, Menz S, Neuhaus R, Christian S, Haegebarth A, Algire C, Pollak M. Are Metformin Doses Used in Murine Cancer Models Clinically Relevant? Cell Metab. 2016; 23: 569-70. doi: 10.1016/j.cmet.2016.03.010.
124. Dowling RJ, Lam S, Bassi C, Mouaaz S, Aman A, Kiyota T, Al-Awar R, Goodwin PJ, Stambolic V. Metformin Pharmacokinetics in Mouse Tumors: Implications for Human Therapy. Cell Metab. 2016; 23: 567-8. doi: 10.1016/j.cmet.2016.03.006.
125. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia. 2009; 52: 1766-77.
126. Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Bmj. 2005; 330: 1304-5.
127. Currie CJ, Poole CD, Jenkins-Jones S, Gale EA, Johnson JA, Morgan CL. Mortality after incident cancer in people with and without type 2 diabetes: impact of metformin on survival. Diabetes Care. 2012; 35: 299-304.
128. Noto H, Goto A, Tsujimoto T, Noda M. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 2012; 7: e33411. doi: 10.1371/journal.pone.0033411.
129. Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006; 29: 254-8.
130. Hsieh MC, Lee TC, Cheng SM, Tu ST, Yen MH, Tseng CH. The influence of type 2 diabetes and glucose-lowering therapies on cancer risk in the Taiwanese. Exp Diabetes Res. 2012; 2012: 413782. doi: 10.1155/2012/413782.
131. Monami M, Lamanna C, Balzi D, Marchionni N, Mannucci E. Sulphonylureas and cancer: a case-control study. Acta Diabetol. 2009; 46: 279-84.
132. Mazzone PJ, Rai H, Beukemann M, Xu M, Jain A, Sasidhar M. The effect of metformin and thiazolidinedione use on lung cancer in diabetics. BMC Cancer. 2012; 12: 410. doi: 10.1186/1471-2407-12-410.
133. Bodmer M, Becker C, Jick SS, Meier CR. Metformin does not alter the risk of lung cancer: a case-control analysis. Lung Cancer. 2012; 78: 133-7. doi: 10.1016/j.lungcan.2012.08.010.
134. Tan BX, Yao WX, Ge J, Peng XC, Du XB, Zhang R, Yao B, Xie K, Li LH, Dong H, Gao F, Zhao F, Hou JM, et al. Prognostic influence of metformin as first-line chemotherapy for advanced nonsmall cell lung cancer in patients with type 2 diabetes. Cancer. 2011; 117: 5103-11. doi: 10.1002/cncr.26151.
135. Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009; 27: 3297-302.
136. Mazzone PJ, Rai HS, Beukemann M, Xu M, Abdallah R, Sasidhar M. The Effect of Metformin and Thiazolidinedione Use on Lung Cancer. Chest. 2010; 138: supp 882A.
137. Skinner HD, McCurdy MR, Echeverria AE, Lin SH, Welsh JW, O’Reilly MS, Hofstetter WL, Ajani JA, Komaki R, Cox JD, Sandulache VC, Myers JN, Guerrero TM. Metformin use and improved response to therapy in esophageal adenocarcinoma. Acta Oncol. 2012.
138. Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012; 18: 290-300.
139. Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, Glicksman R, Chaib S, Zamiara P, Milosevic M, Wouters BG, Bristow RG, Koritzinsky M. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. 2013; 19: 6741-50. doi: 10.1158/1078-0432.CCR-13-1787.
140. Sandulache VC, Skinner HD, Ow TJ, Zhang A, Xia X, Luchak JM, Wong LJ, Pickering CR, Zhou G, Myers JN. Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer. 118: 711-21.
141. Wink KC, Belderbos JS, Dieleman EM, Rossi M, Rasch CR, Damhuis RA, Houben RM, Troost EG. Improved progression free survival for patients with diabetes and locally advanced non-small cell lung cancer (NSCLC) using metformin during concurrent chemoradiotherapy. Radiother Oncol. 2016; 118: 453-9. doi: 10.1016/j.radonc.2016.01.012.
142. Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010; 17: 351-60.
143. Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008; 15: 833-9.
144. Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen—induced lung tumorigenesis. Cancer Prev Res (Phila). 2010; 3: 1066-76.
145. Lai SW, Liao KF, Chen PC, Tsai PY, Hsieh DP, Chen CC. Antidiabetes drugs correlate with decreased risk of lung cancer: a population-based observation in Taiwan. Clin Lung Cancer. 2012; 13: 143-8. doi: 10.1016/j.cllc.2011.10.002.
146. Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N. mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer. 2006; 94: 195-9.
147. Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci. 2009; 122: 3589-94.
148. Guo D, Bell EH, Mischel P, Chakravarti A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr Pharm Des. 2014; 20: 2619-26.
149. Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013; 18: 153-61. doi: 10.1016/j.cmet.2013.05.017.
150. Lettieri Barbato D, Vegliante R, Desideri E, Ciriolo MR. Managing lipid metabolism in proliferating cells: new perspective for metformin usage in cancer therapy. Biochim Biophys Acta. 2014; 1845: 317-24. doi: 10.1016/j.bbcan.2014.02.003.
151. Storozhuk Y, Sanli T, Hopmans SN, Schultz C, Farrell T, Cutz JC, Steinberg GR, Wright J, Singh G, Tsakiridis T. Chronic modulation of AMP-Kinase, Akt and mTOR pathways by ionizing radiation in human lung cancer xenografts. Radiat Oncol. 2012; 7: 71. doi: 10.1186/1748-717X-7-71.