
INTRODUCTION
The epidermal growth factor receptor (EGFR) is constitutively activated either by somatic mutations or by overexpression in multiple cancer entities [1]. Overexpression of the receptor is associated with reduced overall survival and enhanced metastasis in patients with colorectal (CRC) or head and neck squamous cell cancer (HNSCC) [1–3]. Accordingly, the EGFR is attractive for therapeutic targeting by monoclonal antibodies or tyrosine kinase inhibitors (TKI). Large randomized clinical trials have confirmed the efficacy of the anti-EGFR antibodies cetuximab and panitumumab as monotherapy or in combination with chemo- and radiotherapy in patients with CRC and HNSCC [4–10]. The EGFR-TKIs gefitinib, erlotinib and afatinib have dramatically improved therapeutic options for patients with EGFR-mutated non-small cell lung cancer (NSCLC) [11–14]. However, primary and acquired resistances to therapies targeting the EGFR are a clinically relevant problem. Aberrant activation of the mitogen-activated protein kinase (MAPK) or the phosphatidylinositol 3- kinase/protein kinase B (PI3K/AKT) pathway which act downstream of the EGFR is implied in resistance to anti-EGFR therapies. Activation of both pathways may result from (i) oncogenic mutations of signaling mediators or (ii) cross activation by additional growth factor receptors. Several retrospective analyses confirmed that patients with CRC harboring mutations of Kirsten rat sarcoma viral oncogene homolog (KRAS), Neuroblastoma RAS viral oncogene homolog (NRAS), v-Raf murine sarcoma viral oncogene homolog B (BRAF) or phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), or loss of the phosphatase and tensin homolog (PTEN), a negative regulator of the PI3K/AKT pathway, associate with reduced response rates to anti-EGFR antibodies [9, 15–25]. KRAS or PIK3CA mutations are also negative predictors for efficacy of EGFR-TKI therapy in patients with NSCLC [26, 27]. Cross activation of EGFR downstream pathways by the hepatocyte growth factor receptor (c-MET) mediates resistance to EGFR-TKIs and anti-EGFR antibodies in NSCLC and CRC models [28, 29]. However, the relative impact of the PI3K/AKT and MAPK pathways on resistance to EGFR-targeting therapies is unclear. Extensive cross-talk between the MAPK and the PI3K/AKT pathway has been described [30–32]. Activation of RAS by growth factor receptor signaling nor by oncogenic mutation activates the rapidly accelerated fibrosarcoma family (RAF) but also PI3K. Extracellular signal–regulated kinases 1/2 (ERK1/2), which act downstream of RAF in the MAPK pathway, can activate the PI3K/AKT pathway at the level of tuberous sclerosis complex 1 and 2 (TSC1 and 2) or mammalian target of rapamycin complex 1 (mTORC1) [31]. In contrast, constitutively activated PI3K/AKT signaling negatively triggers the MAPK pathway by phosphorylation of inhibitory sites of RAF [32]. So far the exact molecular mechanisms how activation of these central pathways mediates resistance to anti-EGFR targeted therapy are unclear. Better understanding will help to develop therapeutic strategies that more patients can profit from EGFR-targeting drugs.
Against this background we established models to study the impact of isolated activation of the MAPK and PI3K/AKT pathways on the response to anti-EGFR therapy. In addition we correlated markers of pathway activation in tumor biopsies from patients with mCRC treated at the West German Cancer Center with their response to cetuximab.
We find that isolated activation of MAPK- or AKT-signaling equally mediates resistance to cetuximab in vitro and in vivo. While conditional activation of RAF-1 upregulated anti-apoptotic B-cell lymphoma-extra large (BCL-XL) and B-cell lymphoma 2 (BCL-2) proteins conditionally activated AKT stabilized anti-apoptotic myeloid leukemia cell differentiation protein 1 (MCL-1). In tumor samples from patients with mCRC biomarkers of MAPK activation strongly correlated with markers of PI3K/AKT activity, both in KRAS wild type and KRAS-mutated tumors. Pharmacologic inhibition of the MAPK and the PI3K/AKT cascade was most effective in sensitizing RAS-mutated CRC models to anti-EGFR antibody therapy. In conclusion, the MAPK and the PI3K/AKT pathways independently mediate resistance to anti-EGFR therapies.
RESULTS
Coactivation of the MAPK and PI3K/AKT pathways in RAS-mutated CRC
Somatic RAS mutations are negative predictors of the efficacy of anti-EGFR antibodies in patients with mCRC. We have previously shown that oncogenic RAS mediates resistance by upregulation and stabilization of the anti-apoptotic protein BCL-XL [33]. As RAS signaling is coupled to the MAPK and the PI3K/AKT pathways we aimed to develop models for functional dissection of the relative contribution of these pathways to the RAS-mediated resistance phenotype of CRC. To this end we stably expressed HRASG12V in the EGFR-positive, cetuximab-sensitive cancer cell lines A431 and Difi [33]. A431-HRASG12V- and Difi-HRASG12V cells exhibited higher levels of pERK1/2T202/Y204 and pAKTS473 than their RASwt counterparts (Figure 1A and data not shown). This indicates co- or cross-activation of MAPK and PI3K/AKT signaling by oncogenic RAS, which is in line with previous observations [30–32].
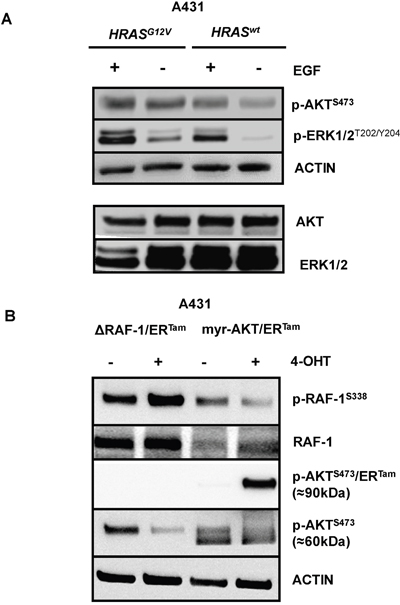
Figure 1: Coactivation of the MAPK and PI3K/AKT pathways in RAS-mutated EGFR-positive cancer cells. (A) A431 cells were retrovirally transduced to stably express the oncogenic RAS mutant HRASG12V. Constitutive and ligand-induced (EGF 10 ng/ml) phosphorylation of PI3K/AKT and MAPK signal transducers AKT and ERK1/2 in A431-HRASG12V cells or controls. ACTIN serve as control for equal loading. (B) A431-RAS wild type cells were retrovirally transduced to stably express a ΔRAF-1/ERTam- or a myristoylated-AKT/ERTam (myr-AKT/ERTam) construct. Phosphorylation of RAF-1 was strongly induced in A431-ΔRAF-1/ERTam cells and phosphorylation of myr-AKT/ERTam was strongly induced in A431-myr-AKT/ERTam cells by the addition of 4-hydroxytamoxifen (4-OHT).
Activated MAPK and PI3K/AKT signaling confers resistance to anti-EGFR targeted therapy
To dissect the relative contribution of each pathway to resistance against anti-EGFR therapy, we stably expressed a ΔRAF-1/ERTam- or a myristoylated-AKT/ERTam (myr-AKT/ERTam) construct in RAS wild type A431 and Difi cancer cell lines. Both transgenes are conditionally activated by addition of hydroxytamoxifen (4-OHT) [34]. Functional transgene expression was confirmed by immunoblot analyses of phosphoepitopes indicating 4-OHT-induced ΔRAF-1/ERTam- or myr-AKT/ERTam activation (Figure 1B and Supplementary Figure 1). Due the higher molecular weight of the myr-AKT/ERTam fusion construct (90kDa) the phosphorylated transgenic protein could be easily separated from endogenous AKT (60kDa). Interestingly, phosphorylation of endogenous RAF-1 was not increased in 4-OHT-treated A431-myr-AKT/ERTam cells, and phosphorylation of endogenous AKT was not enhanced in 4-OHT-treated A431-ΔRAF-1/ERTam cells. In fact, phosphorylation of these signaling mediators was rather reciprocally reduced, which might be explained by the activation of negative feedback regulation as suggested by Zimmermann and Moelling [35] (Figure 1B).
Next, we incubated both transgenic A431 cell lines with EGF, the monoclonal EGFR-antibody cetuximab, and the combination of both. In the absence of 4-OHT EGF dramatically induced the phosphorylation of EGFR, ERK1/2 and AKT indicating activation of the MAPK- and PI3K/AKT pathways (Figure 2A, 2B). In contrast, cetuximab reduced the activation of EGFR signaling. When A431-ΔRAF-1/ERTam cells were pre-incubated with 4-OHT markers of MAPK signaling were strongly activated, independently of incubation with EGF or cetuximab (Figure 2A). In line, 4-OHT pre-incubation of A431-myr-AKT/ERTam cells strongly induced markers of PI3K/AKT pathway activation (Figure 2B). Hence, our models were well suited for isolated functional analysis of either MAPK- or AKT-signaling (Figure 2A, 2B).
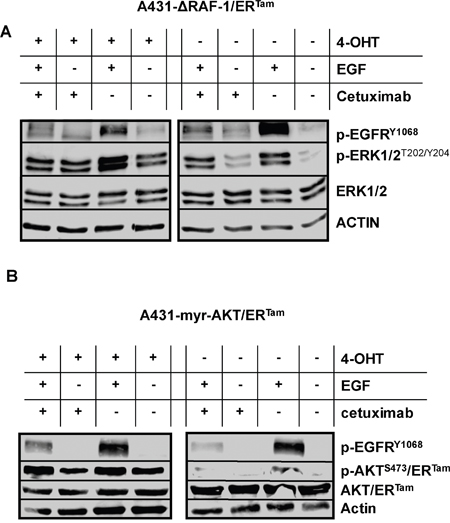
Figure 2: ΔRAF-1/ERTam and myr-AKT/ERTam restores EGFR downstream signaling in cetuximab treated cells. A431-ΔRAF-1/ERTam- (A) and A431-myr-AKT/ERTam (B) cells were incubated with 4-OHT, EGF (10 ng/ml) or cetuximab (1 μg/ml). (A) In the absence of 4-OHT, phosphorylation of EGFR and ERK was strongly induced by EGF. Cetuximab inhibited the ligand induced activation of EGFR downstream signaling. Upon pre-incubation with 4-OHT phosphorylation of ERK1/2 as marker of MAPK signaling was strongly induced, independently of incubation with EGF or cetuximab. (B) In the absence of 4-OHT, phosphorylation of EGFR and AKT/ERTam was strongly induced by EGF. Cetuximab inhibited the ligand induced activation of EGFR downstream signaling. Upon pre-incubation with 4-OHT phosphorylation of AKT/ERTam as marker of PI3K/AKT signaling was strongly induced, independently of incubation with EGF or cetuximab.
Next, we analyzed the impact of conditional pathway activation on the response to anti-EGFR therapy. In the absence of 4-OHT both clinically approved anti-EGFR antibodies, cetuximab and panitumumab, effectively inhibited proliferation and clonogenic survival of Difi cancer cells in vitro. The addition of 4-OHT protected Difi-ΔRAF-1/ERTam- and Difi-myr-AKT/ERTam cells against anti-EGFR antibody therapy in vitro (Figure 3A, 3B and Supplementary Figure 2). Moreover, 4-OHT-activated ΔRAF-1/ERTam and myr-AKT/ERTam protected Difi cells against cetuximab-induced apoptosis (Figure 3C, 3D). To study the impact of PI3K/AKT or MAPK pathway activation on the response to cetuximab in a host context, tumors were established by subcutaneous injection of A431 and Difi cells expressing the myr-AKT/ERTam or the ΔRAF-1/ERTam construct in non-obese diabetic severe combined immunodeficiency (NOD/SCID) mice. Mice were fed with diet with or without tamoxifen for transgene activation in vivo. Immunoblot analyses of explanted myr-AKT/ERTam tumors confirmed activation of myr-AKT/ERTam in tamoxifen-fed mice (Figure 4A). Explanted ΔRAF-1/ERTam tumors were immunohistochemically stained for pERK1/2. Tumors from tamoxifen-fed mice stained strongly positive for pERK1/2 (mean H-Score 129) compared to explanted tumors from mice without tamoxifen diet (mean H-Score 13, p<0.01; Mann-Whitney test) (Figure 4D and Supplementary Figure 3A). For further analyses of pathway activation by the myr-AKT/ERTAM construct we selected mice bearing Difi tumors as they proved highly responsive to cetuximab in vivo [33]. NOD/SCID mice bearing palpable flank tumors were fed with or without tamoxifen diet. Two days after the initiation of tamoxifen diet, mice were treated by twice weekly intraperitoneal antibody injections of cetuximab (0.5 mg) or the anti-CD20 control antibody rituximab (1 mg), and tumor development was monitored. Cetuximab induced a dramatic shrinkage of established Difi-myr-AKT/ERTam tumors in mice not receiving tamoxifen diet. When myr-AKT/ERTam was activated by tamoxifen tumors were significantly protected against cetuximab (Figure 4B). No difference in tumor growth was observed between mice fed with or without tamoxifen diet which were treated with the control antibody rituximab (Figure 4C). To study the impact of pathway activation with the ΔRAF-1/ERTam construct in vivo we applied an “adjuvant treatment setting” and A431 cells as previously published [33]. Mice were fed with or without tamoxifen for one week. Intraperitoneal antibody injections with cetuximab or the anti-CD20 control antibody rituximab (1 mg twice weekly) were initiated one day after subcutaneous implantation of A431-ΔRAF-1/ERTam cells. Tumor development and survival were monitored. Cetuximab completely prevented the outgrowth of A431-ΔRAF-1/ERTam tumors in NOD/SCID mice fed without tamoxifen diet (Figure 4E). In contrast all tamoxifen-fed mice developed tumors despite cetuximab treatment, resulting in a dramatically inferior survival (p=0.002, log rank) (Figure 4E and Supplementary Figure 3B). Treatment with the control antibody rituximab failed to prevent development of A431-ΔRAF-1/ERTam tumors in mice fed with or without tamoxifen diet. Survival times were short in both groups irrespective of tamoxifen diet (p=0.317, log rank) (Figure 4F and Supplementary Figure 3B).
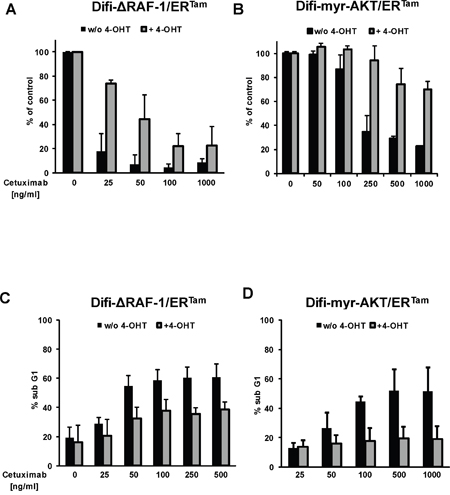
Figure 3: Activated MAPK and PI3K/AKT signaling confers resistance to anti-EGFR targeted therapy in vitro. Clonogenic survival of Difi-ΔRAF-1/ERTam (A) and Difi-myr-AKT/ERTam (B) cells cultured in the presence of cetuximab with (gray columns) or without (black columns) 4-OHT for 10 days. Mean colony numbers (+ SD) normalized to medium control from three independent experiments are given. (C, D) Induction of apoptosis by cetuximab in Difi-ΔRAF-1/ERTam (C) and Difi-myr-AKT/ERTam cells (D) with (gray columns) or without (black columns) 4-OHT for 48h. Mean percentages (+ SD) of cells with subgenomic DNA content from three independent experiments are given.
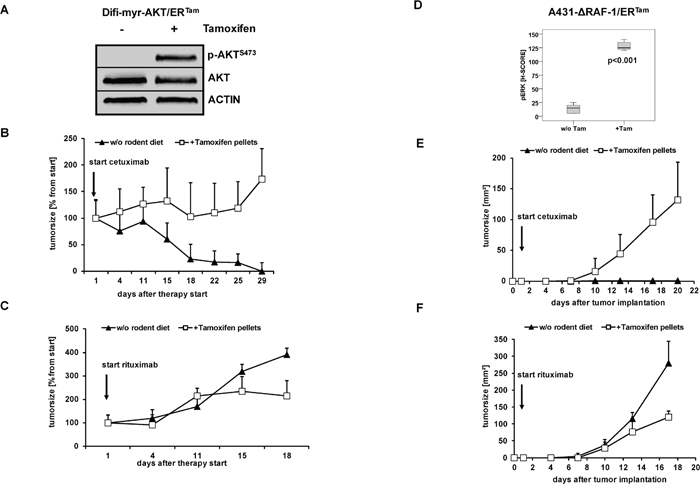
Figure 4: Activated PI3K/AKT and MAPK signaling protects tumors against anti-EGFR antibody-mediated cytotoxicity in vivo. (A) Palpable flank tumors were established by subcutaneous injection of A431-myr-AKT/ERTam cells in NOD/SCID mice. Tumor-bearing mice were fed with diet with or without tamoxifen for transgene activation in vivo for one week. A strong phosphorylation of AKT as marker of PI3K/AKT signaling was detected in protein lysates from explanted tumors by immunoblot analyses. (B, C) Tumor growth following injection of Difi-myr-AKT/ERTam cells in NOD/SCID mice fed with (open boxes) or without (closed triangles) tamoxifen. After tumors were palpable (arrow), mice were treated biweekly with intraperitoneal injections of cetuximab (0.5 mg) (B) or rituximab (1 mg) (C). Mean bidimensional tumor sizes (+ SD) of 5 mice per group are given. (E, F) NOD/SCID mice were fed with diet with or without tamoxifen for one week before A431-myr-ΔRAF-1/ERTam cells were subcutaneously implanted. The day after the tumor implantation mice were treated biweekly with intraperitoneal injections of cetuximab (1 mg) (E) or rituximab (1 mg) (F). Tumor growth in NOD/SCID mice fed with (open boxes) or without (closed triangles) tamoxifen was measured bidimensional twice weekly. Mean bidimensional tumor sizes (+ SD) of 5 mice per group are given. (D) Palpable flank tumors of mice treated with rituximab were explanted and analyzed by immunhistochemistry. A strong phosphorylation of ERK1/2 was detected in explanted tumors of mice fed with tamoxifen diet.
In conclusion, both signaling pathways, MAPK and PI3K/AKT, acting downstream of oncogenic RAS confer resistance of cancer cells to anti-EGFR antibody therapy in vitro and in vivo.
Activated MAPK and PI3K/AKT signaling upregulates anti-apoptotic BCL-2 proteins
Recently, we have shown that oncogenic RAS or activation of RAS downstream signaling mediates resistance by increased expression of anti-apoptotic BCL-2 proteins through transcriptional and posttranscriptional mechanisms [33, 36, 37]. BCL-2-type proteins protect against apoptosis by preventing permeabilization of the mitochondrial outer membrane [38]. Against this background, we analyzed the expression of the anti-apoptotic BCL-2 family members BCL-2, BCL-XL and MCL-1 in our cancer models with conditional pathway activation. Treating A431-ΔRAF-1/ERTam cells with 4-OHT induced the expression of BCL-XL and BCL-2, but not MCL-1. Blocking protein synthesis with cycloheximide revealed significantly enhanced stability of BCL-XL and BCL-2, suggesting a posttranslational mechanism (Figure 5A). In contrast, 4-OHT treatment of A431-myr-AKT/ERTam- and Difi-myr-AKT/ERTam cells had no impact on the expression of BCL-XL or BCL-2, but strongly induced and stabilized MCL-1 (Figure 5B, 5C) [36]. Cetuximab decreased the expression of MCL-1 in a time dependent manner, which was delayed by activation of myr-AKT/ERTam. In contrast, no change in BCL-XL levels was seen (Figure 5B, 5C). Interestingly, basal BCL-2 expression was very low but was strongly induced by the addition of cetuximab in both cell lines. As BCL-2 levels were increased following 4-OHT in the A431-ΔRAF-1/ERTam model (Figure 5A), this observation might be explained by AKT-induced compensatory feed-back activation of MAPK signaling (Figure 5B, 5C). In addition to anti-apoptotic BCL-2 family proteins, programmed cell death is regulated by pro-apoptotic family members. In this process the BH1-2-3 proteins bcl-2 homologous antagonist/killer (BAK) and bcl-2-like protein 4 (BAX) are essential mediators of mitochondrial outer membrane permeabilization [38, 39]. In line, cetuximab treatment induced the RNA expression of pro-apoptotic BAK in A431-myr-AKT/ERTam cells in the absence of 4-OHT. This effect was completely abolished by addition of 4-OHT (Figure 5D).
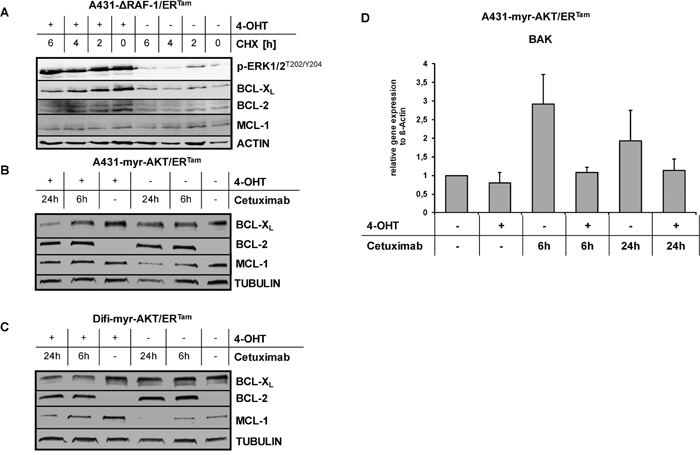
Figure 5: Upregulation of anti-apoptotic BCL2 proteins by activated MAPK and PI3K/AKT pathway. (A) Expression of BCL-XL, BCL-2 and MCL-1 in A431-ΔRAF-1/ERTam cells with or without 4-OHT pre-incubation. To prevent new protein synthesis cells were treated with cycloheximide (CHX, 10 mg/mL) for indicated periods. (B, C) Expression of BCL-XL, BCL-2 and MCL-1 in A431-myr-AKT/ERTam (B) and Difi-myr-AKT/ERTam (C) cells treated with cetuximab (1 μg/ml) for indicated periods. (D) Relative BAK transcript level in A431-myr-AKT/ERTam cells treated with cetuximab (1 μg/ml) for indicated periods with or without 4-OHT. Expression levels of BAK were normalized to the housekeeping gene beta-ACTIN. Mean change (+ SD) of BAK transcript levels from three independent experiments are given.
Taken together, conditional activation of RAF-1 signaling increased the expression of anti-apoptotic BCL-XL and BCL-2, whereas activation of AKT signaling stabilized anti-apoptotic MCL-1 and prevented the expression of pro-apoptotic BAK. The endpoint of both events is protection of cancer cells to apoptosis executed via the intrinsic pathway of caspase activation [39].
Biomarkers of MAPK and PI3K/AKT pathway activation correlate with outcome in cetuximab-treated colorectal cancer patients
To corroborate our preclinical findings and to specifically study the impact of activated MAPK and PI3K/AKT signaling on cetuximab-based therapy we analyzed a retrospective cohort of mCRC patients. Formalin-fixed, paraffin-embedded (FFPE) surplus tumor tissue obtained at diagnostic biopsy or resection was available for analysis from 39 patients with histologically confirmed CRC, who had been treated with cetuximab between 2004 and 2007. This cohort is particularly suited to compare the impact of oncogene mutations and biomarkers of pathway activation, as these patients were treated before cetuximab therapy was restricted to patients with RAS wild typic tumors [9]. Patients’ characteristics are summarized in Supplementary Table 1. Tissue sections were immunohistochemically stained for pERK1/2, pAKT and pp70S6K1 as markers of pathway activation, and tumor-derived DNA was analyzed for mutational status of KRAS exon 2 and BRAF exon 15 by amplicon sequencing.
KRAS exon 2 mutations were detected in 14 patients (39%), and BRAF exon 15 mutations were detected in 3 patients (9%). Almost 50% of tumors stained negatively or only slightly positive for pAKTS473 and thus were grouped as low expressers (0+ and 1+). The other 50% of tumors stained moderately or highly positive for pAKT and were grouped as high expressers (2+ and 3+) (Supplementary Table 2 and Supplementary Figure 4). Inhomogeneous staining patterns were detected for pERK1/2 and p-Ribosomal protein S6 kinase beta-1 (pp70S6K1). Thus, the absolute numbers of IHC-positive cells were counted instead of categorizing into low and high expressers. Samples were subsequently dichotomized according to IHC-positive cell counts below and above the median of the entire population (≤median; >median) (Supplementary Table 2 and Supplementary Figure 4). As expected detection of pAKT correlated with pp70S6K1 positivity as both proteins are activated by canonical PI3K/AKT signaling (Table 1A). In addition, a positive correlation was observed between pERK1/2 and pAKT positivity, and between pERK1/2 and pp70S6K1. This is in line with cross- or co-activation of MAPK and PI3K/AKT signaling in human CRC (Table 1A). Interestingly, this co-activation was equally observed in samples with wild-typic and mutated KRAS (exon 2) (Table 1B). As additional mutations in KRAS exons 3 and 4 and NRAS exon 2 to 4 are observed in mCRC and were described as negative predictors for the efficacy of anti-EGFR antibodies, we reanalyzed residual samples for these additional mutations. In 23 out of 39 samples the “all RAS and BRAF” mutational analyses was feasible, and in total 17 RAS or BRAF mutations were detected. The baseline characteristics of these 23 comprehensively analyzed patients were similar to the entire population (data not shown). Similar overall correlations between pAKT, pp70S6K1 and pERK positivity were observed in this subpopulation (Supplementary Table 3A). However, this was entirely carried by the subgroup of patients with mCRC mutated in RAS or BRAF (Supplementary Table 3B).
Table 1A: Study population (N=39)
All pts |
pAKTS473 low |
pAKTS473 high |
pERK1/2T202/Y204 ≤ median |
pERK1/2T202/Y204 > median |
|---|---|---|---|---|
pp70S6K1T389 < median |
13 |
8 |
18 |
3 |
pp70S6K1T389 > median |
6 |
12 |
3 |
15 |
p = 0.075, chi-square; |
p < 0.0001, chi-square; |
|||
pERK1/2T202/Y204 ≤ median |
14 |
7 |
||
pERK1/2T202/Y204 > median |
5 |
13 |
||
p = 0.015, chi-square; |
||||
Table 1B: KRAS exon 2 population (N=36)
KRAS wt |
KRAS mut |
KRAS wt |
KRAS mut |
|||||
|---|---|---|---|---|---|---|---|---|
pp70S6K1T389 ≤ median |
pp70S6K1T389 > median |
pp70S6K1T389 ≤ median |
pp70S6K1T389 > median |
pAKTS473 low |
pAKTS473 high |
pAKTS473 low |
pAKTS473 high |
|
pERK1/2T202/Y204 |
7 |
3 |
9 |
0 |
8 |
2 |
5 |
4 |
pERK1/2T202/Y204 |
2 |
10 |
1 |
4 |
4 |
8 |
0 |
5 |
p = 0.011, chi-square; |
p = 0.001, chi-square; |
p = 0.029, chi-square; odds ratio (95% CI): 8.000 (1.127-56.793) |
p = 0.038, chi-square; |
|||||
1For pp70S6K1T389 ≤ median cohort; 2for pAKTS473 high cohort.
To validate these findings a second, independent cohort of 88 mCRC patients was immunohistochemically analyzed for expression of pAKT, pERK1/2, pp70S6K1 and the phosphatase PTEN, a negative regulator of the PI3K/AKT pathway. An independent scoring system (H-Score) was applied, and biomarker positivity was correlated with the mutational status of KRAS, NRAS, BRAF and PIK3CA (Supplementary Tables 4A, 4B). Positive correlation between markers of MAPK and PI3K/AKT pathway activation was observed, which confirmed the findings obtained in the first patient cohort (Table 2A). In this validation cohort, RAS mutations were detected in 33% of patients, which associated with positivity for pERK1/2 as biomarker for MAPK pathway activation (Table 2B). PI3KCA mutations were found in 8% of patients which strongly correlated with positivity for pAKT and pp70S6K as biomarkers for PI3K/AKT pathway activation (Table 2B).
Table 2A: Marker correlation
All pts |
pAKTS473 |
pERK1/2T202/Y204 |
pp70S6K1T389 |
PTENloss |
|---|---|---|---|---|
pp70S6K1T389 |
||||
p-value* |
0.015# |
0.020# |
n.a. |
0.018# |
pERK1/2T202/Y204 |
||||
p-value* |
0.207 |
n.a. |
0.020# |
0.012# |
pAKTS473 |
||||
p-value* |
n.a. |
0.207 |
0.015# |
0.121# |
n.a.: not applicable; *Spearman-rho; #significance p < 0.05
Table 2B: RAS, BRAF, PIK3CA and marker correlation
pAKTS473 |
pERK1/2T202/Y204 |
pp70S6K1T389 |
PTENloss |
|
|---|---|---|---|---|
All RAS mutation1 |
||||
p-value* |
0.340 |
0.035# |
0.175 |
0.168 |
BRAF mutation2 |
||||
p-value* |
0.406 |
0.161 |
0.219 |
0.275 |
all RAS or BRAF mutation1,2 |
||||
p-value* |
0.392 |
0.111 |
0.097§ |
0.109 |
PIK3CA mutation3 |
||||
p-value* |
0.001* |
0.055§ |
0.496 |
0.214 |
n.a.: not applicable; 1KRAS exon 2-4, NRAS exon 2-4; 2BRAF exon 15; 3PIK3CA exon 10 and 21; *Spearman-Rho; #denotes significance (p < 0.05); §trend (p < 0.1).
Biomarkers of MAPK and PI3K/AKT pathway activation negatively predict response to cetuximab in patients with mCRC
Next, we correlated KRAS and BRAF mutation status and markers of downstream pathway activation with the clinical outcome of cetuximab-based therapy (Table 3 and Supplementary Table 3C-3F for “all RAS/BRAF” cohort). Patients with KRAS exon 2 wild-typic and “all RAS” wild-typic tumors, respectively, had a numerically higher ORR (Table 4A and Supplementary Table 3D). Formal statistical significance was not established due to the limited sample size. Interestingly, neither median Progression free survival (PFS) nor median Overall survival (OS) differed between patients with KRAS exon 2 wild-typic or “all RAS” wild-typic and mutated tumors (Table 4B, 4C and Supplementary Table 3E, 3F). No patient with a BRAF-mutated tumor objectively responded to cetuximab-based therapy. Median PFS was significantly reduced whereas median OS did not differ (Table 4A-4C and Supplementary Table 3E, 3F). Patients with tumors staining low for pERK1/2T202/Y204 had better ORR, longer median PFS and longer median OS as compared to patients with high pERK1/2T202/Y204 staining (Table 4A-4C, Supplementary Table 3D-3F and Figure 6). This finding was reproducible when patients were grouped by KRAS exon 2/BRAF or “all RAS/BRAF” mutational status. However, formal statistical significance was not established in the “all RAS” population due to limited sample size. Also, ORR of patients with low staining for pAKT or pp70S6K1 was higher than in patients with high staining for these biomarkers of AKT pathway activation, but PFS did not differ (Table 4A, 4B and Supplementary Table 3D-3F). The OS was numerically longer in patients with low pAKT or pp70S6K1 staining (Table 4C and Supplementary Table 3F and Supplementary Figure 5). In summary, these findings in mCRC patients support the negative predictive value of biomarkers of MAPK and PI3K/AKT pathway activation for response to cetuximab-based therapy.
Table 3A: Efficacy data (study population; N=39)
N |
% |
|
|---|---|---|
ORR |
7 |
18 |
CR |
0 |
0 |
PR |
7 |
18 |
SD |
22 |
56 |
PD |
10 |
26 |
Median PFS |
3.5 months (1.9-10.1) |
|
Median OS since start of cetuximab |
11.1 months (1.9-74.0) |
|
Table 4A: ORR
Marker |
ORR (%) |
Odds ratio (95% CI) |
p-value |
|---|---|---|---|
All patients |
18 |
||
KRAS wt vs mut* |
22 vs 14 |
1.7 (0.2-10.6) |
0.43 |
BRAF wt vs mut# |
19 vs 0 |
0.7 (0.1-8.2) |
0.54 |
pERK1/2T202/Y204 < median vs > median |
29 vs 6 |
6.8 (0.7-63.1) |
0.07 |
pAKTS473 0+/1+ vs 2+/3+ |
31 vs 5 |
8.7 (0.9-81.9) |
0.04 |
pp70S6K1T389 < median vs > median |
23 vs 11 |
2.5 (0.4-14.8) |
0.27 |
KRAS wt/BRAF wt and |
40 vs 14 |
4.0 (0.3-40.1) |
0.25 |
KRAS mut* or BRAF mut#
and |
22 vs 0 |
2.1 (1.2-3.7) |
0.16 |
KRAS wt/BRAF wt and |
40 vs 14 |
4.0 (0.3-47.1) |
0.25 |
KRAS mut* or BRAF mut# and |
33 vs 0 |
3.8 (1.6-8.7) |
0.04 |
KRAS wt/BRAF wt and |
43 vs 20 |
3.0 (0.3-25.9) |
0.31 |
KRAS mut* or BRAF mut# and |
17 vs 0 |
1.5 (1.0-2.1) |
0.33 |
*exon 2; #exon 15.
Table 4B: PFS
Marker |
Median PFS (months) |
HR (95% CI) |
p-value |
|---|---|---|---|
All patients |
3.5 |
||
KRAS wt vs mut* |
3.5 vs 4.0 |
0.8 (0.4-1.6) |
0.58 |
BRAF wt vs mut# |
3.5 vs 1.4 |
4.2 (1.1-15.7) |
0.03 |
pERK1/2T202/Y204 < median vs > median |
4.0 vs 3.1 |
1.8 (0.9-3.6) |
0.09 |
pAKTS473 0+/1+ vs 2+/3+ |
3.5 vs 4.1 |
0.7 (0.4-1.4) |
0.32 |
pp70S6K1T389 < median vs > median |
3.5 vs 3.7 |
0.9 (0.5-1.8) |
0.91 |
KRAS wt/BRAF wt and |
3.6 vs 3.0 |
1.9 (0.7-5.5) |
0.22 |
KRAS mut* or BRAF mut# and |
4.0 vs 2.5 |
1.7 (0.6-4.7) |
0.31 |
KRAS wt/BRAF wt and |
3.0 vs 5.0 |
0.9 (0.3-2.4) |
0.76 |
KRAS mut* or BRAF mut# and |
4.0 vs 3.3 |
0.8 (0.3-2.4) |
0.67 |
KRAS wt/BRAF wt and |
3.5 vs 4.1 |
1.0 (0.4-2.7) |
0.97 |
KRAS mut* or BRAF mut# and |
3.5 vs 2.8 |
0.9 (0.3-2.8) |
0.89 |
*exon 2; #exon 15.
Table 4C: OS
Marker |
Median OS (months) |
HR (95% CI) |
p-value |
|---|---|---|---|
All patients |
11.1 |
||
KRAS wt vs mut* |
11.3 vs 10.6 |
0.8 (0.4-1.6) |
0.55 |
BRAF wt vs mut# |
10.2 vs 10.8 |
1.5 (0.5-5.2) |
0.51 |
pERK1/2T202/Y204 < median vs > median |
13.0 vs 9.8 |
2.7 (1.3-5.6) |
<0.01 |
pAKTS473 0+/1+ vs 2+/3+ |
12.2 vs 10.4 |
1.5 (0.8-2.9) |
0.22 |
pp70S6K1T389 < median vs > median |
11.1 vs 10.4 |
1.2 (0.6-2.3) |
0.52 |
KRAS wt/BRAF wt and |
19.6 vs 6.1 |
4.2 (1.2-15.1) |
0.02 |
KRAS mut* or BRAF mut# and |
11.5 vs 7.8 |
3.2 (1.0-9.9) |
0.03 |
KRAS wt/BRAF wt and |
14.7 vs 9.5 |
2.0 (0.7-6.0) |
0.20 |
KRAS mut* or BRAF mut# and |
10.2 vs 10.8 |
1.0 (0.4-2.8) |
0.97 |
KRAS wt/BRAF wt and |
19.6 vs 9.5 |
1.4 (0.5-4.0) |
0.50 |
KRAS mut* or BRAF mut# and |
10.8 vs 7.8 |
1.9 (0.6-5.9) |
0.25 |
*exon 2; #exon 15.
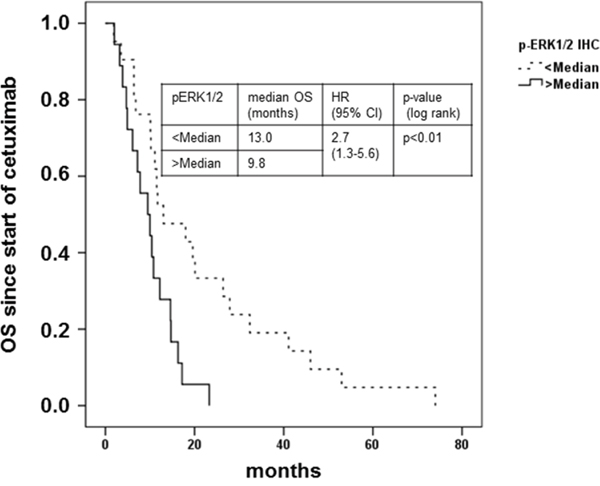
Figure 6: Clinical outcome of patients with metastatic colorectal cancer treated with cetuximab in combination with irinotecan in relation to pERK1/2T202/Y204 expression. Kaplan-Meier plot of overall survival (OS) from start of treatment with cetuximab in combination with irinotecan of patients with mCRC with immunohistochemical pERK1/2T202/Y204 stainig intensity below (dashed line) or above median (solid line). Patients with tumors stained for pERK1/2T202/Y204 below the median demonstrated a prolonged OS (13.0 vs. 9.8 months, HR 2.7; 95% CI 1.3-5.6); p < 0.01 log rank).
Combined pharmacologic targeting of MAPK and PI3K/AKT pathways resensitizes EGFR-positive cancer cells to anti-EGFR therapies
It is expected that rational combination of signaling inhibitors may delay or circumvent resistance, which is frequently encountered with targeted mono-therapies. This is exemplified by the recent approval of combined BRAF and MEK inhibition for therapy of patients with metastatic BRAF-mutated malignant melanoma [40–43]. In BRAF-mutated mCRC the rational use of BRAF inhibitors in combination with anti-EGFR targeting drugs has shown promising efficacy in preclinical models and in early clinical trials [44–46]. To explore whether combined MAPK and/or PI3K/AKT pathway inhibition would overcome RAS-mediated resistance to cetuximab we studied Difi-HRASG12V cells and HCT116 cells, which harbor an endogenous KRAS exon 2 mutation and an additional PIK3CA exon 20 mutation. First, we tested the combination of cetuximab with the MEK inhibitor U0126 in Difi-HRASG12V cells. U0126 inhibited the constitutive as well as EGF-induced phosphorylation of ERK1/2, whereas cetuximab did not inhibit ERK1/2 phosphorylation (Supplementary Figure 6). In line, cetuximab only marginally inhibited the proliferation of Difi-HRASG12V cells. In contrast, U0126 strongly suppressed the proliferation of Difi-HRASG12V cells. The combination of U0126 and cetuximab acted synergistically and completely inhibited proliferation of Difi-HRASG12V cells (Figure 7A).
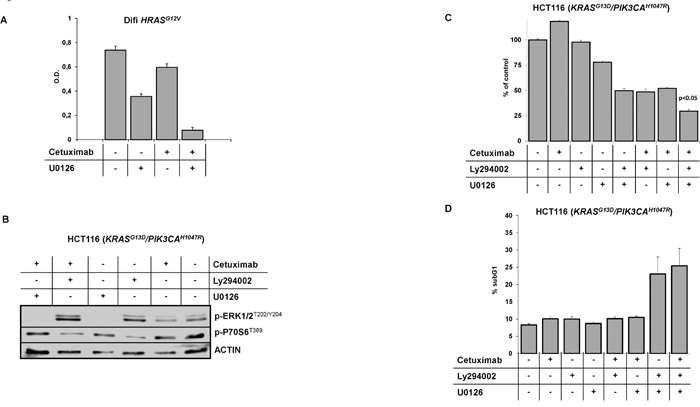
Figure 7: Combined pharmacologic targeting of MAPK and PI3K/AKT signaling resensitizes EGFR positive cells to anti-EGFR therapies. (A) Proliferation of Difi-HRASG12V cells grown in the presence of cetuximab (100 ng/ml), the MEK inhibitor U0126 (1 μM) or in combination. Mean values (± SD) of three independent MTT assays. (B) Immunoblot analysis of HCT116 which harbors an endogenous KRAS exon 2 and an additional PIK3CA exon 20 mutation treated with cetuximab (20 μg/ml), the PI3K inhibitor Ly294002 (10 μM), the MEK inhibitor U0126 (1 μM) and in combination. Inhibition of constitutive phosphorylation of MAPK and PI3K/AKT signal transducers ERK1/2 and p70S6 by the pharmacological inhibitors Ly294002 and U0126, respectively. (C) Proliferation of HCT116 cells grown in the presence of cetuximab (20 μg/ml), the PI3K inhibitor Ly294002 (10 μM), the MEK inhibitor U0126 (1 μM) and in combination. Mean values (± SD) of three independent MTT assays. (D) Fraction of apoptotic HCT116 cells with subgenomic DNA content (sub-G1) following treatment with cetuximab (20 μg/ml], Ly294002 (15 μM), U0126 (1 μM) and in combination (mean ± SD of three independent experiments).
Next, we combined inhibitors of PI3K (Ly294002) and MEK (U0126) in the HCT116 model. HCT116 cells exhibit phosphorylation of ERK1/2 and p70S6K1 ribosomal protein, indicating constitutive activation of MAPK and PI3K/AKT signaling (Figure 7B). Treatment with cetuximab only slightly inhibited the phosphorylation of ERK1/2 and S6 ribosomal protein. Ly294002 markedly inhibited the phosphorylation of S6 ribosomal protein whereas phosphorylation of ERK1/2 was induced, arguing for compensatory feed-back activation. Treatment with U0126 completely prevented ERK1/2 phosphorylation. Treatment of HCT116 cells with cetuximab or the PI3K inhibitor Ly294002 alone failed to inhibit proliferation, while the MEK inhibitor U0126 had some anti-proliferative activity (Figure 7C). Interestingly the combination of Ly294002 with U0126 or the combination of one of the inhibitors with cetuximab dramatically reduced cell proliferation. The combination of all three compounds had the strongest effect with a nearly complete inhibition of proliferation. Neither treatment with cetuximab, Ly294002 or U0126 alone nor the combination of cetuximab with one of the inhibitors induced apoptosis in HCT116 cells (Figure 7D). In contrast, the combination of Ly294002 and U0126 markedly induced apoptosis. This effect could be further enhanced by the addition of cetuximab.
In conclusion, our preclinical and clinical data provide a strong argument to combine inhibitors of the MAPK and the PI3K/AKT pathways to overcome resistance to cetuximab in RAS-mutated colorectal cancer.
DISCUSSION
The monoclonal anti-EGFR antibodies cetuximab and panitumumab are effective as monotherapy or in combination with chemotherapy in patients with mCRC [4–6, 9, 10]. In addition cetuximab is also approved in combination with radiotherapy for patients with locally advanced HNSCC ineligible for platinum based chemotherapy or in combination with platinum in the recurrent or metastatic setting [7, 8]. However, not all patients respond to this targeted therapy or acquire resistance. In patients with mCRC activating mutations of the RAS, BRAF and PIK3CA oncogenes have been identified as key predictors of primary resistance in comprehensive biomarker-analyses of large clinical trials [9, 15–25]. Additional aberrations associated with resistance to anti-EGFR antibody therapy in mCRC comprise amongst others amplification, mutations or overexpression of the human epidermal growth factor receptor 2 (HER2) and HER3, the hepatocyte growth factor receptor (c-MET), the platelet-derived growth factor receptor A (PDGFRA) and the fibroblast growth factor receptor 1 (FGFR1) [55, 56]. While these biomarkers which can be readily assessed in tumor biopsies indicate a clinical resistance phenotype, the actual mechanisms how resistance is mediated is less well defined. Current thinking implies deregulated intracellular signal transduction pathways as the main effectors of primary and acquired resistance to anti-EGFR antibody therapy. In particular the MAPK and PI3K/AKT pathways, which are activated by most growth factor receptor tyrosine kinases, are considered. However, these pathways do not act in linear, monodirectional ways but are closely interconnected with each other and additional signaling pathways. Hence, a clinical resistance phenotype has to be appreciated as the output of a complex interplay of external and internal signaling cues that are integrated by the cancer cell. Further understanding of such cross-talk and the exact molecular mechanisms how pathway activation mediates resistance is necessary to develop more precise therapeutic strategies breaking resistance to anti-EGFR antibodies.
Using transgenic expression of conditionally activatable activators of canonical MAPK and PI3K/AKT signaling we here demonstrate in vitro and in relevant in vivo models that either pathway is independently capable of mediating resistance to EGFR blockade. However, enforced expression of RAS mutants simultaneously activates both pathways, which is in line with cross-activation as previously described [30–32]. The clinical relevance of this observation is supported by our biomarker study of two independent cohorts of patients with mCRC, which revealed a strong correlation between markers of signal transduction pathway activation in primary tumor samples. Interestingly, an association of positivity for biomarkers of activated MAPK or PI3K/AKT signaling with lower likelihood of response to cetuximab and reduced overall survival from start of cetuximab treatment was found in patients with mCRC that was independent of the RAS or BRAF mutational status. This observation underscores the importance of pathway activation as mechanistic basis of resistance, which may result from somatic mutations or RAS or BRAF proto-oncogenes but also other causes. In the light of the limited sample size of our two clinical cohorts, these findings, however, require validation in larger, independent cohorts of patients with mCRC treated with anti-EGFR antibodies.
Conceptually our findings may have implications on therapeutic strategies to overcome resistance to cetuximab or panitumumab. In our preclinical models combined pharmacological modulation of the MAPK and the PI3K/AKT pathway by specific small molecule inhibitors sensitized RAS-mutated cancers to cetuximab. Clinical translation of these findings may be challenging with the toxicities of currently available inhibitors of MEK and PI3K. However, novel compounds specifically targeting ERK and specific subtypes of the RAF and PI3K kinases effective combination therapies may become clinically feasible [59, 60]
Deregulated growth factor signaling frequently impacts on the regulation of apoptotic cells death. In particular, high expression of anti-apoptotic proteins of the BLC-2 family is observed over several cancer entities, which is in line with our present findings in cancer models. Activation of the MAPK pathway resulted in stabilization of BCL-XL and BCL-2, whereas activation of the PI3K/AKT pathway resulted in an induction of MCL-1. We have previously identified BCL-XL upregulation as a main effector in preventing RAS mutant CRC cell lines from anti-EGFR antibody induced apoptosis, and in CD20-positive B-cell Non-Hodgkin Lymphoma (B-NHL) cells from rituximab-induced apoptosis [33, 57]. MCL-1 was previously identified by our group and others as mediator of resistance against cytotoxic drugs in acute myeloid leukemia (AML) but also against antigen-specific cytotoxic T-cells in solid tumor models [36, 58]. Mechanistically, these BCL-2 type proteins inhibit apoptosis by binding of the pro-apoptotic proteins BAX and BAK to prevent permeabilization of the mitochondrial outer membrane and subsequent caspase activation [38, 39]. In our most sensitive cancer models cetuximab induced apoptosis in vitro and shrinkage of established tumors in vivo. Interestingly, this associated with cetuximab-induced downregulation of MCL-1 and transcriptional induction of pro-apoptotic BAK, and both events were prevented by conditional activation of AKT signaling. We previously demonstrated that modulation of BCL-2 type proteins by pharmacological inhibitors or by siRNA sensitized tumor cells to different therapeutic strategies including cytotoxic drugs, immunotherapies and monoclonal antibodies [33, 36, 57, 58]. Thus, these molecular effectors of treatment resistance mediated by aberrant MAPK and PI3K/AKT pathway activation are additional targets of interest strategies to improve EGFR-directed therapies.
MATERIALS AND METHODS
Cell lines and reagents
The human EGFR-positive cancer cell lines A431 and the colorectal cancer cells HCT116 harboring an endogenous KRAS exon 2 and a PIK3CA exon 20 mutation were obtained from DSMZ (Braunschweig, Germany). Difi cells were obtained from R. Coffey (Nashville, Tennessee). All cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS, PAA, Coelbe, Germany), L-glutamine, penicillin and streptomycin (Invitrogen, Frankfurt, Germany). Stable expression of KRASG12V or HRASG12V was achieved by retroviral transduction as described previously [47]. The myristoylated-AKT/ERTam (myr-AKT/ERTam) construct kindly provided by J. Downward cloned into the retroviral vector plasmid pQCxIP (Clonetech) and the ΔRAF-1/ERTam construct subcloned into the retroviral pBabePuro vector kindly provided by S. Cook were stably expressed as described previously [36]. Clinical grade cetuximab (Erbitux, Merck Serono, Darmstadt, Germany) and rituximab (Mabthera, Roche, Grenzach-Wyhlen, Germany) were purchased from the pharmacy of the University Hospital Essen; U0126, Ly294002, 4-hydroxytamoxifen (4-OHT) were purchased from Sigma (Deisenhofen, Germany).
The following primary antibodies were used for immunoblotting following standard protocols: AKT1/2 (H-136), BCL-2 (C2) (all from Santa Cruz Biotechnology, Santa Cruz, CA), phospho-ERK1/2T204Y204, ERK1/2, phospho-AKTS473, BCL-XL (54H6), EGFR, phospho-EGFRY1068, phospho-S6 ribosomal protein (all from Cell Signaling Technology, Danvers, MA), actin (C4, ICN, Irvine, CA), MCL-1 (Epitomics, Burlingame, CA).
Gene expression analysis
For RNA expression analysis, total RNA was isolated (High Pure RNA Isolation Kit, Roche Diagnostics, Mannheim, Germany) and reversely transcribed into cDNA (Transcription High Fidelity cDNA Synthesis Kit, Roche Diagnostics, Mannheim, Germany) following the manufacturer’s instructions. Quantitative RT-PCR analysis was performed on a LC480 instrument using SYBR Green 1 Master chemistry (Roche Diagnostics, Mannheim, Germany) and primers for human BAK: 5'-AACCGACGCTATGACT-3', 5'-TCGTACCACAAACTGGC-3' and human ACTIN: 5'-TCA GCT GTG GGG TCC TGT-3', 5'-GAA GGG ACA GGC AGT GAG-3' as previously described [33].
Animal models
All animal studies were conducted in compliance with institutional guidelines and German Animal Protection Law, and were approved by the responsible regulatory authority (Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein-Westfalen, Az. G969/08). NOD/SCID mice (Charles River Laboratories, France) received single subcutaneous flank injections of 2x106 A431 or 1x107 Difi cells suspended in 200 μl saline. Tamoxifen rodent diet (400 mg/kg) was obtained by Harlan Teklad (Teklad CRD Tam 400/CreER, irradiation by 21 kGy/min). Animals were monitored for tumor development twice weekly, and tumor growth was bidimensionally quantified using a caliper. Antibodies were dissolved in 200 μl saline and administered as biweekly intraperitoneal injections. Protein lysates of representative tumors were prepared as described previously and analyzed by Immunoblotting [36]. After mice were scarified, tumors were explanted and fixed over night in 4% formalin. Sections were subjected to hematoxilin & eosin staining and immunohistochemical analyses following diagnostic protocols of the Institute of Pathology. The pERK1/2T202/Y204 staining was categorized according to the H-Score as described previously [53]. For statistical analysis, the Mann-Whitney test was used.
Cellular assays
For clonogenic survival analysis, 5x103 A431 or Difi cells were seeded in 6-well plates in the presence of the indicated antibodies or pharmacological inhibitors. Following incubation for 7 to 14 days colonies were fixed with ethanol (70% v/v) for 30 minutes, stained with Coomassie brilliant blue and automatically counted using an Infinity-100 System and the Vision Capt software (Vilber Lourmat, Eberhardzell, Germany). Proliferation was quantified by means of the MTT assay according to the manufacturer’s instruction (Roche, Mannheim, Germany). Apoptosis was quantified by flow cytometric determination of cells with subgenomic DNA content following hypotonic lysis and staining with propidium iodide as previously described [44]. All results were obtained from at least three independent experiments.
Analysis of primary tumor samples
Clinical data and surplus tumor specimens were retrieved from 39 patients with metastatic CRC. All patients were heavily pretreated and had a documented disease progression before initiation of irinotecan based chemotherapy. Subsequently, patients were switched to irinotecan in combination with cetuximab as established by the phase III “Bond”-trial [4]. All patients except two received irinotecan and cetuximab on a weekly schedule, whereas two patients received irinotecan every three weeks and cetuximab weekly. Baseline characteristics are summarized in Supplementary Table 1. Overall response rate (ORR) was determined by RECIST 1.1 [48]. Differences between the groups were calculated using the chi-square test and odds ratios (OD). Progression free survival (PFS) was defined as time from start of irinotecan and cetuximab to progressive disease or death, overall survival (OS) was defined as time from start of therapy to death. For statistical analyses, univariate Cox proportional hazards and log rank tests were used. Tissue microarrays were prepared from formalin-fixed, paraffin-embedded (FFPE) tumor samples. Sections were subjected to hematoxilin & eosin staining and immunohistochemical analyses following diagnostic protocols of the Institute of Pathology. The pAKTS473 staining was categorized into negative (0+), weak (1+), moderate (2+) and strong (3+) [49]. For the quantification of pERK1/2T202/Y204 and pp70S6K1T389 positive cells were counted due to an inhomogeneous staining pattern [50, 51]. An independent scoring system according to the H-Score was used to analyze pERK1/2T202/Y204, pAKTS473 and pp70S6K1T389 staining of the second patient cohort with newly diagnosed metastatic colorectal cancer as described previously [53]. In addition the immunhistochemical expression of PTEN was analyzed as described [52]. Baseline characteristics of the second cohort are summarized in Supplementary Table 4A.
For statistical analysis, chi-square test was used. Tumor DNA was isolated from FFPE tumor sections following microdissection. KRAS and BRAF mutation status was determined in the patient cohort treated with cetuximab and irinotecan by PCR amplification of the relevant exons followed by Sanger sequencing as described previously [52]. DNA from patients without KRAS Exon 2 or BRAF mutations and DNA from the second patient cohort were analyzed by a 15-gene/62-exon panel read including exon 2-4 of KRAS and NRAS, Exon 15 of BRAF and exon 10 and 21 of PIK3CA on the MiSeq platform (Illumina, San Diego, CA) as described [52]. All studies of patient data or samples were approved by the Ethics Committee of the Medical Faculty of the University Duisburg-Essen (AZ.: 05-2882).
Author contributions
Study design: SK, SB, TT, FB, MS. Data Acquisition: SK, HR, SZ, SN, AM, MG, LP, ST, SW, AE, KW, MP, MW. Analysis and data interpretation: SK, HR, AM, AE, MG, KW, MW, JM, AP, KWS, FN, MS. Material support: AP, KWS. Manuscript writing: SK, MS. Critical revision of the manuscript: SK, FB, SN, SW, MS.
ACKNOWLEDGMENTS
We thank Sabine Harde and the staff of the Central Animal Facility, and the Pathology and Molecular Pathology Laboratories, University Hospital Essen, for their support. This work was funded by grants from the Wilhelm Sander-Stiftung (2005.136.3, M.S.), the Deutsche Forschungsgemeinschaft (SCHU 1541/5-1, M.S.), the “Stiftung Kopf-Hals Tumor” (S.K.), Mercator Research Center Ruhr-MERCUR (An-2011-0031, S.K.) and the IFORES program of the Medical Faculty of the University Duisburg-Essen (M.S.). The West German Cancer Center at University Hospital Essen is supported by an Oncology Center of Excellence grant of the Deutsche Krebshilfe (number 110534).
CONFLICTS OF INTEREST
Stefan Kasper has served as consultant and received speakers’ honoraria from Amgen, Bayer, BMS, Celgene, Lilly, Merck, MSD, Roche and Sanofi. Johannes Meiler has served as consultant and received speakers’ honoraria from Novartis, Sanofi, Amgen and SOBI. Martin Schuler has served as consultant to AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Lilly, Novartis, Roche, and his institution has received research grants from Boehringer Ingelheim, Bristol-Myers Squibb, and Novartis; Tanja Trarbach has received research funding, consulting and speaker honoraria from Merck and Amgen.
REFERENCES
1. Mayer A, Takimoto M, Fritz E, Schellander G, Kofler K, Ludwig H. The prognostic significance of proliferating cell nuclear antigen, epidermal growth factor receptor, and mdr gene expression in colorectal cancer. Cancer. 1993; 71:2454-60.
2. Maurizi M, Almadori G, Ferrandina G, Distefano M, Romanini ME, Cadoni G, Benedetti-Panici P, Paludetti G, Scambia G, Mancuso S. Prognostic significance of epidermal growth factor receptor in laryngeal squamous cell carcinoma. Br J Cancer. 1996; 74:1253-7.
3. Hemming AW, Davis NL, Kluftinger A, Robinson B, Quenville NF, Liseman B, LeRiche J. Prognostic markers of colorectal cancer: an evaluation of DNA content, epidermal growth factor receptor, and Ki-67. J Surg Oncol. 1992; 51:147-52.
4. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004; 351:337-45.
5. Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2009; 27:663-71.
6. Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D'Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009; 360:1408-17.
7. Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, Erfan J, Zabolotnyy D, Kienzer HR, Cupissol D, Peyrade F, Benasso M, Vynnychenko I, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008; 359:1116-27.
8. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, Jones CU, Sur R, Raben D, Jassem J, Ove R, Kies MS, Baselga J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006; 354:567-78.
9. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26:1626-34.
10. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010; 28:4697-705.
11. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009; 361:947-57.
12. Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, Chao TY, Nakagawa K, Chu DT, Saijo N, Duffield EL, Rukazenkov Y, Speake G, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011; 29:2866-74.
13. Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011; 12:735-42.
14. Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai CM, Boyer M, Su WC, Bennouna J, Kato T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013; 31:3327-34.
15. Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008; 359:1757-65.
16. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-62.
17. Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011; 22:1535-46.
18. Peeters M, Oliner KS, Parker A, Siena S, Van Cutsem E, Huang J, Humblet Y, Van Laethem JL, André T, Wiezorek J, Reese D, Patterson SD. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013; 19:1902-12.
19. Peeters M, Douillard JY, Van Cutsem E, Siena S, Zhang K, Williams R, Wiezorek J. Mutant KRAS codon 12 and 13 alleles in patients with metastatic colorectal cancer: assessment as prognostic and predictive biomarkers of response to panitumumab. J Clin Oncol. 2013; 31:759-65.
20. Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013; 369:1023-34.
21. Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon JL, Hecht JR, Yu H, Oliner KS, Go WY. PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild-type KRAS exon 2 metastatic colorectal cancer. J Clin Oncol. 2014; 32:2240-7.
22. Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014; 15:1065-75.
23. Van Cutsem E, Lenz HJ, Köhne CH, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, Ciardiello F. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J Clin Oncol. 2015; 33:692-700.
24. Peeters M, Oliner KS, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, André T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, et al. Analysis of KRAS/NRAS mutations in a phase III study of panitumumab with FOLFIRI compared with FOLFIRI alone as second-line treatment for metastatic colorectal cancer Res. 2015; 21:5469-79.
25. Hsu HC, Thiam TK, Lu YJ, Yeh CY, Tsai WS, You JF, Hung HY, Tsai CN, Hsu A, Chen HC, Chen SJ, Yang TS. Mutations of KRAS/NRAS/BRAF predict cetuximab resistance in metastatic colorectal cancer patients. Oncotarget. 2016; 7:22257-70. doi: 10.18632/oncotarget.8076.
26. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, Bekele BN, Herbst RS, Wistuba II. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007; 13:2890-6.
27. Ludovini V, Bianconi F, Pistola L, Chiari R, Minotti V, Colella R, Giuffrida D, Tofanetti FR, Siggillino A, Flacco A, Baldelli E, Iacono D, Mameli MG, et al. Phosphoinositide-3-kinase catalytic alpha and KRAS mutations are important predictors of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2011; 6:707-15.
28. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007; 316:1039-43.
29. Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011; 17:472-82.
30. Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Bäsecke J, Stivala F, Donia M, Fagone P, Malaponte G, Mazzarino MC, Nicoletti F, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011; 2:135-64. doi: 10.18632/oncotarget.240.
31. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011; 36:320-8.
32. Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002; 277:31099-106.
33. Kasper S, Breitenbuecher F, Reis H, Brandau S, Worm K, Köhler J, Paul A, Trarbach T, Schmid KW, Schuler M. Oncogenic RAS simultaneously protects against anti-EGFR antibody-dependent cellular cytotoxicity and EGFR signaling blockade. Oncogene. 2013; 32:2873-81.
34. Kohn AD, Barthel A, Kovacina KS, Boge A, Wallach B, Summers SA, Birnbaum MJ, Scott PH, Lawrence JC Jr, Roth RA. Construction and characterization of a conditionally active version of the serine/threonine kinase Akt. J Biol Chem. 1998; 273:11937-43.
35. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999; 286:1741-4.
36. Hahnel PS, Thaler S, Antunes E, Huber C, Theobald M, Schuler M. Targeting AKT signaling sensitizes cancer to cellular immunotherapy. Cancer Res. 2008; 68:3899-906.
37. Marques CA, Hahnel PS, Wolfel C, Thaler S, Huber C, Theobald M, Schuler M. An immune escape screen reveals Cdc42 as regulator of cancer susceptibility to lymphocyte-mediated tumor suppression. Blood. 2008; 111:1413-9.
38. Green DR. Apoptotic pathways: ten minutes to dead. Cell. 2005; 121:671-4.
39. Um HD. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget. 2016; 7:5193-203. doi: 10.18632/oncotarget.6405.
40. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandalà M, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014; 371:1877-88.
41. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015; 372:30-9.
42. Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion-Sileni V, Lebbe C, Mandalà M, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015; 386:444-51.
43. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014; 371:1867-76.
44. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012; 483:100-3.
45. Van Geel R, Elez E, Bendell JC, Faris JE, Lolkema MP, Eskens F, Spreafico A, Kavan P, Delord JP, Schuler MH, Wainberg ZA, Yamada Y, Yoshino T, et al. Phase I study of the selective BRAFV600 inhibitor encorafenib (LGX818) combined with cetuximab and with or without the α-specific PI3K inhibitor BYL719 in patients with advanced BRAF-mutant colorectal cancer. J Clin Oncol. 2014; 32:5s.
46. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015; 373:726-36.
47. Schuler M, Maurer U, Goldstein JC, Breitenbucher F, Hoffarth S, Waterhouse NJ, Green DR. p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ. 2003; 10:451-60.
48. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version1.1). Eur J Cancer. 2009; 45:228-47
49. Scartozzi M, Giampieri R, Maccaroni E, Mandolesi A, Biagetti S, Alfonsi S, Giustini L, Loretelli C, Faloppi L, Bittoni A, Bianconi M, Del Prete M, Bearzi I, Cascinu S. Phosphorylated AKT and MAPK expression in primary tumours and in corresponding metastases and clinical outcome in colorectal cancer patients receiving irinotecan-cetuximab. J Transl Med. 2012; 10:71.
50. Lugli A, Zlobec I, Minoo P, Baker K, Tornillo L, Terracciano L, Jass JR. Role of the mitogen-activated protein kinase and phosphoinositide 3-kinase/AKT pathways downstream molecules, phosphorylated extracellular signal-regulated kinase, and phosphorylated AKT in colorectal cancer—a tissue microarray-based approach. Hum Pathol. 2006; 37:1022-31.
51. Baba HA, Wohlschlaeger J, Cicinnati VR, Hilgard P, Lang H, Sotiropoulos GC, Takeda A, Beckebaum S, Schmitz KJ. Phosphorylation of p70S6 kinase predicts overall survival in patients with clear margin-resected hepatocellular carcinoma. Liver Int. 2009; 29:399-405.
52. Wiesweg M, Ting S, Reis H, Worm K, Kasper S, Tewes M, Welt A, Richly H, Meiler J, Bauer S, Hense J, Gauler TC, Köhler J, et al. Feasibility of preemptive biomarker profiling for personalised early clinical drug development at a Comprehensive Cancer Center. Eur J Cancer. 2013; 49:3076-82.
53. Reis H, Herold T, Ting S, Worm K, Huber U, Christoph DC, Eberhardt WE, Kostbade K, Kasper S, Stamatis G, Welter S, Darwiche K, Karpf-Wissel R, et al. HER2 expression and markers of phosphoinositide-3-kinase pathway activation define a favorable subgroup of metastatic pulmonary adenocarcinomas. Lung Cancer. 2015; 88:34-41.
54. Wiesweg M, Eberhardt WE, Reis H, Ting S, Savvidou N, Skiba C, Herold T, Christoph DC, Meiler J, Worm K, Kasper S, Theegarten D, Hense J, et al. High prevalence of concomitant oncogene mutations in prospectively identified patients with ROS1-positive metastatic lung cancer. J Thorac Oncol. 2017; 12:54-64.
55. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015; 526:263-7.
56. Cushman SM, Jiang C, Hatch AJ, Shterev I, Sibley AB, Niedzwiecki D, Venook AP, Owzar K, Hurwitz HI, Nixon AB. Gene expression markers of efficacy and resistance to cetuximab treatment in metastatic colorectal cancer: results from CALGB 80203 (Alliance). Clin Cancer Res. 2015; 21:1078-86.
57. Stolz C, Hess G, Hähnel PS, Grabellus F, Hoffarth S, Schmid KW, Schuler M. Targeting Bcl-2 family proteins modulates the sensitivity of B-cell lymphoma to rituximab-induced apoptosis. Blood. 2008; 112:3312-21.
58. Kasper S, Breitenbuecher F, Heidel F, Hoffarth S, Markova B, Schuler M, Fischer T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J. 2012; 2:e60.
59. Soulières D, Faivre S, Mesía R, Remenár É, Li SH, Karpenko A, Dechaphunkul A, Ochsenreither S, Kiss LA, Lin JC, Nagarkar R, Tamás L, Kim SB, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017; 18:323-35.