
INTRODUCTION
During the last years immunotherapies of cancer particularly immune checkpoint blockade inhibitors and adoptively transferred T cells have improved the outcome of tumor patients. Since the monitoring of tumor samples and immune responses prior and following these therapies are essential for the optimization of the mode of immunotherapy and for the selection of patients suitable for a specific immunotherapy the identification and systematic assessment of biomarkers has become an important issue for immunotherapy of cancer. In principle, liquid biopsies represent suitable samples not only for early detection, monitoring residual diseases, but also for monitoring immunotherapy responses and development of resistances, because multiple samples can be easily obtained prior, during and after immune and other therapies [1].
Liquid biopsies as a source for tumor-derived information can be analyzed at the cellular, DNA, RNA, epigenetic, protein and metabolome levels. With the development and implementation of high-throughput technologies personalized targeted and/or tumor immunotherapies have become feasible. In the future, an individual antibody binding pattern of patients might be also used for the development of personalized immunotherapy and for the monitoring of immune responses. The ultimate goal to predict immunotherapeutic success is the identification of reliable prognostic and predictive biomarkers, which link immunity with patients’ outcome thereby allowing the selection of a group of patients, who will most likely benefit from a respective treatment regimen.
The NIH/WHO international program on chemical safety has defined a biomarker as “any substance, structure, or process that can be measured in the body or its products and that can influence or predict the incidence of outcome or disease” [2].
What will be used/monitored?
For monitoring immune cell responses with long term follow-up, body fluids, such as e.g. peripheral blood and serum/plasma, should be collected at different time points prior, during and after treatment until disease progression. In addition to conventional analysis of serum markers, the absolute lymphocyte count (ALC), subtypes of lymphocytes, composition of the immune cell repertoire, activity of different immune cell subpopulations, exosome and extracellular microvesicle (EV) profiles, expression of genes/microRNAs/proteins, cytokines, metabolites, chemokines, putative tumor-associated antigens (TAA), polymorphisms in Fc receptors, antibodies and the proliferative capacity of adoptively transferred immune cells could be also analyzed in primary tumors and/or body fluids (Figure 1) [3–9].
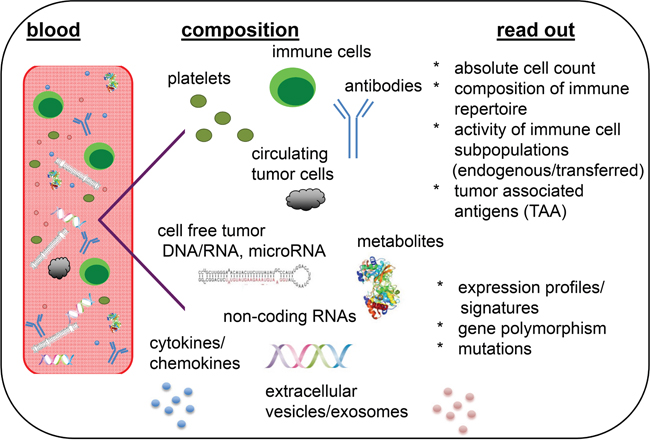
Figure 1: Liquid biopsies as representatives of the molecular heterogeneity and immunologic phenotype of tumors. Blood serves here as representative liquid biopsy containing the highest variety on biological analytes.
The integration of multiple high-throughput “omics” technologies including cancer genome sequencing studies that have catalogued the mutational landscape of human tumors (e.g. single nucleotide variants, copy-number alterations, somatic events), transcriptomics for analysis of peripheral blood mononuclear cells (PBMCs) and DNA/RNA analysis in circulating tumor cells (CTC) has helped to define biomarkers. Furthermore, different proteome-based technologies, such as the serologic evaluation of proteins and antibodies (PROTEOMEX), “bottom up” and “top down” proteomics, multi-parameter ELISAs, bead or chip based affinity proteomics and multispectral imaging have been implemented for diagnosis, immune monitoring, immune response assays and/or identification of novel (immuno) therapeutic targets. However, these different profiling methods have some advantages and disadvantages (Table 1), which significantly depend on the sample preparation and handling.
Table 1: Advantages and disadvantages of technologies used for preclinical biomarker identification and for liquid biopsies
techniques |
advantages |
disadvantages |
|---|---|---|
Blood count |
Quick and reproducible, cheap |
Limited information |
Flow cytometry |
Numbers, frequency and activity of specific immune cell subpopulations |
Restricted to max. 30 parameters determined in parallel, data analysis requires strong expertise |
NGS |
High throughput analysis, identification of mutations, polymorphisms, expression profiles |
Large data set, strong bioinformatics required, expensive |
mRNA andmiRNA arrays |
High throughput analysis, mRNA and miRNA expression pattern |
Large data sets, targets not always identified |
Proteomics |
High throughput analysis, protein expression pattern |
Large data set, strong bioinformatics required, depending on the method very challenging |
IHC |
Frequency and localization, comparison to liquid biopsy data |
Not always antibodies available for paraffin-embedded tissues |
MSI |
Localisation and distance of immune cells |
Not an established procedure for different immune markers |
Metabolomics |
High through put analysis |
Large data sets, bioinformatics required, high dependence on patients life style |
Luminex/ELISA |
Stable, reproducible assay |
Limited information due to limited parallel analytes |
Early biomarker screening prior and during therapy could help to timely identify non-responders, and accordingly discontinue time- and cost-sensitive therapies in order to give patients the chance to switch to alternative treatment regimens. Such screening is mainly based on the identification of a biomarker signature rather than one particular biomarker, since a single biomarker cannot encompass all informations needed. A set of distinct biomarkers should be able to (i) discriminate between different tumor entities rather than merely between tumor patients and healthy individuals, (ii) discriminate different (early and advanced) tumor stages or subtypes and (iii) guide selection of efficient treatment options.
What is currently discussed to serve as (immune) biomarker in the blood?
Cell-free nucleic acids (cfNA) including DNA, RNA and Ago2-bound microRNAs (miRNAs), as well as CTC, exosomes and other EVs, protein panels, (auto)-antibodies, diverse metabolites, soluble mediators and changes in the frequency, composition and/or activity of (immune) cell populations are currently discussed as minimally-invasive biomarkers, which are easily accessible in blood samples from tumor patients to detect premalignant and early-stage cancers, to monitor response to treatment and development of therapy resistance [9–16].
Recently, exosomes have gained interest to advance biomarker research, since these nanometer-scale vesicles are secreted by living cells and carry the full range of molecules, e.g. DNA, RNA, miRNAs, proteins, cytokines and chemokines from their originating cells [6]. Due to their encapsulation in the bi-lipid membrane envelope their cargo is protected from enzymatic degradation and thus possesses a high biomarker potential [17]. Different methods [19] allowing the purification of high quality exosomal RNA (ExoLution™) or the simultaneous co-isolation of both cell-free DNA and exosomal RNA (ExoLution™ Plus) have been recently developed [20], which result in the isolation of EVs independent of their cellular origin. While almost all cell types secrete such vesicles, large number of vesicles will be also obtained from normal, non-cancerous cells, a problem, which has to be solved by the identification of cancer-derived exosomes as recently suggested for pancreatic cancer patients [21].
Circulating cell-free and exosomal DNA and RNA in human blood is derived from different cellular sources, including blood and tumor cells. It is postulated that circulating tumor DNA (ctDNA) reflects the molecular changes occurring at the tumor site in real time [22]. Indeed, the identification of somatic mutations within the epidermal growth factor receptor (EGF-R) gene using ctDNA isolated from plasma with the therascreen EGF-R Plasma RGQ PCR Kit [23] was successful. The kit was originally developed for tissue biopsies and was successfully implemented for non-small cell lung carcinoma (NSCLC) patients. The EGF-R mutational analyses of tissue biopsies provided evidence for predicting patients that respond to and clinically benefit from treatment with afatinib, erlotinib or gefitinib [24] and can also be applied for analyses of EGF-R mutations in blood samples [23]. CtDNA analysis is able to give a global view of cancer genomes across tumor sites [24–26] and can be used to monitor therapy response [27]. Due to the short half-life of ctDNA (± 2 hrs) and early changes following cytotoxic treatment ctDNA may provide an indication of tumor response [30, 31]. Therefore ctDNA analysis may prove to be useful for molecular stratification (e.g. exome-seq for mutation load or neo-antigen prediction), monitoring response (e.g. individualized TAA-seq for early kinetics and relapse) and for molecular profiling of relapsing patients (e.g. exome-seq to study tumor evolution) undergoing immunotherapy. Due to the chromosomal instability of tumors, analysis of plasma ctDNA is a suitable tool for determination of copy number profiles in comparison to patients’ biopsies [32]. Analogous to exosomes also cfNA might be tracked back to their origin (e.g. tumor, immune or cells of other origin) as recently investigated in plasma samples derived from gastric cancer patients using MYC and HER2/neu genes known to be amplified in this cancer type [29, 30].
Circulating miRNAs in serum or plasma might be also used as biomarkers due to their high stability under storage, easy handling conditions and emerging expression signatures that are associated with cancer survival and therapy response [35]. Plasma/serum miRNAs exist as cell-free, protein-bound molecules that are released by apoptotic and necrotic cells into the blood circulation, but are also actively released in exosomes. In HER-2/neu-positive breast cancer patients, high serum levels of cell-free miR-21 correlate with a shorter overall survival prior and after neoadjuvant therapy [32]. In ovarian cancer patients elevated serum concentrations of cell-free miR-429 are associated with advanced FIGO stages, higher values of the tumor marker CA125 and a poor overall survival rate [33]. In breast cancer patients the serum levels of exosomal miR-373 are significantly higher than those of cell-free miR-373 and associated with estrogen-negative receptor status and apoptosis [34]. It is still discussed whether the majority of circulating miRNAs is packaged into exosomes [39] or rather exists as Ago2-bound complexes [36], but both populations can be readily detected in plasma [20].
Although more than 50 years ago CTCs in the periphery of solid tumor patients have been identified, CTCs are still under critical review concerning their usefulness as biomarkers in cancer patients [41]. The biggest challenge is the low number of 1-10 CTCs/ml of blood thereby making their analysis both in terms of sensitivity and specificity very difficult.
Single or panels of proteins (≥ 10 analytes) from serum that address soluble immune-regulatory molecules, such as cytokines, chemokines and co-stimulators [38], acute phase proteins, complement and innate mediators as well as growth/apoptosis markers have been proven as biomarkers for the stratification of patients. These markers can be determined by multiplex analysis e.g. Luminex.
Potentially of high importance are the absolute numbers and relative proportions of immune cell populations within the patients’ tumor and immune cell populations, in particular in blood, lymph nodes and the bone marrow. The significance of the immune cell composition, the frequency as well as the localization of immune cell subpopulations within the tumor quantified in the “Immunoscore” [43] has been established in the last years and might be at least for colorectal cancer (CRC) superior than TNM grading and staging. However, since it requires tumor tissues and high skilled manpower alternative measurements with similar power are currently developed, such as multiplexing with centralized digital pathology (multispectral imaging) and/or multiplex tumor flow cytometry. Features of immune cells in terms of their differentiation and activation status as well as their effector potential are currently analyzed, in particular in blood of tumor patients undergoing diverse immunotherapies. This led for example to the identification of the prognostic value of peripheral blood CD8highCD57+ lymphocytes regarding recurrence-free survival of patients with non-muscle-invasive bladder carcinoma after transurethral resection and intra-vesical IL-2 treatment [40]. Other studies suggest that quantities and proportions of T (mainly CD8+) lymphocyte subsets in PBMCs are related to the increased survival of cancer patients already serving as biomarkers in myeloma [45]. Functional analyses for CD8+ T cell cytokine responsiveness as measured by changes in the phosphorylation status of STAT molecules might serve as biomarker discriminating glioblastoma patients benefiting from a DC vaccine [46].
Several studies imply that cancers with higher mutation burdens, such as microsatellite instability associated with a greater likelihood of expressing neo-antigens, are most likely to respond to checkpoint blockade inhibitors [7]. However, a broader picture of the patients’ tumor mutational load along with the status of the immune system as shown for the therapeutic success of anti-CTLA-4 (ipilimumab) [43] is needed to predict a patients’ response to immunotherapy [44]. So far, this information was obtained by the use of difficult to access and repeated sampling from tumor tissues. This type of analyses is currently translated to liquid biopsies using the potential of circulating immune cells, ctDNA, CTCs, or exosomal RNA to identify patients’ overall tumoral mutation load or specific TAAs from blood. Additional, analyses of protein levels for immune markers would increase the knowledge of the patients’ blood immune contexture.
While the induction of immune-related adverse events (irAEs) is a serious concern of immune-checkpoint blockade inhibition observing an inflamed tumor environment as well as increased preexisting autoantibodies, parameters that might act as markers of immune responsiveness bear the potential as clinical biomarkers [45, 46]. Thus monitoring auto-reactivity and anti-tumor reactivity by screening of antibodies should be included in a comprehensive biomarker strategy.
Sample preparation and difficulties in harmonizing biomarker detection
Sample preparation and generation is an important issue, since significant differences have been observed between distinct methods and consumables used for serum, plasma and immune cells obtained from PBMCs. In detail, the standby times until centrifugation (1 – 48 hrs), the blood collection method, nucleic acid preparation methods, exosome purification methods, methodical aspects for flow cytometry and the IgG purification from these samples could affect the analysis and result in reproducibility problems. Furthermore, if immune parameters are monitored, information about chemotherapy, anti-inflammatory drugs and hormone therapy is crucial, since these will most definitely affect the immune signatures. International consortia are currently establishing standard practices for blood collection and processing for circulating biomarkers (e.g. SPEDIA [51] and CANCER-ID [52]).
Other often neglected preclinical variables are the condition of the patients at the time of the specimen collection and by the specimen collection process itself. Common examples of patients’ condition include the duration of a fasting state/diet for blood lipids, glucose and other parameters. In addition, the time of day for blood collection affects cortisol and albumin levels [53]. The challenges with blood as for any other material undergoing biomarker analyses are the differences in the routine handling procedure, assay performance and inter-individual variability.
Two distinct strategies to synchronize biomarker detection are currently discussed in the literature: (i) the standardization of assays and (ii) the harmonization of assays. The first requires the exact same assay with the same standard operating procedures (SOP) and well-trained personal is needed in all laboratories, which is hard in terms of costs and feasibility. The second, more accepted way of handling this issue is the harmonization that requires trained personal and standardized laboratory equipment, not the exact same assay kits, but positive quality controls that have to be implemented in every assay in all cooperating/participating laboratories and research centers [9]. Successful harmonization of assays has been achieved exemplarily for the enzyme-linked immune spot (ELISPOT) technology aimed in recognizing antigen specific T cells in blood samples. After the stepwise introduction of harmonizing workflow guidelines the success rate of the participating centers in detection of rare antigen specific T cells within the central distributed standardized quality control samples increased dramatically from 53% to 93% [50].
Preclinical biomarker identification
Biomarker development following a traditional stepwise process from cell culture via mouse models to patient’s samples has helped, but also often failed in generating novel biomarkers with clinical relevance [55]. To identify new/improved biomarkers different well-known, but also new experimental approaches have to be implemented. Therefore, techniques and strategies like 3D culture, humanized mouse models and plasma protein profiling using recombinant antibody microarrays are discussed in this section [56, 57].
3D models in vitro
To explore cell biology and drug efficacy, cell-based assays were employed, in which tumor cells were typically grown on two-dimensional plastic surfaces or as single cell suspensions. However, since cell biology is profoundly influenced by its microenvironment, cell-based assays should be rather performed using 3D cultures, since they reflect both extracellular matrix and cell-cell contacts, such as cell-matrix interactions. These techniques were successfully implemented to study tumor-stroma interactions in NSCLC or tumor immune cell infiltration [51, 52]. This innovative method has the potential to improve the search for new immune biomarkers that could predict the patients’ responsiveness to immunotherapy. 3D culture models could be used to test the patients’ antitumor immune system and its modulation by immunotherapies in an ex vivo tumor micro tissue. A potential biomarker in this sense would have the capacity of a particular immune cell type to enter the tumor micro tissue or to change the effector function inside the tumor micro tissue upon a given immunotherapeutic intervention.
Experimental animal models
Very old, but still valid tools to study human diseases are experimental mouse models. Initially murine syngeneic mouse models or immune deficient human xenograft models were used to study new (immuno) therapeutic strategies to treat cancer. These models have major drawbacks since the tumors are either not of human origin or are human or even patient derived xenografts (PDX), but lack the immune system and the human stroma environment. In particular immunotherapies, such as checkpoint inhibitors like anti-PD1/anti-PD-L1 cannot be tested in immune deficient mice, since their intervention relies on the existing immune system. To circumvent these problems humanized mouse models have been established, in which the murine immune system is replaced by the human immune system using PBMCs or bone marrow stem cell transplantations [60]. These humanized mouse models are still under development, since they are difficult to handle and they do not reconstitute a complete human immune system [60]. However, in such humanized mice PDX can be established, which allow to study any of the new (immuno) therapeutic strategies in preclinical in vivo immunocompetent models over time. Thus, such model systems have the potential to lead to the identification of blood-borne biomarkers that predict patients’ responsiveness to immunotherapy.
Proteome/metabolome-based technologies
Other technologies are proteome-based, such as e.g. the Global Proteome Survey GPS that employs an oriented single chain antibody fragment immobilization approach with known antibodies and with context-independent motif specific (CIMS) antibodies that recognize peptides of hundreds of proteins allowing for a target discovery [61]. One big advantage of this technology is the low amount of blood in the μl range required for analysis. Using this technology, breast cancer has been graded with a significantly higher resolution when compared to conventional pathology [55]. Furthermore, using recombinant antibody microarrays serum protein signatures of immune regulatory molecules (cytokines, chemokines and co-stimulators) and unidentified peptides have been identified in a multicenter trial for pancreatic ductal adenocarcinoma, which allowed discriminating cancer patients from controls [38, 56]. Even more importantly, a large validation set of over 1300 pancreatic carcinoma patients and controls from two centers in Denmark identified a similar serum signature, which discriminated non-cancer patients from stage I and II pancreatic cancer patients. These data are of high significance for a potential early cancer diagnostic kit, resulting in a dramatically increase in patients’ survival chance for this particular cancer type normally displaying a dismal outcome with 5 year survival of 5-7 %. However, if early stage I/II disease can be detected the 5-year survival could increase to over 50 % (Japanese Pancreas Society). Currently, prospective studies are on the way in the USA. This demonstrates the potential in precision diagnostics [63].
A very interesting additional family of analytes with the potency to serve as biomarkers in blood samples are metabolites [64] including immune modulating amino acids, (anti)-oxidants and members of different lipid classes. In a study with breast cancer patients receiving endocrine therapy with tamoxifen several metabolites from different classes were altered in patients under tamoxifen when compared to cancer patients not receiving this treatment.
Auto-antibody detection
Furthermore, the use of auto-antibodies as biomarkers was exemplified in dissecting prostate cancer patients using a combination of different techniques such as protein arrays and Luminex that are spiked with auto-antigens (4012 or 3061). These results were validated on tissue microarrays (TMAs) and by qPCR of tissue samples. A different auto-antibody panel comprised of 165 antibodies (Ab) was found in serum of high when compared to low inflammation prostate cancer patients [59]. Of note, among the respective auto-antigens only Spastin (SPAST) was significantly increased in high versus low inflammation patients, while mRNA levels for all auto-antigens were unaltered. Thus no direct correlations between antibody titers in serum and their protein/mRNA expression levels exist in prostate cancer.
In vitro diagnostic (IVD)
IVD development in companies for biomarker identification is time consuming due to an inadequate understanding of the modes of action of therapeutic targets, inadequate preclinical models for discovery and validation in addition to the separation of the noise from real data and the cross-validation of a specific marker in an independent large cohort of patients. Another big challenge is the broad diversity in the patients’ cohort due to inter- and intra-heterogeneity of a particular disease [66–68]. One or a panel of identified biomarker(s) might not hold true for a complete patients’ cohort giving the need for individual consideration and the definition of inclusion/exclusion criteria. To figure this out, a close collaboration between physicians, academic research institutes and pharmaceutical industry partners is needed.
Liquid biopsies from cerebrospinal fluid, saliva, urine and other sources
In addition to blood as the most abundant liquid body fluid there are other body liquids to study biomarkers, such as ascites/pleural fluid, cerebrospinal fluid (CSF), bronchial alveolar lavage (BAL), urine and saliva/sputum (Table 2). Among these alternative liquids, saliva and urine are non-invasive and therefore of high interest to clinicians as current and future biomarker source. Collecting body fluids proximal to the tumor site of interest can significantly increase sensitivity for detection and monitoring [60]. Common to all alternative liquids for biomarker analysis and different to blood is the appearance of immune cells only in the case of inflammation or presumably late stage organ damage. All other (ctDNA, exosomes, proteins, metabolites and antibodies) mentioned blood biomarker will be testable to varying degrees in these alternative liquids.
Table 2: Non-invasive and invasive body fluids for the identification of tumor-derived information
body fluids |
non-invasive |
Invasive |
|---|---|---|
Peripheral blood |
✓ |
|
Serum/plasma |
✓ |
|
Saliva |
✓ |
|
CSF |
✓ |
|
Urine |
✓ |
|
BAL |
✓ |
|
Pleural effusion |
✓ |
Compared to blood saliva contains a similar variety of DNA, RNA, proteins, metabolites, and microbiota that can be compiled into a multiplex of cancer detection markers [61]. Urine has already been a well-studied source for biomarker analysis in different cancer types, such as, bladder cancer [62], kidney cancer [63], ovarian cancer [63] and prostate cancer [74]. Recently, urine samples were used in a study of more than 1500 patients of prostate cancer to predict high-grade prostate cancer by analysis of RNA from urinary exosomes [65]. Ascites has been analyzed in ovarian cancer patients for biomarkers associated with disease prognosis and patients’ outcome. In these patients high IFN-γ levels correlated with an increased overall survival [66]. This list could be extended to multiple other references on different liquid samples, which is not the prime focus of this article.
Individualized diagnostic and therapy
The most cost and effort effective way would certainly be to use standardized assays for biomarker analysis for a large number of patients. Since one faces a high patients’ cohort diversity, these standardized assays for a particular type of cancer disease might not be applicable. The profiling of blood samples might allow reducing the number of patients’ subgroups that require analyses of tissue biopsies [77, 78]. However, these patients’ subgroups would not allow predicting the clinical performance of the given patients’ group according to the current paradigm of “personalized medicine”. Thus, the logical extension of personalized medicine would tailor treatment to the specific individual without prior subgrouping of patients.
Novel individual therapeutic options include the alteration of the epigenetic features of tumors, in particular histone modifications and DNA methylation. Interestingly, the methylation status of melanoma patients does correlate with the overall survival [67]. Furthermore, in vitro data and in vivo preclinical mice data show re-expression of cancer testis antigens, improved anti-tumor immunity and better tumor control upon treatment with hypomethylating agents and/or histone deacetylation inhibitors. These data provide the rational for the use of hypomethylating agents and histone deacetylation inhibitors in melanoma patients. In the future, the methylation status should be investigated in liquid biopsies prior to individual therapy decision.
Properly controlled tissue acquisition and multiplex immunohistochemical analysis might overcome issues related to pre-analytical and analytical variability leading to a more reproducible assessment of clinically meaningful biomarkers. However, this technology still has to be improved and optimized due to the limited tissue material available in the clinical setting. The analysis of multiple immune parameters in tissue biopsies by multispectral fluorescent immunohistochemistry is an important advancement and has recently been successfully implemented in some clinical studies [68, 69]. This technique has several advantages, since it allows (i) a combination of different markers on a single histology section, (ii) the calculation of distances between individual cell types and (iii) the usage of already established antibodies for immunohistochemistry by only adding an additional labeling step [82].
Other non-invasive methods to image therapy success
The success rate of cancer immunotherapy is difficult to predict, as its efficacy often depends not only upon characteristics of the tumor lesions, but also of the tumor microenvironment involving immune cells and soluble mediators. Molecular and functional imaging with Positron Emission Tomography (PET) and Magnetic Resonance Imaging (MRI) allows repeated non-invasive in vivo measurement of many critical molecular features of tumor lesions and microenvironment, such as metabolism, proliferation, hypoxia, cell death and immune cell infiltrate, which can assist the knowledge of how cancer immunotherapy works and also facilitates clinical decision making [84].
More conventional PET imaging tracers such as 18F-FLT and 18F-FDG-PET were successfully used to study the kinetics and involvement of lymphocyte subsets in response to vaccination. These tracers incorporate in highly proliferative cells and are therefore not specific to cell types, but when locally applied e.g. in the tumor draining lymph node in conjunction with a DC vaccine, they reflect lymphocyte proliferation to vaccination. This technique allows an early discrimination between responding and non-responding patients in anti-cancer vaccination protocols and aids physicians in individualized decision-making processes [84].
New imaging tracers specifically targeting subsets of immune cells by the use of radiolabeled fragmented specific antibodies to CD8+ and CD4+ T cells (use of cys-diabody and minibody) and CD11b+ myeloid cells (use of variable domain of a camelid heavy-chain antibody) are successfully used in preclinical mouse models and currently being developed for advanced PET imaging in clinical monitoring applications to cancer immunotherapy [85–87].
From biopsy to peripheral blood
One of the major challenges remains to establish the biological connection of peripheral biomarkers and the complex tumor immune contexture. There is still a debate of whether peripheral immune cells are representative for the anti-tumor immunity observed in the tumor microenvironment (TME). Particularly, the link between peripheral blood immune cells and the prognostic and predictive relevance of the spatial distribution of immune cells in the tumor tissue is still unclear [76]. This is related to the fact that activated immune cells detected in the blood, primary or secondary lymphoid organs may be silenced locally by tumor cells or by immune suppressive cells e.g. regulatory T cells (Treg), myeloid derived suppressor cells (MDSC), tumor-associated macrophages (TAM) or cancer associated fibroblast [76]. Further, it is still not fully understood whether cancer cells release an equal amount of CTC, exosomes or ctDNA during disease progression and in response to treatment with a given therapy into the periphery. Overall, more interventional studies with integrated blood and tissue biomarkers combining proteomic and genomic multiplex approaches are needed to identify the clinical relevance of blood-based markers. However, due the invasive nature of tumor biopsies and the limitation to obtain multiple biopsies for serial assessments in the clinical routine, further investigation of blood based markers as a relevant surrogate tissue is needed.
The cellular sources of blood not only include the “classical” immune cells, but also CTCs of diverse tumor entities, hematopoietic stem cells, erythrocytes and platelets. In hemostasis, platelets have been demonstrated to be more present than effector cells and represent the major inflammatory cells mediating inflammasome signaling thereby affecting both innate and adaptive immune responses [89]. This led to the assumption that platelets are involved in the development of cancer and in success/failure of cancer therapies, which is a rather new and interesting field for basic researchers and clinicians [78].
Controversial discussion of tissue PD-L1 expression as biomarker: alternatives may predict treatment success to PD-1/PD-L1 axis using liquid biopsies
Recently, the use of antibodies directed against different checkpoint targets, such as anti-PD-1 (Nivolumab, Pembrolizumab and Pidilizumab), anti-PD-L1 (MS-936559, MEDI4736, and MPDL3280A) and anti-CTLA4 (Ipilimumab) has been successfully implemented for the treatment of a number of different tumor entities [79]. For anti-PD1/-PD-L1 treatment, the immunohistochemical analysis of PD-L1 expression on the surface of tumor cells was suggested as a suitable biomarker to predict therapy success. It was postulated that blocking PD1/PD-L1 could be used in patients’ with tumors expressing PD-L1. PD-L1 positive tumors are known to cause impaired T cell effector function by binding to PD-1 on T cells. However, the use of PD-L1 as biomarker for melanoma and lung cancer [80, 81] is still under debate. This is due to conflicting results in different patients’ cohorts by lack of standardized methodology, lack of consensus on the “best” antibody associated with a lack of standardized cut off values for sample positivity or values of intensity and lack of agreement on cells to analyze. Additionally, PD-L1 and PD1 are not exclusively expressed on tumor tissues or effector T cells, but are rather widely distributed among immune cells and other tissue cells, present in the TME. Furthermore, it is noteworthy that at least one known additional ligand for PD-1, the PD-L2, also known as B7-DC, exists, which is not blocked by anti-PD-L1 antibodies [94]. Even more complexity arises from the fact that the existence of soluble forms of PD-L1 has been confirmed, which might affect the efficacy of an anti-PD-L1-based therapy [83, 84].
Alternatively to PD-L1 expression on tumor cells as a biomarker, one study found a significant correlation of high tumor PD-L1 expression on immune cells infiltrating the tumor as a predictive marker for therapy success [85]. The success of anti-PD-1 therapy appears to rely on the pre-existence of intra-tumoral CD8+ T cells with proliferative capacity [86, 87]. Other factors, such as increased density and decreased diversity in antigen specificity of T cells within the tumor, may also provide predictive value, as observed in melanoma patients treated with PD-1 mAb [87]. However, these alternative profiling strategies have not yet been validated on different cohorts or/and tumor entities. This is in line with a number of other putative novel published biomarkers, which often has either not yet been validated or the validation attempts from other laboratories with another patients’ cohort failed. However, a multi-parameter serum analysis demonstrated differentially expressed analytes in responder and non-responder patients upon nivolumab treatment in melanoma patients (personal communication R. Hockett). These promising data have already been validated in a second patient cohort and are currently analyzed in a prospective study.
Pros and cons of blood biopsies
There exist a number of “pros” and “cons” in terms of biomarker analysis for both blood samples and the current gold standard, tumor biopsies. The advantages of tumor biopsies are (i) the in situ analysis of the primary site of interest as well as (ii) the spatial resolution of the TME known as the tumor landscape. Disadvantages of tissue biopsies are (i) the invasiveness of the method, (ii) lack of willingness of patients for the necessary surgery, (iii) serial assessment and of high importance, (iv) the tumor heterogeneity (intra and primary versus metastases). The advantage of blood samples are (i) minimal invasive, (ii) serial assessment, (iii) live monitoring and (iv) systemic view that may account of tumor heterogeneity (v) the speed and reduced cost of sample preparation and (vi) the easy access of blood material from healthy volunteers or patients’ with different diseases for comparison. The disadvantages on the other hand are (i) no spatial resolution of tumor microenvironment (ii) lack of knowledge whether the blood reflects the “true” host tumor interaction and (iii) uncertainty whether the blood sample overcomes the tumor heterogeneity issue (Figure 2). However, clinical data to address some of these concerns are starting to accumulate, and some ctDNA-based assays have already been found to be as predictive as tissue in recent retrospective clinical studies [88]. As a definitive sign that liquid biopsies are starting to enter clinical practice several liquid biopsy IVD products are already available, e.g. FDA recently approved Roche cobas EGF-R mutation test for detection of EGF-R mutations in plasma of NSCLC patients as a Companion Diagnostic test to Tarceva [101].
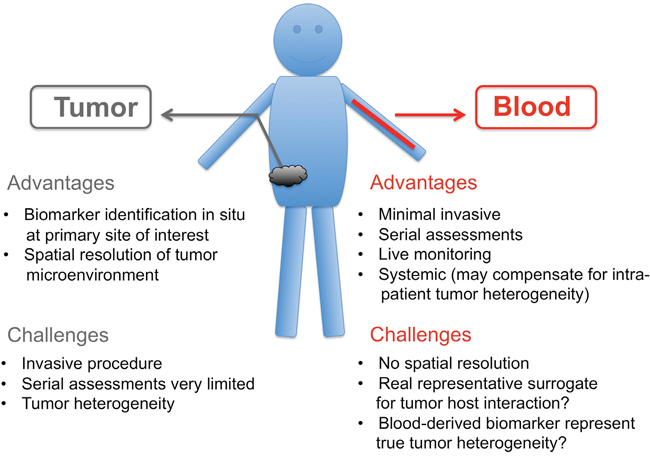
Figure 2: “Pros” and “Cons” of tumor biopsies versus blood biopsies.
More broadly, a comprehensive and integrated bio-sampling approach of tissues and PBMCs in the clinical setting and preclinical models may allow the identification of less invasive biomarkers that reflect clinically meaningful aspects of the immune tumor microenvironment, improving the triage and management of patients in cancer immunotherapy. Overall, new tools for tissue analysis, rigorous validation and standardization of methods will help to better understand the dynamic nature of immune-tumor interaction.
CONCLUSIONS
The clinical implementation of blood-derived biomarkers to predict tumor progression, response to therapy, therapy resistance or even early diagnosis of tumor occurrence is certainly wanted and urgently required. Recent progress is promising and the identification of the “right” biomarker/biomarker panel to define different tumor entities, if possible at distinct disease stages, is emerging, but needs to overcome current challenges of sample preparation, standardization of techniques, and acceptance in clinical practice. Even though, prospective studies that validate the liquid biopsy data in independent patient cohorts are crucial and are still missing in most cases, the so far obtained biomarkers from retrospective studies using liquid biopsies are very encouraging Despite these advances there is still an urgent need of blood analysis as a potential surrogate for tissues samples as summarized in Figure 2. Some of the different biomarkers presented here might serve as diagnostic tools, such as tumor specific auto-antibodies, which are long lasting. Others might be implemented for real-time monitoring of therapy success, such as DNA and RNA mutations, since these alterations are reflecting disease burden. Blood- or tumor-derived miRNAs seem to illuminate the topic of therapy responder/non-responder and also could timely uncover therapy success. Protein biomarker panels have shown accurate diagnostic abilities, although further research is needed to identify their true nature in the context of different cancer indications. Here, exosomes may allow biofluid-based detection of membrane surface proteins from the tumor microenvironment. Liquid biopsies are a powerful tool that fosters efficient new (immuno) therapeutic implementations to treat cancer and have the potential to significantly reduce tumor biopsies.
Abbreviations
Ab: antibody; ALC: absolute lymphocyte count; BAL: broncho alveolar lavage; cfNA: cell-free nucleic acid; CIMS: context independent motif specific; CRC: colorectal cancer; CSF: cerebrospinal fluid; ctDNA: circulating tumor DNA; CTC: circulating tumor cells; EGF-R: epidermal growth factor receptor; EV: extracellular vesicle; miRNA: microRNA; MSI: multispectral imaging; NSCLC: non-small cell lung carcinoma; PBMC: peripheral blood mononuclear cells; PDX: patients’ derived xenografts; SNP: single nucleotide polymorphisms; SOP: standard operation procedures; TAA: tumor associated antigen; TMA: tissue micro array: TME: tumor microenvironment.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Bardelli A, Pantel K. Liquid biopsies, what we do not know (yet). Cancer Cell. 2017; 31:172-179.
2. WHO. International Programme on Chemical Safety Biomarkers in Risk Assessment: Validity and Validation. 2001; http://www.inchem.org/documents/ehc/ehc/ehc222.htm.
3. Characiejus D, Jacobs JJ, Pasukoniene V, Kazlauskaite N, Danileviciute V, Mauricas M, Den Otter W. Prediction of response in cancer immunotherapy. Anticancer Res. 2011; 31:639-647.
4. Amur SG, Sanyal S, Chakravarty AG, Noone MH, Kaiser J, McCune S, Buckman-Garner SY. Building a roadmap to biomarker qualification: challenges and opportunities. Biomark Med. 2015; 9:1095-1105.
5. Bernicker E. Next-generation sequencing and immunotherapy biomarkers: a medical oncology perspective. Arch Pathol Lab Med. 2016; 140:245-248.
6. Feller SM, Lewitzky M. Hunting for the ultimate liquid cancer biopsy - let the TEP dance begin. Cell Commun Signal. 2016; 14:24.
7. Gubin MM, Schreiber RD. CANCER. The odds of immunotherapy success. Science. 2015; 350:158-159.
8. Schumacher TN, Kesmir C, van Buuren MM. Biomarkers in cancer immunotherapy. Cancer Cell. 2015; 27:12-14.
9. van der Burg SH, Kalos M, Gouttefangeas C, Janetzki S, Ottensmeier C, Welters MJ, Romero P, Britten CM, Hoos A. Harmonization of immune biomarker assays for clinical studies. Sci Transl Med. 2011; 3:108ps144.
10. Deng X, Nakamura Y. Cancer precision medicine: from cancer screening to drug selection and personalized immunotherapy. Trends Pharmacol Sci. 2017; 38:15-24.
11. Hofman P. Liquid biopsy for early detection of lung cancer. Curr Opin Oncol. 2017; 29:73-78.
12. Hofman P, Popper HH. Pathologists and liquid biopsies: to be or not to be? Virchows Arch. 2016; 469:601-609.
13. Huang WL, Chen YL, Yang SC, Ho CL, Wei F, Wong DT, Su WC, Lin CC. Liquid biopsy genotyping in lung cancer: ready for clinical utility? Oncotarget. 2017; 8:18590-18608. doi: 10.18632/oncotarget.14613.
14. Shigeyasu K, Toden S, Zumwalt TJ, Okugawa Y, Goel A. Emerging role of micrornas as liquid biopsy biomarkers in gastrointestinal cancers. Clin Cancer Res. 2017 Jan 31. doi: 10.1158/1078-0432.CCR-16-1676. [Epub ahead of print].
15. Thiele JA, Bethel K, Kralickova M, Kuhn P. Circulating tumor cells: fluid surrogates of solid tumors. Annu Rev Pathol. 2017; 12:419-447.
16. Torrano V, Royo F, Peinado H, Loizaga-Iriarte A, Unda M, Falcon-Perez JM, Carracedo A. Vesicle-MaNiA: extracellular vesicles in liquid biopsy and cancer. Curr Opin Pharmacol. 2016; 29:47-53.
17. Yi X, Ma J, Guan Y, Chen R, Yang L, Xia X. The feasibility of using mutation detection in ctDNA to assess tumor dynamics. Int J Cancer. 2017; 140:2642-2647.
18. Nawaz M, Fatima F, Nazarenko I, Ekstrom K, Murtaza I, Anees M, Sultan A, Neder L, Camussi G, Valadi H, Squire JA, Kislinger T. Extracellular vesicles in ovarian cancer: applications to tumor biology, immunotherapy and biomarker discovery. Expert Rev Proteomics. 2016; 13:395-409.
19. Abramowicz A, Widlak P, Pietrowska M. Proteomic analysis of exosomal cargo: the challenge of high purity vesicle isolation. Mol Biosyst. 2016; 12:1407-1419.
20. Enderle D, Spiel A, Coticchia CM, Berghoff E, Mueller R, Schlumpberger M, Sprenger-Haussels M, Shaffer JM, Lader E, Skog J, Noerholm M. Characterization of RNA from exosomes and other extracellular vesicles isolated by a novel spin column-based method. PLoS One. 2015; 10:e0136133.
21. Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, LeBleu VS, Mittendorf EA, Weitz J, Rahbari N, Reissfelder C, Pilarsky C, Fraga MF, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015; 523:177-182.
22. Karachaliou N, Mayo-de-Las-Casas C, Molina-Vila MA, Rosell R. Real-time liquid biopsies become a reality in cancer treatment. Ann Transl Med. 2015; 3:36.
23. Menzel C, Kazinski M. Liquid biopsy: its impact on cancer diagnostics. MLO Med Lab Obs. 2015; 47:27-28.
24. Syed YY. therascreen ((R)) EGFR RGQ PCR Kit: a companion diagnostic for afatinib and gefitinib in non- small cell lung cancer. Mol Diagn Ther. 2016; 20:191-198.
25. Douillard JY, Ostoros G, Cobo M, Ciuleanu T, Cole R, McWalter G, Walker J, Dearden S, Webster A, Milenkova T, McCormack R. Gefitinib treatment in EGFR mutated caucasian NSCLC: circulating-free tumor DNA as a surrogate for determination of EGFR status. J Thorac Oncol. 2014; 9:1345-1353.
26. Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012; 4:136ra168.
27. Murtaza M, Dawson SJ, Pogrebniak K, Rueda OM, Provenzano E, Grant J, Chin SF, Tsui DW, Marass F, Gale D, Ali HR, Shah P, Contente-Cuomo T, et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat Commun. 2015; 6:8760.
28. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108-112.
29. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. New Engl J Med. 2013; 368:1199-1209.
30. Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008; 14:985-990.
31. Qin Z, Ljubimov VA, Zhou C, Tong Y, Liang J. Cell-free circulating tumor DNA in cancer. Chin J Cancer. 2016; 35:36.
32. Vanderstichele A, Busschaert P, Smeets D, Landolfo C, Van Nieuwenhuysen E, Leunen K, Neven P, Amant F, Mahner S, Braicu E, Zeilinger R, Coosemans A, Timmerman D. Chromosomal instability in cell-free DNA as a highly specific biomarker for detection of ovarian cancer in women with adnexal masses. Clin Cancer Res. 2016; 23:2223-2231.
33. Park KU, Lee HE, Park DJ, Jung EJ, Song J, Kim HH, Choe G, Kim WH, Lee HS. MYC quantitation in cell-free plasma DNA by real-time PCR for gastric cancer diagnosis. Clin Chem Lab Med. 2009; 47:530-536.
34. Shoda K, Masuda K, Ichikawa D, Arita T, Miyakami Y, Watanabe M, Konishi H, Imoto I, Otsuji E. HER2 amplification detected in the circulating DNA of patients with gastric cancer: a retrospective pilot study. Gastric Cancer. 2015; 18:698-710.
35. Schwarzenbach H, Nishida N, Calin GA, Pantel K. Clinical relevance of circulating cell-free microRNAs in cancer. Nat Rev Clin Oncol. 2014; 11:145-156.
36. Muller V, Gade S, Steinbach B, Loibl S, von Minckwitz G, Untch M, Schwedler K, Lubbe K, Schem C, Fasching PA, Mau C, Pantel K, Schwarzenbach H. Changes in serum levels of miR-21, miR-210, and miR-373 in HER2-positive breast cancer patients undergoing neoadjuvant therapy: a translational research project within the Geparquinto trial. Breast Cancer Res Treat. 2014; 147:61-68.
37. Meng X, Joosse SA, Muller V, Trillsch F, Milde-Langosch K, Mahner S, Geffken M, Pantel K, Schwarzenbach H. Diagnostic and prognostic potential of serum miR-7, miR-16, miR-25, miR-93, miR-182, miR-376a and miR-429 in ovarian cancer patients. Br J Cancer. 2015; 113:1358-1366.
38. Eichelser C, Stuckrath I, Muller V, Milde-Langosch K, Wikman H, Pantel K, Schwarzenbach H. Increased serum levels of circulating exosomal microRNA-373 in receptor-negative breast cancer patients. Oncotarget. 2014; 5:9650-9663. doi: 10.18632/oncotarget.2520.
39. Gallo A, Tandon M, Alevizos I, Illei GG. The majority of microRNAs detectable in serum and saliva is concentrated in exosomes. PLoS One. 2012; 7:e30679.
40. Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, Gibson DF, Mitchell PS, Bennett CF, Pogosova-Agadjanyan EL, Stirewalt DL, Tait JF, Tewari M. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci U S A. 2011; 108:5003-5008.
41. Alix-Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer discovery. 2016; 6:479-491.
42. Gerdtsson AS, Malats N, Sall A, Real FX, Porta M, Skoog P, Persson H, Wingren C, Borrebaeck CA. A multicenter trial defining a serum protein signature associated with pancreatic ductal adenocarcinoma. Int J Proteomics. 2015; 2015:587250.
43. Angell H, Galon J. From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol. 2013; 25:261-267.
44. Characiejus D, Pasukoniene V, Jacobs JJ, Eidukevicius R, Jankevicius F, Dobrovolskiene N, Mauricas M, Van Moorselaar RJ, Den Otter W. Prognostic significance of peripheral blood CD8highCD57+ lymphocytes in bladder carcinoma patients after intravesical IL-2. Anticancer Res. 2011; 31:699-703.
45. Sze DM, Brown RD, Yuen E, Gibson J, Ho J, Raitakari M, Basten A, Fazekas de St Groth J, Fazekas de St Groth B. Clonal cytotoxic T cells in myeloma. Leuk Lymphoma. 2003; 44:1667-1674.
46. Everson RG, Jin RM, Wang X, Safaee M, Scharnweber R, Lisiero DN, Soto H, Liau LM, Prins RM. Cytokine responsiveness of CD8(+) T cells is a reproducible biomarker for the clinical efficacy of dendritic cell vaccination in glioblastoma patients. J Immunother Cancer. 2014; 2:10.
47. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015; 350:207-211.
48. Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The “cancer immunogram”. Science. 2016; 352:658-660.
49. Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H. Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol. 2014; 11:509-524.
50. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016; 16:275-287.
51. SPIDIA. http://www.spidia.eu.
52. CANCER. http://www.cancer-id.eu.
53. Miller WG, Tate JR, Barth JH, Jones GR. Harmonization: the sample, the measurement, and the report. Ann Lab Med. 2014; 34:187-197.
54. Janetzki S, Panageas KS, Ben-Porat L, Boyer J, Britten CM, Clay TM, Kalos M, Maecker HT, Romero P, Yuan J, Kast WM, Hoos A, Elispot Proficiency Panel of the CVCIAWG. Results and harmonization guidelines from two large-scale international Elispot proficiency panels conducted by the Cancer Vaccine Consortium (CVC/SVI). Cancer Immunol Immunother. 2008; 57:303-315.
55. Maurel J, Postigo A. Prognostic and predictive biomarkers in colorectal cancer. From the preclinical setting to clinical practice. Curr Cancer Drug Targets. 2015; 15:703-715.
56. Halfter K, Mayer B. Bringing 3D tumor models to the clinic-predictive value for personalized medicine. Biotechnol J. 2017; 12. doi: 10.1002/biot.201600295. Epub 2017 Jan 18.
57. Sia D, Moeini A, Labgaa I, Villanueva A. The future of patient-derived tumor xenografts in cancer treatment. Pharmacogenomics. 2015; 16:1671-1683.
58. Amann A, Zwierzina M, Gamerith G, Bitsche M, Huber JM, Vogel GF, Blumer M, Koeck S, Pechriggl EJ, Kelm JM, Hilbe W, Zwierzina H. Development of an innovative 3D cell culture system to study tumour--stroma interactions in non-small cell lung cancer cells. PLoS One. 2014; 9:e92511.
59. Koeck S, Zwierzina M, Huber JM, Bitsche M, Lorenz E, Gamerith G, Dudas J, Kelm JM, Zwierzina H, Amann A. Infiltration of lymphocyte subpopulations into cancer microtissues as a tool for the exploration of immunomodulatory agents and biomarkers. Immunobiology. 2016; 221:604-617.
60. Sanmamed MF, Chester C, Melero I, Kohrt H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann Oncol. 2016; 27:1190-1198.
61. Sall A, Persson H, Ohlin M, Borrebaeck CA, Wingren C. Advancing the global proteome survey platform by using an oriented single chain antibody fragment immobilization approach. N Biotechnol. 2016; 33:503-513.
62. Olsson N, Carlsson P, James P, Hansson K, Waldemarson S, Malmstrom P, Ferno M, Ryden L, Wingren C, Borrebaeck CA. Grading breast cancer tissues using molecular portraits. Mol Cell Proteomics. 2013; 12:3612-3623.
63. Gerdtsson AS, Wingren C, Persson H, Delfani P, Nordstrom M, Ren H, Wen X, Ringdahl U, Borrebaeck CA, Hao J. Plasma protein profiling in a stage defined pancreatic cancer cohort: implications for early diagnosis. Mol Oncol. 2016; 10:1305-1316.
64. Wishart DS, Mandal R, Stanislaus A, Ramirez-Gaona M. Cancer metabolomics and the human metabolome database. Metabolites. 2016; 6:pii: E10.
65. Schlick B, Massoner P, Lueking A, Charoentong P, Blattner M, Schaefer G, Marquart K, Theek C, Amersdorfer P, Zielinski D, Kirchner M, Trajanoski Z, Rubin MA, et al. Serum autoantibodies in chronic prostate inflammation in prostate cancer patients. PLoS One. 2016; 11:e0147739.
66. Halim AB. Some imminent but overlooked preanalytical and analytical challenges currently facing biomarkers and companion diagnostics. Ann N Y Acad Sci. 2015; 1346:63-70.
67. Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017.
68. Wan JC, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R, Rosenfeld N. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017; 17:223–238.
69. Wang Y, Springer S, Mulvey CL, Silliman N, Schaefer J, Sausen M, James N, Rettig EM, Guo T, Pickering CR, Bishop JA, Chung CH, Califano JA, et al. Detection of somatic mutations and HPV in the saliva and plasma of patients with head and neck squamous cell carcinomas. Sci Transl Med. 2015; 7:293ra104.
70. Mishra S, Saadat D, Kwon O, Lee Y, Choi WS, Kim JH, Yeo WH. Recent advances in salivary cancer diagnostics enabled by biosensors and bioelectronics. Biosens Bioelectron. 2016; 81:181-197.
71. Frantzi M, Latosinska A, Fluhe L, Hupe MC, Critselis E, Kramer MW, Merseburger AS, Mischak H, Vlahou A. Developing proteomic biomarkers for bladder cancer: towards clinical application. Nat Rev Urol. 2015; 12:317-330.
72. Rodrigues D, Monteiro M, Jerónimo C, Henrique R, Belo L, Bastos ML, Guedes de Pinho P, Carvalho M. Renal cell carcinoma: a critical analysis of metabolomic biomarkers emerging from current model systems. Transl Res. 2017; 180:1-11.
73. Liao JB, Yip YY, Swisher EM, Agnew K, Hellstrom KE, Hellstrom I. Detection of the HE4 protein in urine as a biomarker for ovarian neoplasms: clinical correlates. Gynecolo Oncol. 2015; 137:430-435.
74. Locke JA, Black PC. Next generation biomarkers in prostate cancer. Front Biosci. 2016; 21:328-342.
75. McKiernan J, Donovan MJ, O'Neill V, Bentink S, Noerholm M, Belzer S, Skog J, Kattan MW, Partin A, Andriole G, Brown G, Wei JT, Thompson IM Jr, Carroll P. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. JAMA Oncol. 2016; 2:882-889.
76. Chen YL, Cheng WF, Chang MC, Lin HW, Huang CT, Chien CL, Chen CA. Interferon-gamma in ascites could be a predictive biomarker of outcome in ovarian carcinoma. Gynec Oncol. 2013; 131:63-68.
77. Collins DC, Sundar R, Lim JS, Yap TA. Towards precision medicine in the clinic: from biomarker discovery to novel therapeutics. Trends Pharmacol Sci. 2017; 38:25-40.
78. Riquelme I, Saavedra K, Espinoza JA, Weber H, Garcia P, Nervi B, Garrido M, Corvalan AH, Roa JC, Bizama C. Molecular classification of gastric cancer: towards a pathway-driven targeted therapy. Oncotarget. 2015; 6:24750-24779. doi: 10.18632/oncotarget.4990.
79. Sigalotti L, Covre A, Fratta E, Parisi G, Sonego P, Colizzi F, Coral S, Massarut S, Kirkwood JM, Maio M. Whole genome methylation profiles as independent markers of survival in stage IIIC melanoma patients. J Transl Med. 2012; 10:185.
80. Feng Z, Puri S, Moudgil T, Wood W, Hoyt CC, Wang C, Urba WJ, Curti BD, Bifulco CB, Fox BA. Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. J Immunother Cancer. 2015; 3:47.
81. Nghiem PT, Bhatia S, Lipson EJ, Kudchadkar RR, Miller NJ, Annamalai L, Berry S, Chartash EK, Daud A, Fling SP, Friedlander PA, Kluger HM, Kohrt HE, et al. PD-1 blockade with pembrolizumab in advanced merkel-cell carcinoma. N Engl J Med. 374:2542-2552.
82. Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods. 2014; 70:46-58.
83. Juergens RA, Zukotynski KA, Singnurkar A, Snider DP, Valliant JF, Gulenchyn KY. Imaging biomarkers in immunotherapy. Biomark Cancer. 2016; 8:1-13.
84. Aarntzen EH, Srinivas M, De Wilt JH, Jacobs JF, Lesterhuis WJ, Windhorst AD, Troost EG, Bonenkamp JJ, van Rossum MM, Blokx WA, Mus RD, Boerman OC, Punt CJ, et al. Early identification of antigen-specific immune responses in vivo by [18F]-labeled 3'-fluoro-3'-deoxy-thymidine ([18F]FLT) PET imaging. Proc Natl Acad Sci U S A. 2011; 108:18396-18399.
85. Tavare R, Escuin-Ordinas H, Mok S, McCracken MN, Zettlitz KA, Salazar FB, Witte ON, Ribas A, Wu AM. An effective immuno-pet imaging method to monitor cd8-dependent responses to immunotherapy. Cancer Res. 2016; 76:73-82.
86. Rashidian M, Keliher EJ, Bilate AM, Duarte JN, Wojtkiewicz GR, Jacobsen JT, Cragnolini J, Swee LK, Victora GD, Weissleder R, Ploegh HL. Noninvasive imaging of immune responses. Proc Natl Acad Sci U S A. 2015; 112:6146-6151.
87. Tavare R, McCracken MN, Zettlitz KA, Salazar FB, Olafsen T, Witte ON, Wu AM. Immuno-PET of murine T cell reconstitution postadoptive stem cell transplantation using anti-CD4 and anti-CD8 Cys-diabodies. J Nucl Med. 2015; 56:1258-1264.
88. Kirilovsky A, Marliot F, El Sissy C, Haicheur N, Galon J, Pages F. Rational bases for the use of the Immunoscore in routine clinical settings as a prognostic and predictive biomarker in cancer patients. Int Immunol. 2016; 28:373-382.
89. Hottz ED, Monteiro AP, Bozza FA, Bozza PT. Inflammasome in platelets: allying coagulation and inflammation in infectious and sterile diseases? Mediators Inflamm. 2015; 2015:435783.
90. Franco AT, Corken A, Ware J. Platelets at the interface of thrombosis, inflammation, and cancer. Blood. 2015; 126:582-588.
91. Lipson EJ, Forde PM, Hammers HJ, Emens LA, Taube JM, Topalian SL. Antagonists of PD-1 and PD-L1 in cancer treatment. Semin Oncol. 2015; 42:587-600.
92. Aguiar PN Jr, Santoro IL, Tadokoro H, de Lima Lopes G, Filardi BA, Oliveira P, Mountzios G, de Mello RA. The role of PD-L1 expression as a predictive biomarker in advanced non-small-cell lung cancer: a network meta-analysis. Immunotherapy. 2016; 8:479-488.
93. Fusi A, Festino L, Botti G, Masucci G, Melero I, Lorigan P, Ascierto PA. PD-L1 expression as a potential predictive biomarker. Lancet Oncol. 2015; 16:1285-1287.
94. Seliger B, Quandt D. The expression, function, and clinical relevance of B7 family members in cancer. Cancer Immunol Immunother. 2012; 61:1327-1341.
95. Finkelmeier F, Canli O, Tal A, Pleli T, Trojan J, Schmidt M, Kronenberger B, Zeuzem S, Piiper A, Greten FR, Waidmann O. High levels of the soluble programmed death-ligand (sPD-L1) identify hepatocellular carcinoma patients with a poor prognosis. Eur J Cancer. 2016; 59:152-159.
96. Rossille D, Gressier M, Damotte D, Maucort-Boulch D, Pangault C, Semana G, Le Gouill S, Haioun C, Tarte K, Lamy T, Milpied N, Fest T, Groupe Ouest-Est des Leucémies et Autres Maladies du Sang. High level of soluble programmed cell death ligand 1 in blood impacts overall survival in aggressive diffuse large B-Cell lymphoma: results from a French multicenter clinical trial. Leukemia. 2014; 28:2367-2375.
97. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014; 515:563-567.
98. Gajewski TF. The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Semin Oncol. 2015; 42:663-671.
99. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568-571.
100. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. Assessment of EGFR mutation status in matched plasma and tumor tissue of NSCLC patients from a Phase I study of rociletinib (CO-1686). Clin Cancer Res. 2016; 22:2386-2395.
101. FDA. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm504540.htm.