
INTRODUCTION
Cancer is a short term evolutionary process. Tumor cells acquiring cancer driver mutations give rise to sub-clonal populations that undergo Darwinian selection that can lead to clonal expansion or eradication [1, 2]. Two major models of cancer evolution have been described, linear and branched [1]. In linear evolution a clone acquiring a beneficial mutation undergoes expansion and eventually eliminates the ancestral clone, whereas in branched evolution, different sub-clones expand in parallel. Both models are compatible with the presence of sub-clonality, where some of the mutations are present only in a subset of the tumor cell population.
Sub-clonality has already been reported in multiple malignancies [3–6]. Specifically, in lung adenocarcinoma high throughput sequencing analysis of different tumor lesions in a small set of patients showed evidence of sub-clonality and branched evolution for driver events [7–9]. As for the common driver events (i.e. EGFR mutation and ALK rearrangement) the presence of sub-clonality in lung adenocarcinoma is under debate. Comparison between different lung nodules and between primary and metastatic lesions showed up to 30% discordance for EGFR mutation [10, 11] and 50% for ALK immunohistochemistry [12], in some reports. Additionally, even within the same lesion, multi-region sampling provided evidence for sub-clonality for EGFR mutation status [13, 14] and for ALK rearrangement [15]. In one study, EGFR sub-clonality was specifically linked to micropapillary histological variant [16]. On the other hand, an analysis of 862 cases with EGFR mutations did not find dual mutations and different areas of the tumor as well as paired primary and metastasis were concordant for the mutation [17].
Several studies in recent years have linked sub-clonality to poor prognosis and the development of treatment resistance [18–20]. This is presumably because sub-clonal populations acquire resistance to therapies via different and parallel mechanisms. In lung cancer, several studies reported that sub-clonal mutations were associated with worse prognosis [21] and shorter time to disease relapse [9]. Sub-clonality specific for EGFR was also associated with shorter progression free survival [22, 23] and the presence of sub-clonal resistance EGFR T790M mutation was also linked shorter time to progression [24]. On the other hand, analysis of a large cohort of lung adeno- and squamous cell carcinomas did not find an association between high sub-clonality and patients' survival [25].
The disagreement between the different studies with regard to the presence of sub-clonality in lung cancer and its clinical significance might be linked to differences in the methodologies for determining sub-clonality. Currently, the most widely used approach to determine sub-clonality is based on sampling different lesions or multi-region sampling within the same lesion. Using this approach sub-clonality is defined by the presence of a certain mutation only in a subset of the areas examined. One potential limitation of the multi-region sampling method is that it assumes spatial clustering of the different sub-clones and therefore might miss sub-clonality if the different sub-clones are inter-mixed [26]. We have developed a molecular-morphometric approach to determine sub-clonality, which can overcome this limitation, and applied it successfully to the study of sub-clonality in colon, pancreas and thyroid carcinomas [27–29]. In the present study we applied this approach to determine the presence of sub-clonality and its clinical significance in early stage lung cancer.
RESULTS
Three hundred and forty seven cases were analyzed in the study. The average age at diagnosis was 70±10 and 42% of cases were females. Seventy five percent had stage I disease at diagnosis (Table 1). Molecular analysis identified KRAS mutation in 100 (29%) cases and EGFR mutations in 82 (23%) cases. Six cases (2%) had both KRAS and EGFR mutation. The most common KRAS mutation was c.34G>T and the most common EGFR mutation was exon 19 deletion (Supplementary Table 1).
Table 1: Clinical-pathological characteristics of the patients
Age |
70±10 |
|---|---|
Sex |
|
Female |
147 (42%) |
Male |
200 (58%) |
Smoking |
|
Yes |
226 (76%) |
No |
70 (24%) |
Location1 |
|
LUL |
77 (23%) |
LLL |
58 (17%) |
RUL |
118 (35%) |
RML |
15 (4%) |
RLL |
72 (21%) |
Size (cm) |
2.8±2 |
Stage2 |
|
I |
255 (75%) |
II |
52 (15%) |
III |
35 (10%) |
Histology |
|
Acinar |
103 (29%) |
Solid |
93 (27%) |
Lepidic |
38 (11%) |
Papillary |
65 (19%) |
Micropapillary |
21 (6%) |
Mucinous |
24 (7%) |
Fetal |
3 (1%) |
Molecular |
|
KRAS |
100 (29%) |
EGFR |
82 (23%) |
KRAS&EGFR |
6 (2%) |
WT |
159 (46%) |
LUL: left upper lobe; LLL: left lower lobe; RUL: right upper lobe; RML: right middle lobe; RLL: right lower lobe.
1 Seven cases not included in the location - 2 pleura, 5 involving multiple lobes.
2 Five cases with no stage data available.
Cases that were positive for EGFR mutation tended to be slightly older than wild type (WT) cases (72±9 vs 69±11 in the EGFR vs WT groups, respectively, p=0.04, Table 2). Smoking was more common in KRAS mutation positive cases and in wild-type (WT) compared to EGFR mutation positive cases (88% and 78% vs 56% in KRAS, WT and EGFR, respectively, p=0.0002). Additionally, solid histological variant was less common in EGFR mutation positive cases (12% in EGFR positive cases vs 27% and 34% in KRAS positive and WT cases, respectively, p=0.002), whereas mucinous histologic variant was more common in KRAS positive cases (16% vs 0% and 4% in EGFR positive and WT cases, respectively p=0.0003). Survival analysis showed increased survival in the EGFR mutation positive cases compared to the KRAS mutation positive and the WT groups (p=0.02, Supplementary Figure 1). No statistically significant difference was found between the different mutation groups with regard to sex, location of the lesion, tumor size or disease stage (Table 2).
Table 2: Clinical-pathological differences between the different mutation groups
KRAS |
EGFR |
Wild-type |
P-value |
|
|---|---|---|---|---|
Age |
70±10 |
72±9 |
69±11 |
0.041 |
Sex |
0.06 |
|||
Female |
37 (37%) |
44 (54%) |
60 (38%) |
|
Male |
63 (63%) |
38 (46%) |
98 (62%) |
|
Smoking |
0.00022 |
|||
Yes |
81 (88%) |
32 (56%) |
109 (78%) |
|
No |
11 (12%) |
27 (44%) |
30 (22%) |
|
Location |
0.28 |
|||
LUL |
24 (24%) |
23 (28%) |
27 (18%) |
|
LLL |
20 (20%) |
14 (17%) |
24 (16%) |
|
RUL |
36 (37%) |
26 (32%) |
54 (35%) |
|
RML |
3 (3%) |
1 (1%) |
10 (7%) |
|
RLL |
16 (16%) |
18 (22%) |
37 (24%) |
|
Size |
2.7±2.3 |
2.7±1.5 |
2.9±2 |
0.6 |
Stage |
0.6 |
|||
I |
75 (76%) |
57 (71%) |
118 (76%) |
|
II |
17 (17%) |
14 (17%) |
21 (13%) |
|
III |
7 (7%) |
10 (12%) |
17 (11%) |
|
Histology |
<0.0001 |
|||
Acinar |
26 (26%) |
31 (38%) |
43 (27%) |
0.16 |
Solid |
27 (27%) |
10 (12%) |
54 (34%) |
0.0022 |
Lepidic |
9 (9%) |
13 (16%) |
15 (9%) |
0.15 |
Papillary |
14 (14%) |
20 (24%) |
32 (20%) |
0.25 |
Micropapillary |
8 (8%) |
6 (7%) |
7 (4%) |
0.46 |
Mucinous |
16 (16%) |
0 (0%) |
7 (4%) |
0.00033 |
Fetal |
0 (0%) |
2 (2%) |
1 (1%) |
0.19 |
LUL: left upper lobe, LLL: left lower lobe; RUL: right upper lobe; RML: right middle lobe; RLL: right lower lobe; WT: wild-type.
1 EGFR vs WT.
2 EGFR vs KRAS/WT.
3 EGFR vs KRAS vs WT.
Of the 188 mutation positive cases, 144 were available for both molecular and morphometric analysis. The average mutant allele frequency was 34%±19.7 (range 3-95%) and the average tumor cell fraction in the areas used for analysis was 44%±19.4 (range 3-88%). Bland-Altman analysis performed on repeated measurements of the morphometric analysis showed high agreement with percent difference up to 33% (Figure 1A). Due to the difference between repeated measurements and to avoid misclassification of cases as sub-clonal, only cases with a mutant allele frequency of less than 60% of expected, based on the morphometric analysis, were defined as sub-clonal. Additionally, cases with mutant allele frequency of more than 150% of expected were defined as mutant allele amplification. Following calculation of the fraction of tumor cells carrying a mutation 37 (26%) cases (31 cases with less than 60% of expected mutant allele fraction, 6 cases with double mutation) were defined as carrying sub-clonal mutation and 34 (23%) cases were defined as carrying mutant allele specific amplification (Figure 1B). Chromogenic in-situ hybridization with EGFR probe, performed on several cases, confirmed EGFR copy gains in cases that showed higher than expected mutant allele frequency (Figure 1C and 1D).
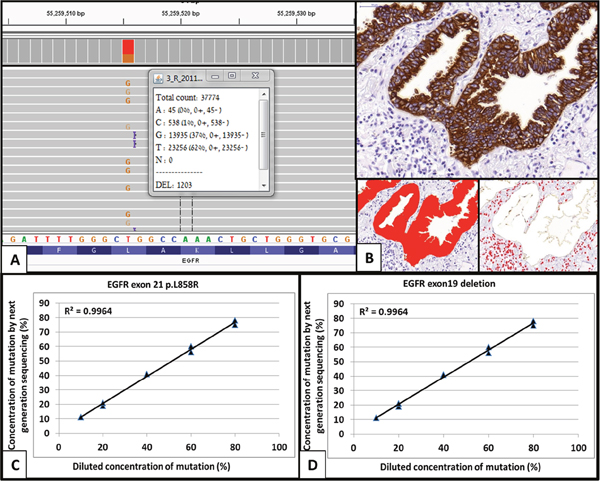
Figure 1: Molecular and morphometric analysis of sub-clonality for KRAS and EGFR mutations. (A) A screen shot from the integrative genome browser software; the middle panel represents the DNA sequences the upper panel shows the summary of changes in each position and the lower panel is the reference genomic sequence. In this sample there is a specific T>G mutation (leading to EGFR p.L858R missense mutation). Using the program we were able to determine the mutation and fraction of mutant copies (inset). (B) For the morphometric analysis the slide was immunohistochemically stained with anti-cytokeratin antibody, and several representative high magnifications images were taken (upper panel). In each image tumor (left lower panel) and normal cell (right lower panel) numbers were counted using the Image Pro Plus program. (C and D) Standardized curve for validation of mutant fraction detection method. As detailed in the methods section, synthetic DNA harboring EGFR exon 21 (C) and exon 19 (D) mutations were diluted in wild-type synthetic DNA to concentrations ranging 10-80%. The different mixtures served as references for the standardized curve. Analysis of the data showed very high correlation between the predicted concentration of mutant EGFR and the concentration measured by next generation sequencing, confirming the reliability of our method. Using the morphometric and molecular results we were able to calculate the fraction of tumor cells harboring mutations and determine clonality status for the mutations.
Sub-clonal cases were associated with earlier disease stage (89% vs 66% stage I disease in sub-clonal vs clonal cases, respectively, p=0.01, Figure 2A) and less lymph node involvement (8% vs 25% in sub-clonal vs clonal cases, respectively, p=0.02, Figure 2C). No association was found between clonality status and tumor size (Figure 2B and 2D). Additionally, mucinous histologic subtype appeared at higher frequency in the cases with sub-clonal mutations (22% vs 7% in sub-clonal vs clonal cases, respectively, p=0.009, Table 3). Surprisingly, sub-clonality for EGFR mutation was associated with shorter survival time (p=0.03, Figure 3B) whereas sub-clonality for KRAS mutation only showed a trend toward shorter survival time (p=0.1, Figure 3A). The presence of sub-clonality for either KRAS or EGFR mutation status was associated with shorter survival time (p=0.02, Figure 3C). Cox multivariate regression analysis showed that disease stage and sub-clonality status were independent predictors of survival, and hence stage I disease with clonal mutation status had the best prognosis whereas cases with stage II or III disease with sub-clonality had the shortest survival time (p<0.05, Figure 3D). No association was found between the presence of mutant allele amplification and any of the clinical or pathological variables measured.
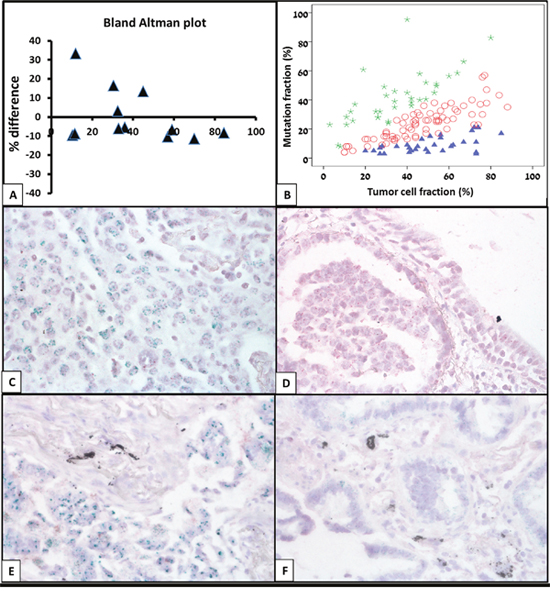
Figure 2: Identification of cases with KRAS or EGFR mutation sub-clonality. (A) Bland-Altman analysis. To determine the reproducibility of the morphometric analysis the entire analysis process was re-performed on 12 cases. Bland-Altman analysis showed high agreement with maximal percent difference of 33%. To avoid misclassification of cases as sub-clonal, only cases that had mutant allele frequency of less than 60% of expected based on the morphometric analysis were defined as sub-clonal. (B) Based on the morphometric analysis, 37 cases were found to be sub-clonal (blue triangle) and 34 cases were found to harbor mutant allele specific amplification. (C-F) To validate the reliability of our molecular-morphometric approach several cases calculated to carry (C, E) and not carry (D, F) EGFR amplification underwent chromogenic in-situ hybridization (CISH) with anti EGFR probe. As demonstrated by CISH, multiple green dots in tumor nuclei, indicating EGFR amplification could be demonstrated in cases with predicted EGFR amplification (C, E) but not in cases with expected EGFR levels (D, F).
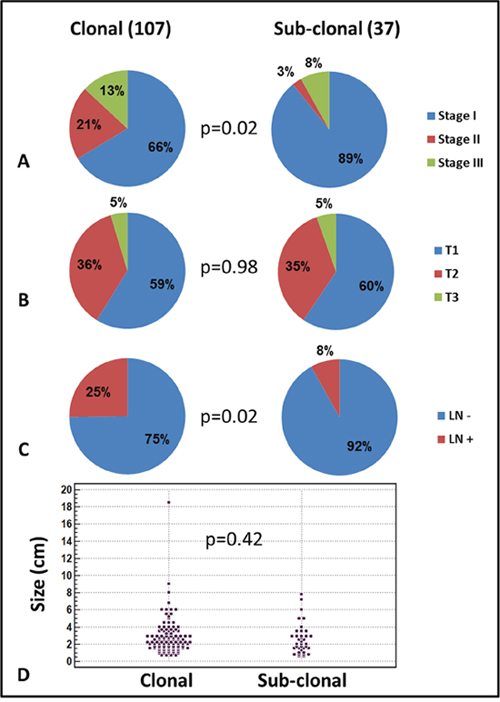
Figure 3: Clinical significance of KRAS or EGFR sub-clonality in lung adenocarcinoma. Sub-clonality was associated with earlier disease stage (A), and less lymph node (LN) involvemet (C). No association was found between clonality status and tumor T stage (B) or diameter (D).
Table 3: Clinical-pathological differences between cases with and without sub-clonality
sub-clonal |
Clonal |
P-value |
|
|---|---|---|---|
Age |
72±10 |
71±9 |
0.66 |
Sex |
0.9 |
||
Female |
18 (49%) |
52 (49%) |
|
Male |
19 (51%) |
55 (51%) |
|
Smoking |
0.6 |
||
Yes |
24 (73%) |
67 (77%) |
|
No |
9 (27%) |
20 (23%) |
|
Size |
2.6±1.8 |
2.9±2.2 |
0.47 |
LN involvement |
3 (8%) |
27 (25%) |
0.02 |
Stage |
0.011 |
||
I |
33 (89%) |
71 (66%) |
|
II |
1 (3%) |
22 (21%) |
|
III |
3 (8%) |
14 (13%) |
|
Histology |
|||
Acinar |
9 (24%) |
39 (36%) |
0.17 |
Solid |
9 (24%) |
21 (20%) |
0.54 |
Lepidic |
2 (5%) |
14 (13%) |
0.2 |
Papillary |
8 (22%) |
14 (13%) |
0.21 |
Micropapillary |
1 (3%) |
11 (10%) |
0.15 |
Mucinous |
8 (22%) |
7 (7%) |
0.009 |
Fetal |
0 (0%) |
1 (1%) |
0.5 |
LN – lymph nodes.
1 between stages I and II+III.
DISCUSSION
Sub-clonality is an inevitable consequence of the dynamic nature of cancer progression. Tumor cells continuously acquire mutations, hence, at any given moment, at least those recently acquired mutation are sub-clonal. Supporting this concept, many research articles looking at single drivers or using comprehensive sequencing technologies were able to demonstrate sub-clonality in various malignancies [3, 30, 31]. There is also accumulating data suggesting that sub-clonality has clinical significance. The association between sub-clonality and treatment failure is biologically plausible. Small sub-clones with acquired resistance to therapy can undergo selection and cause disease progression [19]. Indeed, many studies found an association between sub-clonality and shorter time to disease progression in different malignancies such as chronic lymphocytic leukemia [32] and head and neck cancer [33]. Nevertheless, this subject is under debate with other studies linking sub-clonality to early disease stage for BRAF mutation in thyroid cancer [27, 34] and KRAS mutation in pancreatic [28] and colon cancer [35].
In the present study we found that sub-clonality for KRAS or EGFR mutation was associated with shorter survival times. The presence of sub-clonality for the common drivers in lung adenocarcinoma is in accordance with some previous reports [7, 8, 10, 15, 16]. However, different cohorts report lack of sub-clonality [17] or claim that it is probably a rare condition [36]. Interestingly, even among the studies that did identify sub-clonality, there is disagreement regarding its clinical significance with some associating sub-clonality with shorter time to progression [9, 22], whereas others report lack of clinical associations [11, 14, 25]. Moreover, low frequency TP53 mutant allele fraction, interpreted as sub-clonality, was associated with better survival in cases of lung carcinoma [37].
One potential reason for the disagreement between different studies with regard to the clinical effect of sub-clonality might be related to the method used to measure sub-clonality. The most widely used approach to determine sub-clonality is based on comparison of different tumor lesions (e.g. primary tumor and metastasis) or multi-region sampling of a single lesion [4, 6, 7, 10, 11, 13, 15]. Using this approach, a tumor is defined as being heterogeneous if some mutations are present only in some of the samples. However, an intrinsic assumption of this approach is that different sub-clones are spatially separated, which might not be the case in many tumors. Interestingly, a recently reported model supports the rapid intra-tumor cell mixing during cancer evolution [38]. Considering the possibility of admixing of sub-clones, it is possible that multiple samples from different regions will be positive for a specific mutation despite it being present only in a subset of cells from each region. Mutant allele fraction in the entire sample being analyzed has also been evaluated as a tool to determine sub-clonality, with low allele frequency being defined as sub-clonality [39, 40]. The discordant results of these two studies looking into the clinical significance of sub-clonality for BRAF mutation in thyroid cancer might be the result of lack of correction for tumor cell fraction in each sample. Evaluation of the tumor cell content in the sample is essential to differentiate between true sub-clonality and low tumor cell content. A study linking high frequency TP53 mutations with worse prognosis [37] is also limited by the lack of correction for tumor cell fraction. A molecular morphometric approach, like the one used in the present study should be able to accurately determine sub-clonality in intermixed tumor and also account for the tumor cell content in the sample. An alternative approach is to use laser microdissection and isolate only the tumor cells from the sample [41].
Another finding in our research is the association between sub-clonality and early disease stage. This finding stands in some contradiction to the association between sub-clonality and worse prognosis. This phenomenon can be potentially explained by understanding the different modes of evolution that can present as sub-clonality. Basically, the two major evolutionary models are linear evolution and branched evolution, and both can lead to sub-clonality [1]. In linear evolution model, if a sample is being analyzed before a new sub-clone eradicated the previous clone the result would be sub-clonality for the mutations carried by the new sub-clone. In a branched model, different sub-clones evolve in parallel leading to sub-clonality. We propose that the different types of sub-clonality are related to different associations with the clinical phenotype. Sub-clonality related to linear evolution would be expected in early disease stage and smaller lesions where nutrient and blood supply are abundant. This could explain the association reported between sub-clonality and early disease stage and smaller lesions [27, 28]. On the other hand, sub-clonality associated with branched evolution might be the type of sub-clonality that is associated with increased tumor fitness, development of treatment resistance and poor survival [9, 19, 20] (Figure 4).
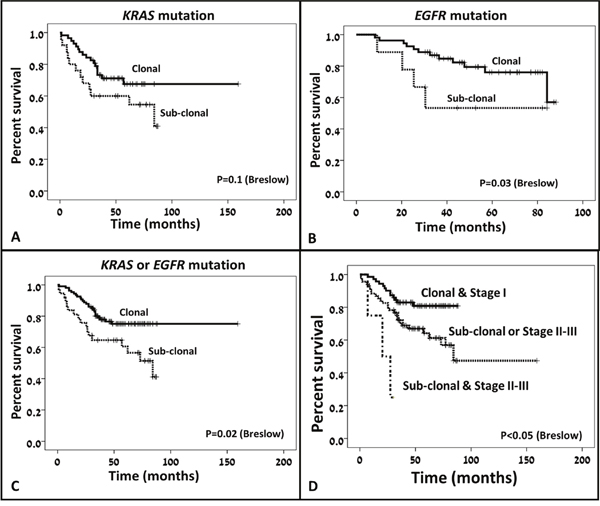
Figure 4: Clonality status effect on survival. While sub-clonality for KRAS mutation showed only trend toward statistically significant association with reduced survival (A), sub-clonality for EGFR mutation (B) and sub-clonality for either KRAS or EGFR mutations (C) were significantly associated with shorter survival times. A combined score including disease stage and clonality status could stratify the patients into 3 prognostic groups, where cases with clonal disease at stage I had the longer survival and cases with sub-clonal disease and more advanced disease stage had the shortest survival time (D).
Copy number gain of oncogenes including EGFR, KRAS and c-MYC has been reported in lung carcinoma, where it was associated with advanced stage and poor clinical outcome [42–45]. In the present study we did not find any association between mutant allele amplification and clinic-pathological variables. One potential explanation is that oncogene amplification is not restricted to the mutant allele copy and that amplifications could be present in the wild-type copy [42] thereby masking the clinical difference between the groups. Additionally, oncogene amplification pattern was reported to be heterogeneous within the tumor tissue [44] and it is possible that some cases with focal amplification were unidentified by our approach. Lastly, our sample set includes cases with early resectable disease, in which the associations between oncogene amplification and clinic-pathological variables might be less pronounced.
In conclusion, using a molecular morphometric approach we were able to demonstrate sub-clonality for KRAS and EGFR mutations in lung adenocarcinoma. We found that sub-clonality was associated with both early disease stage and with poor prognosis, suggesting that sub-clonality should not be regarded as a single entity, and could represent different evolutionary stages in tumor development.
MATERIALS AND METHODS
DNA extraction from formalin fixed paraffin embedded (FFPE) tissue samples
DNA extraction from 347 surgical specimens of lung adenocarcinomas, taken between 2007 and 2011, was performed as previously described [46]. Briefly, an area containing a high fraction of tumor cells was marked by a pathologist, microscopically dissected and DNA was extracted using the QuickExtract FFPE DNA Extraction kit (Epicentre, Madison, WI) according to manufacturer instructions. Following treatment with RNase A (Qiagen, Hilden, Germany), DNA was purified using the DNA Clean and Concentrator kit (Zymo Research, Orange, CA). The study was approved by the local ethics committee.
Molecular morphometric approach
The tumor mass in solid tumors such as adenocarcinoma of the lung are composed of tumor cells as well as stromal, blood vessels and inflammatory cells. While the tumor cells carry the tumor promoting mutations, the non-tumor cells of the mass contain a wild-type copy of the gene. To determine the fraction of tumor cells carrying a specific gene mutation we used a dual approach, combining molecular and computerized morphometry tools. As detailed below, molecular tools including next generation sequencing were used to determine the relative number of mutated DNA copies in each sample. Additionally, the area from which DNA was extracted was scrutinized using computerized morphometry, to determine the fraction of tumor cells in each sample. Combining both results we were able to calculate the fraction of tumor cells carrying a mutation in each sample, or, in other words, the degree of sub-clonality of each sample with regard to the mutations examined (Figure 5).
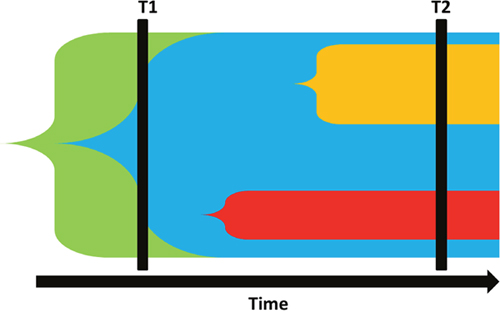
Figure 5: Tumor evolutionary scenarios associated with sub-clonality. The horizontal axis represents time and the different colors represent different mutational events leading to new clones. Cancer driver mutations can be sub-clonal in both linear and branched evolutionary model. In linear evolution model (T1) sub-clonality can be identified it the tumor is sampled before the new mutation resulted in eradication of the ancestral clone (clonal sweep). Alternatively, in branched evolution different clones expand in parallel, also resulting in sub-clonality (T2). Depending on sampling time, sub-clonality can either represent early linear model associated with early stage disease or more advanced stage with different sub-clones associated with shorter survival times.
EGFR and KRAS mutation screening
EGFR mutation analysis was performed using the Cobas EGFR mutation test (RocheMolecular System Inc., Branchburg, NJ), that can detect mutations present in as little as 10% of the sample. Additionally, samples were screened for KRAS exon 2 mutations using high resolution melting technology [46], another sensitive method, that in our hands could detect mutation present in as little as 1% of the sample.
Library generation and determining the fraction of mutated copies
In cases that screened positive for KRAS or EGFR mutation further analysis was performed to determine mutant allele fraction in each case. Toward this aim we used the Ion Torrent Personal Genome Machine (PGM) sequencer platform. DNA extracted from the tumor samples was PCR amplified using primers forward 5'-GGCCTGCTGAAAATGACTGAA-3' and reverse 5'-GGTCCTGCACCAGTAATATGCA-3' for KRAS, forward 5'- AGCATGTGGCACCATCTCAC -3' and reverse 5'- AGACATGAGAAAAGGTGGGC -3' for EGFR exon 19 and forward 5'- AATTCGGATGCAGAGCTTC -3' and reverse 5'- GCATGGTATTCTTTCTCTTCCG -3' for EGFR exon 21. Each primer pair was supplemented with Ion-Torrent adapters P1 and A, to allow binding to the Ion Sphere Particles (ISPs). Additionally, 20-30 different forward primers, each with a different barcode, were used for every genomic area amplified to allow the analysis of multiple samples in a single reaction. Amplicons were purified using the Qiagen PCR purification kit (Qiagen, Hilden, Germany) and were then sequenced using an Ion 314 chip and sequenced on the PGM for 65 cycles. We aimed for X1000 coverage to allow accurate determination of mutant allele fraction in each sample. Data from the PGM runs was initially processed using the Ion Torrent platform-specific pipeline software Torrent Suite v1.3.1 to generate sequence reads, trim adapter sequences, filter, and remove poor signal-profile reads. Generated sequence files were aligned to the genomic sequence of KRAS exon 2 and EGFR exons 19 and 21 and we determined the fraction of the mutation and the wild type copies of the gene in each sample using the Integrative Genomic Viewer (IGV 2.3) free software [47, 48].
Establish standard curve to determine mutant allele fraction
To determine the mutant allele fraction for each case from the next generation sequencing results we built standard curves for point mutations and for deletions involving KRAS and EGFR mutation “hot-spots”. Toward this aim we synthesized gBlock DNA sequences ~450bps long that include the mutation/deletion area. Additionally, the wild-type sequence was also synthesized. We then mixed wild-type and mutant sequences to generate synthetic samples with mutant allele frequency ranging between 10% and 90%. These samples underwent PCR amplification and next generation sequencing and the results served as standard curve for validation and standardization of the molecular results of the study.
Quantitative image analysis
In order to establish the proportion of tumor versus non-tumor cells, histological slides retrieved from the archives of the pathology department were immunohistochemically stained with anti-cytokeratin antibody (Lab Vision, Fremont, CA) thus enhancing the observer’s ability to visually separate between tumor cells and non-tumor elements (such as stroma, vessels and inflammation). For area measurements, stained slides were entirely scanned at a magnification of X20 using the dotSlide 2.0 virtual microscopy system (Olympus, Germany & Japan). The digital virtual images were loaded in an image analysis system (Image Pro Plus 6.3, MediaCybernetics, MA, USA). The total area of the tumor was segmented from other elements (stroma, lymphatic aggregates and normal colonic crypts) and measured. For cell number counts, representative images of the tumor and other tissue elements were further captured at a magnification of X400 and cells were counted using the Image Pro Plus program. The total number of cells within each tissue element (tumor, stroma, lymphocytes, normal) was then calculated by using a mathematical extrapolation. This information was used to calculate the fraction of tumor cells in the samples. To determine the reproducibility of the morphometric analysis the entire analysis process was re-performed on 12 cases. Bland-Altman analysis [49] was used to determine the agreement between repeated measurements and the percent difference was calculated.
Determining KRAS and EGFR mutation sub-clonality
In order to determine the fraction of tumor cells carrying a specific gene mutation, we combined the data obtained from the molecular and morphometric analysis. Assuming that KRAS or EGFR mutations are present in one allele, the fraction of tumor cells carrying the mutation was calculated using the formula:
2*(mutant allele frequency)/(tumor cell fraction).
Validation of amplification
To validate the findings in cases predicted to have EGFR mutant allele amplification based on the molecular morphometric approach we performed chromogenic in-situ hybridization (CISH) using the digoxigenylated ZytoDot SPEC EGFR Probe (CE-Marked), the ZytoDot pretreatment kit, and the ZytoDot CISH polymer detection kit, according to the supplier’s protocol (Zytovision, Clinisciences, Montrouge, France). Briefly, the DNA probe and sections were denatured at 95°C and hybridized at 37°C overnight using a HYBrite instrument (Vysis, Downers Grove, IL).
Statistical method
In order to identify the presence of sub-clonality in as little as 10% of the tissue samples, with a statistical power of 90% (beta 0.1) and alpha of 0.05, the calculated sample size needs to be 95 cases at least. Our cohort size meets the power analysis criteria.
Association between the presence of sub-clonality and patients' clinical and histological variables were tested using the Chi square test for categorical variables and Student T test or the Mann-Whitney U test for continuous parametric or non-parametric variables as needed. The impact of sub-clonality on patients' prognosis was calculated using the Kaplan Meier product limit method and the Log-Rank test for detecting significant differences between the groups. Two tailed p values of 0.05 or less were considered statistically significant.
Author contributions
Study conception and design: E.Si., E.Sa., O.B, D.H.
Data acquisition and analysis: E.Si., T.B., S.J., T.S., E.P., L.Y., D.H.
Manuscript drafting and revisiting: E.Si., T.B., S.J., T.S., E.P., L.Y., E.Sa., O.B, D.H.
Approval of final version: E.Si., T.B., S.J., T.S., E.P., L.Y., E.Sa., O.B, D.H.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare regarding the manuscript.
FUNDING
The study was supported by a research grant from the Israel Cancer Association.
REFERENCES
1. Burrell RA, Swanton C. The evolution of the unstable cancer genome. Curr Opin Genet Dev. 2014; 24: 61-7. doi: 10.1016/j.gde.2013.11.011.
2. Hiley C, de Bruin EC, McGranahan N, Swanton C. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol. 2014; 15: 453. doi: 10.1186/s13059-014-0453-8.
3. Beca F, Schmitt F. Growing indication for FNA to study and analyze tumor heterogeneity at metastatic sites. Cancer Cytopathol. 2014; 122: 504-11. doi: 10.1002/cncy.21395.
4. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366: 883-92. doi: 10.1056/NEJMoa1113205.
5. Ledgerwood LG, Kumar D, Eterovic AK, Wick J, Chen K, Zhao H, Tazi L, Manna P, Kerley S, Joshi R, Wang L, Chiosea SI, Garnett JD, et al. The degree of intratumor mutational heterogeneity varies by primary tumor sub-site. Oncotarget. 2016; 7: 27185-98. doi: 10.18632/oncotarget.8448.
6. Lu YW, Zhang HF, Liang R, Xie ZR, Luo HY, Zeng YJ, Xu Y, Wang LM, Kong XY, Wang KH. Colorectal cancer genetic heterogeneity delineated by multi-region sequencing. PLoS One. 2016; 11: e0152673. doi: 10.1371/journal.pone.0152673.
7. de Bruin EC, McGranahan N, Mitter R, Salm M, Wedge DC, Yates L, Jamal-Hanjani M, Shafi S, Murugaesu N, Rowan AJ, Gronroos E, Muhammad MA, Horswell S, et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science. 2014; 346: 251-6. doi: 10.1126/science.1253462.
8. Tan Q, Cui J, Huang J, Ding Z, Lin H, Niu X, Li Z, Wang G, Luo Q, Lu S. Genomic alteration during metastasis of lung adenocarcinoma. Cell Physiol Biochem. 2016; 38: 469-86. doi: 10.1159/000438644.
9. Zhang J, Fujimoto J, Zhang J, Wedge DC, Song X, Zhang J, Seth S, Chow CW, Cao Y, Gumbs C, Gold KA, Kalhor N, Little L, et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science. 2014; 346: 256-9. doi: 10.1126/science.1256930.
10. Chen ZY, Zhong WZ, Zhang XC, Su J, Yang XN, Chen ZH, Yang JJ, Zhou Q, Yan HH, An SJ, Chen HJ, Jiang BY, Mok TS, et al. EGFR mutation heterogeneity and the mixed response to EGFR tyrosine kinase inhibitors of lung adenocarcinomas. Oncologist. 2012; 17: 978-85. doi: 10.1634/theoncologist.2011-0385.
11. Kim EY, Cho EN, Park HS, Kim A, Hong JY, Lim S, Youn JP, Hwang SY, Chang YS. Genetic heterogeneity of actionable genes between primary and metastatic tumor in lung adenocarcinoma. BMC Cancer. 2016; 16: 27. doi: 10.1186/s12885-016-2049-z.
12. Li Y, Li Y, Yang T, Wei S, Wang J, Wang M, Wang Y, Zhou Q, Liu H, Chen J. Clinical significance of EML4-ALK fusion gene and association with EGFR and KRAS gene mutations in 208 Chinese patients with non-small cell lung cancer. PLoS One. 2013; 8: e52093. doi: 10.1371/journal.pone.0052093.
13. Bai H, Wang Z, Chen K, Zhao J, Lee JJ, Wang S, Zhou Q, Zhuo M, Mao L, An T, Duan J, Yang L, Wu M, et al. Influence of chemotherapy on EGFR mutation status among patients with non-small-cell lung cancer. J Clin Oncol. 2012; 30: 3077-83. doi: 10.1200/JCO.2011.39.3744.
14. Tomonaga N, Nakamura Y, Yamaguchi H, Ikeda T, Mizoguchi K, Motoshima K, Doi S, Nakatomi K, Iida T, Hayashi T, Nagayasu T, Tsukamoto K, Kohno S. Analysis of intratumor heterogeneity of EGFR mutations in mixed type lung adenocarcinoma. Clin Lung Cancer. 2013; 14: 521-6. doi: 10.1016/j.cllc.2013.04.005.
15. Cai W, Lin D, Wu C, Li X, Zhao C, Zheng L, Chuai S, Fei K, Zhou C, Hirsch FR. Intratumoral heterogeneity of ALK-rearranged and ALK/EGFR coaltered lung adenocarcinoma. J Clin Oncol. 2015; 33: 3701-9. doi: 10.1200/JCO.2014.58.8293.
16. Cai YR, Dong YJ, Wu HB, Liu ZC, Zhou LJ, Su D, Chen XJ, Zhang L, Zhao YL. Micropapillary: a component more likely to harbour heterogeneous EGFR mutations in lung adenocarcinomas. Sci Rep. 2016; 6: 23755. doi: 10.1038/srep23755.
17. Yatabe Y, Matsuo K, Mitsudomi T. Heterogeneous distribution of EGFR mutations is extremely rare in lung adenocarcinoma. J Clin Oncol. 2011; 29: 2972-7. doi: 10.1200/JCO.2010.33.3906.
18. Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014; 8: 1095-111. doi: 10.1016/j.molonc.2014.06.005.
19. Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015; 21: 1258-66. doi: 10.1158/1078-0432.CCR-14-1429.
20. McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015; 27: 15-26. doi: 10.1016/j.ccell.2014.12.001.
21. Pelosi G, Pellegrinelli A, Fabbri A, Tamborini E, Perrone F, Settanni G, Busico A, Picciani B, Testi MA, Militti L, Maisonneuve P, Valeri B, Sonzogni A, et al. Deciphering intra-tumor heterogeneity of lung adenocarcinoma confirms that dominant, branching, and private gene mutations occur within individual tumor nodules. Virchows Arch. 2016; 468: 651-62. doi: 10.1007/s00428-016-1931-z.
22. Taniguchi K, Okami J, Kodama K, Higashiyama M, Kato K. Intratumor heterogeneity of epidermal growth factor receptor mutations in lung cancer and its correlation to the response to gefitinib. Cancer Sci. 2008; 99: 929-35. doi: 10.1111/j.1349-7006.2008.00782.x.
23. Zhou Q, Zhang XC, Chen ZH, Yin XL, Yang JJ, Xu CR, Yan HH, Chen HJ, Su J, Zhong WZ, Yang XN, An SJ, Wang BC, et al. Relative abundance of EGFR mutations predicts benefit from gefitinib treatment for advanced non-small-cell lung cancer. J Clin Oncol. 2011; 29: 3316-21. doi: 10.1200/JCO.2010.33.3757.
24. Lee Y, Lee GK, Hwang JA, Yun T, Kim HT, Lee JS. Clinical likelihood of sporadic primary EGFR T790M mutation in EGFR-mutant lung cancer. Clin Lung Cancer. 2015; 16: 46-50. doi: 10.1016/j.cllc.2014.09.002.
25. Morris LG, Riaz N, Desrichard A, Senbabaoglu Y, Hakimi AA, Makarov V, Reis-Filho JS, Chan TA. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget. 2016; 7: 10051-63. doi: 10.18632/oncotarget.7067.
26. Fisher Y, Hershkovitz D. Molecular and morphometric tools for next-generation pathology diagnosis of colon carcinoma. Isr Med Assoc J. 2016; 18: 426-32. doi:.
27. Finkel A, Liba L, Simon E, Bick T, Prinz E, Sabo E, Ben-Izhak O, Hershkovitz D. Subclonality for BRAF Mutation in Papillary Thyroid Carcinoma Is Associated With Earlier Disease Stage. J Clin Endocrinol Metab. 2016; 101: 1407-13. doi: 10.1210/jc.2015-4031.
28. Nagawkar SS, Abu-Funni S, Simon E, Bick T, Prinz E, Sabo E, Ben-Izhak O, Hershkovitz D. Intratumor heterogeneity of kras mutation status in pancreatic ductal adenocarcinoma is associated with smaller lesions. Pancreas. 2016; 45: 876-81. doi: 10.1097/MPA.0000000000000562.
29. Farber L, Efrati E, Elkin H, Peerless Y, Sabo E, Ben-Izhak O, Hershkovitz D. Molecular morphometric analysis shows relative intra-tumoural homogeneity for KRAS mutations in colorectal cancer. Virchows Arch. 2011; 459: 487-93. doi: 10.1007/s00428-011-1158-y.
30. Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013; 8: 277-302. doi: 10.1146/annurev-pathol-020712-163923.
31. Crockford A, Jamal-Hanjani M, Hicks J, Swanton C. Implications of intratumour heterogeneity for treatment stratification. J Pathol. 2014; 232: 264-73. doi: 10.1002/path.4270.
32. Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, Wan Y, Zhang W, Shukla SA, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013; 152: 714-26. doi: 10.1016/j.cell.2013.01.019.
33. Mroz EA, Tward AD, Hammon RJ, Ren Y, Rocco JW. Intra-tumor genetic heterogeneity and mortality in head and neck cancer: analysis of data from the Cancer Genome Atlas. PLoS Med. 2015; 12: e1001786. doi: 10.1371/journal.pmed.1001786.
34. de Biase D, Cesari V, Visani M, Casadei GP, Cremonini N, Gandolfi G, Sancisi V, Ragazzi M, Pession A, Ciarrocchi A, Tallini G. High-sensitivity BRAF mutation analysis: BRAF V600E is acquired early during tumor development but is heterogeneously distributed in a subset of papillary thyroid carcinomas. J Clin Endocrinol Metab. 2014; 99: E1530-8. doi: 10.1210/jc.2013-4389.
35. Hershkovitz D, Simon E, Bick T, Prinz E, Noy S, Sabo E, Ben-Izhak O, Vieth M. Adenoma and carcinoma components in colonic tumors show discordance for KRAS mutation. Hum Pathol. 2014; 45: 1866-71. doi: 10.1016/j.humpath.2014.05.005.
36. Jakobsen JN, Sorensen JB. Intratumor heterogeneity and chemotherapy-induced changes in EGFR status in non-small cell lung cancer. Cancer Chemother Pharmacol. 2012; 69: 289-99. doi: 10.1007/s00280-011-1791-9.
37. Lee SY, Jeon HS, Hwangbo Y, Jeong JY, Park JY, Lee EJ, Jin G, Shin KM, Yoo SS, Lee J, Lee EB, Cha SI, Kim CH, Park JY. The influence of TP53 mutations on the prognosis of patients with early stage non-small cell lung cancer may depend on the intratumor heterogeneity of the mutations. Mol Carcinog. 2013; 54: 93-101. doi: 10.1002/mc.22077.
38. Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015; 525: 261-4. doi: 10.1038/nature14971.
39. Cheng SP, Hsu YC, Liu CL, Liu TP, Chien MN, Wang TY, Lee JJ. Significance of allelic percentage of BRAF c.1799T > A (V600E) mutation in papillary thyroid carcinoma. Ann Surg Oncol. 2014; 21: S619-26. doi: 10.1245/s10434-014-3723-5.
40. Gandolfi G, Sancisi V, Torricelli F, Ragazzi M, Frasoldati A, Piana S, Ciarrocchi A. Allele percentage of the BRAF V600E mutation in papillary thyroid carcinomas and corresponding lymph node metastases: no evidence for a role in tumor progression. J Clin Endocrinol Metab. 2013; 98: E934-42. doi: 10.1210/jc.2012-3930.
41. Lin J, Marquardt G, Mullapudi N, Wang T, Han W, Shi M, Keller S, Zhu C, Locker J, Spivack SD. Lung cancer transcriptomes refined with laser capture microdissection. Am J Pathol. 2014; 184: 2868-84. doi: 10.1016/j.ajpath.2014.06.028.
42. Sasaki H, Hikosaka Y, Kawano O, Moriyama S, Yano M, Fujii Y. Evaluation of Kras gene mutation and copy number gain in non-small cell lung cancer. J Thorac Oncol. 2011; 6: 15-20. doi: 10.1097/JTO.0b013e31820594f0.
43. Seo AN, Yang JM, Kim H, Jheon S, Kim K, Lee CT, Jin Y, Yun S, Chung JH, Paik JH. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. Br J Cancer. 2014; 110: 2688-99. doi: 10.1038/bjc.2014.218.
44. Sholl LM, Yeap BY, Iafrate AJ, Holmes-Tisch AJ, Chou YP, Wu MT, Goan YG, Su L, Benedettini E, Yu J, Loda M, Janne PA, Christiani DC, et al. Lung adenocarcinoma with EGFR amplification has distinct clinicopathologic and molecular features in never-smokers. Cancer Res. 2009; 69: 8341-8. doi: 10.1158/0008-5472.CAN-09-2477.
45. Yatabe Y, Takahashi T, Mitsudomi T. Epidermal growth factor receptor gene amplification is acquired in association with tumor progression of EGFR-mutated lung cancer. Cancer Res. 2008; 68: 2106-11. doi: 10.1158/0008-5472.CAN-07-5211.
46. Efrati E, Elkin H, Peerless Y, Sabo E, Ben-Izhak O, Hershkovitz D. LNA-based PCR clamping enrichment assay for the identification of KRAS mutations. Cancer Biomark. 2010; 8: 89-94. doi: 10.3233/CBM-2011-0203.
47. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013; 14: 178-92. doi: 10.1093/bib/bbs017.
48. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011; 29: 24-6. doi: 10.1038/nbt.1754.
49. Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet. 1986; 1: 307-10. doi:.