
INTRODUCTION
Glioblastoma multiforme (GBM) is the most fre-quent and lethal form of primary brain tumor, with an annual incidence of 5.26 per 100,000 people, or 17,000 new cases diagnosed yearly worldwide [1]. GBM is more common among Caucasian men and is typical of advanced age. Clinical management consists of maximal surgical resection followed by radiation and chemotherapy [2–4]. As a result of its infiltrative growth, in the majority of cases, GBM generally reoccurs within 7 months to 10 months after surgical intervention [5–7]. Despite treatment, most patients succumb to the disease some 12 months to 18 months after diagnosis, while for recurrent GBM, life expectancy is ~6 months [1, 8–12]. Due to this short survival with currently available therapies, alternative treatments are under extensive exploration [2, 13–16].
Among the major issues in GBM management are rapid tumor growth, tumor location, and late diagnosis. The general late diagnosis of GBM is a consequence of the asymptomatic nature of the early disease stages, which present with nausea, headache, and cognition changes. To better define GBM, a number of proteins have been proposed as possible biomarkers, including the EGFR/EGFRvIII pair, and the glycoprotein CD133 [17–22]. Further studies have investigated patient serum and cerebrospinal fluid for possible biomarkers, although the specificities of all of these candidate proteins remain to be validated [23–26].
An alternative approach for discovering proteins with specificity for GBM is offered by using nanobodies, which are antigen-binding fragments that are derived from naturally occurring heavy-chain-only antibodies in camelids [27, 28]. Importantly, the complete antigen-binding fragment of a nanobody is encoded by a single gene fragment, the heavy-chain variable region, or VHH. Thus this avoids gene splitting and scrambling during cloning, which happens with single-chain variable fragments [29]. In addition, their extended H3 loop that is responsible for antigen binding predicts the specificity for novel epitopes that can be hidden from classical antibodies and conventional proteomic techniques. Furthermore, nanobodies possess exceptional and beneficial features including small size (~14 kDa), high resistance to non-physiological pH and elevated temperature, water solubility, resistance to aggregation, and simple and inexpensive production in bacteria.
This straightforward anti-proteomic approach led to the identification of seven candidate biomarkers that conventional techniques have failed to uncover. This finding illustrates that the nanobody technology is a suitable alternative approach for identification of GBM-specific proteins and it has provided a good starting point for further investigation of their roles. Potential use of DPYSL2 as a “glioma versus reference” tissue biomarker, and the roles of some of the suggested biomarkers for class differentiation have been confirmed with qPCR on a small scale study, but need to be validated with a larger sample number.
RESULTS
Glioblastoma multiforme target-specific nanobodies
The nanobody library against GBM cells comprised 108 individual transformants, which is consistent with the average size of a high quality immune nanobody library [30]. Phage enrichment during panning on protein extracts of GBM stem-like cell lines was good, as there were at least two-fold more bacteria infected with viral particles retrieved from GBM samples than from reference samples. After the second and third round of panning, large numbers of bacteria were grown and their periplasmic proteins were screened by ELISA. Proteins from the periplasm that showed at least 1.5-fold higher ELISA signals in wells with GBM lysate than in wells with reference lysate were considered positive. Numerous ELISA screenings led to the identification of seven nanobodies with specificity for GBM proteins: Nb10, Nb79, Nb179, Nb225, Nb314, Nb394, and Nb395, with GBM/ reference ELISA ratios of 1.54, 2.27, 1.68, 2.17, 2.25, 1.53, and 3.29, respectively. The nanobody genes obtained after Sanger sequencing were translated in silico to their amino acid sequence and revealed the characteristic starting (i.e., QVQL, DVQL) and ending (i.e., TVSS) amino acid sequences [31, 32]. A unique H3 region for each nanobody suggested that they might recognize different antigens (Figure 1).

Figure 1: Nanobody sequences. The selected nanobodies show the characteristic starting (QVQL or DVQL) and ending (TVSS) nanobody sequences. Different H3 loops imply that all of these nanobodies bind to different antigens; i.e., different proteins of interest. The presence of the GLEW sequence motif in the FR2 region of Nb10 indicates its VH germline origin during the V-D-J recombination, whilst the rest of the nanobodies definitely have a VHH germline origin, as e.g. for the FR2 sequences that have the VHH-typical Arg50. Amino acid sequences of the H3 loops are given in alphabetical order.
Antigens recognized by nanobodies
The purified nanobodies were used to immune-capture their cognate targets in protein lysates from GBM stem-like cell lines. Using a ≤5% false-discovery rate, the captured antigens were identified by mass spectrometry, as: Nb10: β-actin/nucleolin (ACTB/NUCL) complex; Nb79: vimentin (VIM); Nb179: nucleosome assembly protein 1 like (NAP1L1); Nb225: Tu translation elongation factor, mitochondrial (TUFM); Nb314: dihydropyrimidinase-related protein 2 (DPYSL2) and/or methylenetetrahydrofolate dehydrogenase 1 (MTHFD1); Nb394: collapsin response mediator protein 1 (CRMP1); and Nb395: ALY/REF export factor (ALYREF).
Differential protein occurrence in glioblastoma, lower grade glioma, and reference samples
Western blot quantification showed that with the exception of NUCL, all of the other target proteins were over-represented in the cytosolic protein fraction of GBM tissues, compared to the reference samples (Figure 2). Western blotting of the ACTB/NUCL complex, the antigen for Nb10, showed similar expression trends for both NUCL and ACTB, with particularly lower protein expression in the GBM cytosolic protein fraction (Figure 3, GBMc) versus the reference cytosolic protein fraction (Figure 3, REFc), and increased protein expression in the GBM membrane protein fraction (Figure 3, GBMm) versus the reference membrane protein fraction (Figure 3, REFm). The ACTB/NUCL complex was validated in cytosolic and membrane protein fractions because of the reported existence of two NUCL types in GBM for cytosolic and surface occurrence [33].
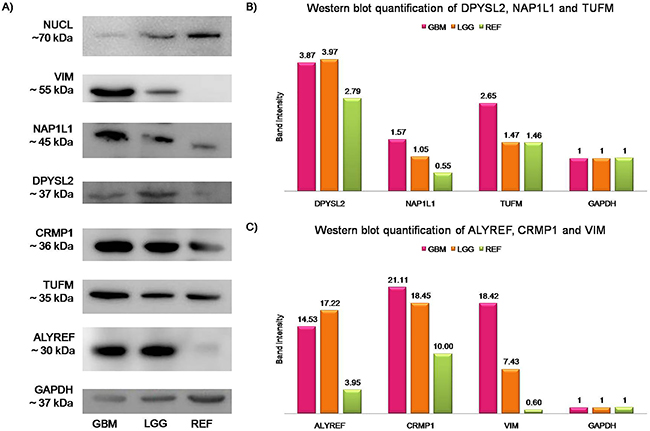
Figure 2: Western blotting validation and quantification of the identified proteins. The band intensity of each protein was plotted after normalization to the GAPDH signal of the same lane, as band intensity . GAPDH was the loading control. (A) Representative Western blotting of the antigens identified. GBM, cytosolic protein extract from GBM tissues; LGG, cytosolic protein extract from lower grade gliomas; REF, cytosolic protein extract from reference samples. (B) Protein expression for DPYSL2, NAP1L1, and TUFM, with 1.38, 2.86, and 1.82 higher expression levels in GBM compared to reference samples, respectively. (C) Protein expression for ALYREF, CRMP1, and VIM, that exhibit up-regulation in GBM with 3.68-, 2.11-, and 30-fold increases compared to the reference samples, respectively.
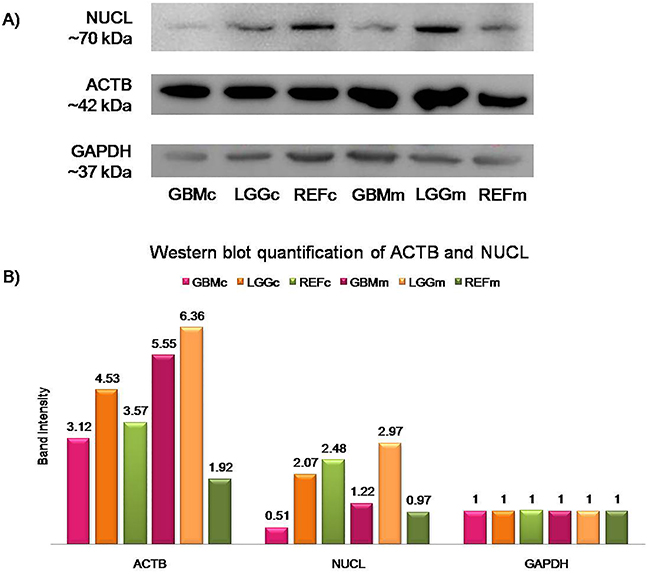
Figure 3: Quantification of NUCL and ACTB. (A) Western blotting for NUCL and ACTB, with GAPDH as the loading control. GBMc, cytosolic protein extract from GBM tissues; LGGc, cytosolic protein extract from lower grade gliomas; REFc, cytosolic protein extract from reference samples; GBMm, membrane protein extract from GBM tissues; LGGm, membrane protein extract from lower grade gliomas; REFm, membrane protein extract from reference samples. (B) Quantification of ACTB and NUCL protein expression in cytosolic and membrane protein fractions of pooled GBM, LGG, and reference samples showed 1.27-fold higher NUCL expression in the membrane protein extract from GBM tissues compared to the membrane protein extract from reference tissues, and almost 3-fold higher ACTB expression in the membrane protein extract from GBM versus the membrane protein extract from reference tissues. Band intensity of each protein was plotted after normalization to GAPDH signal of the same lane.
To determine the specific antigen for Nb314, Western blotting was performed using antigen-free protein samples (Figure 4). The absence of a band in the antigen-free protein sample (Figure 4, GSC) with the appropriate size for DPYSL2 (~37 kDa) demonstrated DPYSL2 as the antigen for Nb314. Bands with the appropriate size for MTHFD1 (~36 kDa) in all of the analyzed protein samples excluded MTHFD1 as the antigen for Nb314.
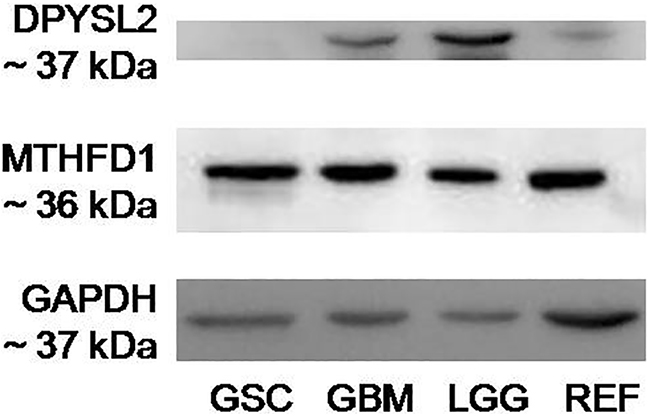
Figure 4: Western blotting for specific binding of Nb314 and its antigen, DPYSL2. Representative Western blotting of the two possible antigens for Nb314, including GAPDH as the loading control. GSC, protein extract from GBM stem-like cell lines after incubation with immobilized Nb314; GBM, cytosolic protein extract from GBM tissues; LGG, cytosolic protein extract from lower (Grade II and III) gliomas; REF, cytosolic protein extract from reference samples. Absence of a band with the appropriate size for DPYSL2 (~37 kDa) in the protein sample that was incubated with immobilized Nb314 prior to performing the analysis shows that Nb314 bound its antigen, which is thus no longer present in the sample, and therefore cannot be detected. Detection of the membrane probed for MTHFD1 showed bands with the appropriate size for this protein (~36 kDa) in all lanes of the analyzed membranes. The particular band in the protein samples incubated with Nb314 prior to Western blotting excludes the possibility for this protein to be an antigen for Nb314.
In-silico analysis and antigen validation for class prediction
The molecular interactions database OncoFinder was used to retrieve and explore the roles of these proteins in cells, and their possible contributions to tumor formation and/or progression [34]. An interaction network was created that showed the functional links among these proteins (Supplementary Figure 1). The pathways in which these proteins are involved are given in Table 1.
Table 1: Interacting proteins and the pathways in which they are involved
Protein 1 |
Protein 2 |
Pathway |
|---|---|---|
CRMP1 |
DPYSL2 |
CRMPs in Sema3A signaling |
ACTB |
ITGB3 |
Platelet activation |
ITGB3 |
NUCL |
Urokinase type plasminogen activator uPA and uPAR mediated signaling |
CRMP1 |
FYN |
CRMPs in Sema3A signaling |
FYN |
ITGAV |
Signaling events mediated by focal adhesion kinase |
ITGAV |
NUCL |
Urokinase type plasminogen activator uPA and uPAR mediated signaling |
FYN |
CTNNB1 |
E-cadherin signaling in keratinocytes |
CTNNB1 |
VIM |
WNT pathway |
NUCL |
NPM1 |
Aurora B signaling |
NPM1 |
H2AFX |
Deposition of new CENPA-containing nucleosomes at the centromere |
H2AFX |
TRIM28 |
ATM |
ALYREF |
SMC1A |
Pre mRNA splicing |
SMC1A |
H2AFX |
Meiotic synapsis |
MTHFD1 |
ATIC |
One carbon pool by folate |
ATIC |
ENTPD1 |
Purine metabolism |
ENTPD1 |
POLR2E |
Purine metabolism |
POLR2E |
ALYREF |
Pre mRNA splicing |
CENPA, centromere protein A
To determine whether the target genes have any value as class predictors, the mRNA expression data of these genes as retrieved from The Cancer Genome Atlas was used to calculate the area-under-the-curve (AUC) values for all of these genes in various ways. For DPYSL2, the “glioma versus reference” samples (Figure 5A) defined an AUC score of 0.985, indicating that it is a promising biomarker. Moreover, NUCL, TRIM28, VIM, and NAP1L1 were indicated as good markers to distinguish glioma tissues from reference tissues (Figure 5A). For “GBM versus lower grade glioma (LGG)” samples, Figure 5B shows nonrandom distribution and enrichment for the CRMP1, NAP1L1, NUCL, ACTB, and VIM gene products with high AUC scores of 0.723, 0.848, 0.852, 0.862, and 0.886, respectively.
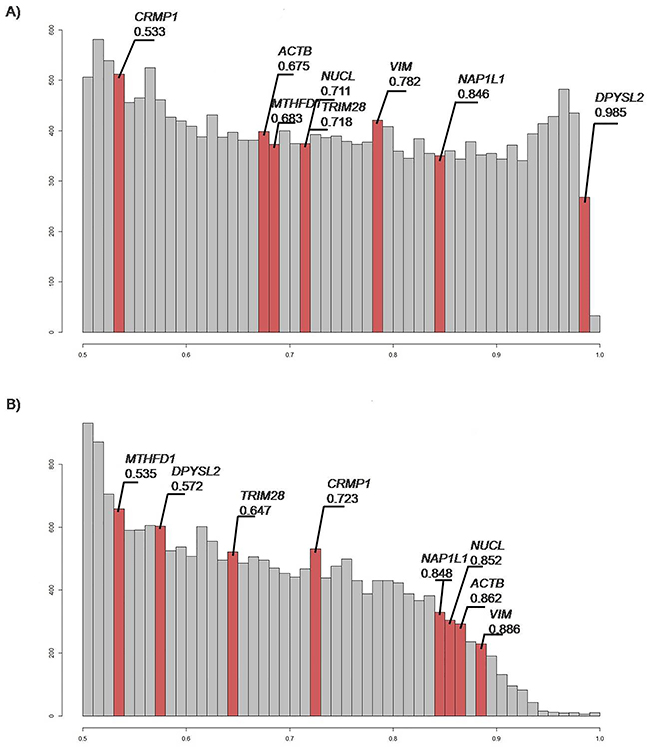
Figure 5: Histograms of area under curve (AUC) values. AUC correlates positively with biomarker quality and varies from 0.5 to 1.0. AUC threshold for discriminating good and bad biomarkers is typically 0.7 or 0.75. Those with greater AUC are considered good-quality biomarkers, and vice-versa [87]. Red bars, genes of interest; gray bars, overall distribution. (A) Gene distribution analyzed, as “glioma versus reference”. This suggests that DPYSL2 is a promising marker, and NUCL, TRIM28, VIM, and NAP1L1 are good markers for distinguishing glioma from reference samples. (B) Gene distribution analyzed as “GBM versus LGG”. CRMP1, NAP1L1, NUCL, ACTB and VIM are indicated as genes that can be used as biomarkers to distinguish between tumor classes.
Statistical analysis of gene expression in glioblastoma, lower grade glioma, and reference samples
Following OncoFinder analysis, we conducted a small scale confirmatory study on 13 GBM and 10 LGG tissue samples from Caucasian patients (Table 2) and compared them to 22 reference non-tumor brain samples. Results from our previous study, TRIM28 and ACTB, were also included in the analysis [35].
Table 2: Clinical data for the glioma patients
Gender |
Age (years) |
Pathological grade |
Anatomic location |
|---|---|---|---|
M |
75 |
IV |
Occipital lobe, right |
F |
53 |
II |
Parietal lobe, right |
M |
44 |
II |
Insular cortex, right |
M |
34 |
IV |
Frontal lobe, right |
F |
48 |
II |
Insular cortex, right |
M |
28 |
II |
Frontal lobe, right |
M |
51 |
II |
Frontal lobe, right |
M |
34 |
III |
Frontal lobe, left |
M |
28 |
III |
Insular cortex, left |
M |
68 |
IV |
Frontal lobe, right |
F |
58 |
IV |
Temporal lobe, left |
F |
72 |
IV |
Frontal lobe, left |
F |
50 |
II |
Frontal lobe, left |
M |
45 |
IV |
Temporal lobe, left |
F |
81 |
IV |
Frontal lobe, left |
M |
48 |
IV |
Temporal lobe, right |
M |
68 |
IV |
Parietal-occipital lobe, left |
M |
25 |
IV |
Temporal lobe, right |
M |
33 |
II |
Temporal lobe, right |
F |
52 |
II |
Frontal lobe, right |
M |
64 |
IV |
Parietal-occipital lobe, right |
M |
59 |
IV |
Parietal lobe, right |
F |
72 |
IV |
Frontal lobe, left |
M |
52 |
IV |
Temporal lobe, left |
M |
35 |
II |
Frontal lobe, right |
M |
41 |
IV |
Temporal lobe, right |
Three genes (TRIM28, DPYSL2 and VIM) were able to successfully distinguish gliomas from reference samples (Figure 6A). In addition, ACTB, TRIM28 and VIM were able to distinguish GBM from LGG (Figure 6B). Results for genes without significant changes in expression either in “glioma versus reference” or in “GBM versus LGG” are presented in Supplementary Figure 2.
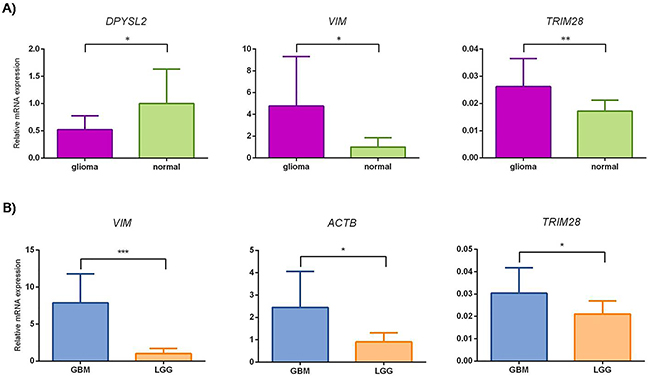
Figure 6: Small scale confirmatory study of relative mRNA expression levels for DPYSL2, VIM, TRIM28 and ACTB. The mean gene expression value corresponds to column height, with error bars representing SD. (A) glioma, gliomas WHO grade II, III and GBM; normal, reference samples. *, P <0.05, **, P <0.01. Mann-Whitney test showed statistically different change in expression for DPYSL2 and VIM with P values 0.0158 and 0.0107, respectively. Student's t-test showed statistically different change in expression for TRIM28 with P value 0.0033. (B) GBM, glioblastoma; LGG, lower grade gliomas. *, P <0.05, ***, P <0.001. Mann-Whitney test for ACTB, TRIM28 and VIM showed statistically different changes in expression with P values 0.0258, 0.0237 and 0.0002, respectively.
Multiple group comparisons showed strong up-regulation for VIM, TRIM28 and ACTB in GBM compared to LGG and reference samples. DPYSL2 showed slightly increased mRNA levels in the reference samples compared to both GBM and LGG groups. The other genes (i.e., NUCL, NAP1L1, TUFM, CRMP1, and ALYREF) did not differ significantly among these sample groups (Supplementary Figure 3).
DISCUSSION
The aim of the present study was to increase our knowledge of the properties that distinguish GBM from LGG and from reference samples. We report a selection of nanobodies that specifically recognize GBM proteins with altered expression. Adapted expression of their target antigens in GBM samples was confirmed at mRNA and protein levels, and possible interactions among these proteins were evaluated, which suggested their involvement in different signaling pathways.
To generate an immune VHH library, initially an adult alpaca was immunized with whole GBM cells to promote the heavy-chain antibody immune response against the abundant and immunogenic GBM-specific molecules. Contrary to antigen-binding fragments of conventional antibodies, the antigen-binding fragments of heavy-chain antibodies are encoded by the single VHH gene fragments that do not get scrambled during cloning [29]. This simplifies their use, because the complete in-vivo matured VHH repertoire is amplified immediately in one amplicon and can be readily cloned. Then, nanobodies that recognize enriched antigens in tumor cells can be retrieved. Seven nanobodies were identified to recognize more antigen in diseased samples than in reference samples. Using a pull-down protocol, each selected nanobody captured its corresponding antigen, which was subsequently identified by mass spectrometry. With the exceptions of DPYSL2, ACTB, TRIM28 and VIM, the genes for the antigens of the other nanobodies showed similar mRNA expression levels between diseased and healthy samples. This suggests that over-representation of the other proteins in GBM samples may have been due to post-translational modifications, epigenetic changes, or slower turn-over.
Prior to starting the bioinformatic analysis, the literature was screened to acquire information about these proteins, as given in Table 3 [36–39]. Bioinformatic analysis of genes of interest against TCGA RNA sequencing data proposed TRIM28, VIM, NAP1L1 and DPYSL2 as promising “glioma versus reference” biomarkers, and suggested CRMP1, NAP1L1, NUCL, ACTB and VIM for discrimination between GBM and LGG. These results were confirmed by analyzing the relative mRNA expression levels of the genes of interest on a small patient cohort. Our confirmatory qPCR study showed differential gene expression levels for DPYSL2, VIM and TRIM28 in “glioma versus reference” samples. Moreover, from the suggested biomarkers to distinguish between GBM and LGG, qPCR confirmed the potential roles of VIM, TRIM28 and ACTB as markers for class differentiation.
Table 3: Antigen roles, as indicated from the literature
Antigen |
Role |
Literature |
|---|---|---|
NUCL |
Abundant in exponentially growing cells Cytoplasmic and surface NUCL in gliomas Surface NUCL is dependent on association with actin cytoskeleton Cell-surface NUCL is a novel angiogenesis marker |
|
VIM |
Role in formation of lamellipodia and invadipodia during cell invasion and migration Typical for infiltrative gliomas with poor prognosis Marker for epithelial to mesenchymal transition Differentially expressed in glioblastomas |
|
NAP1L1 |
Human NAP1L1 locates within the nucleus of dividing cells Co-existence of NAP1L1 and NAP1L4 in neural stem cells and neurons Transcriptional regulation NAP1L1 is overexpressed in small-intestinal carcinoids |
|
TUFM |
Chaperone-like properties in protein folding and renaturation under stress conditions Translational expression of mitochondrial DNA Involved in head and neck squamous cell carcinoma Overexpressed in colorectal carcinoma |
|
DPYSL2 |
Highly expressed during brain development, rarely in post-developmental brain Over-expressed in colorectal carcinoma Altered expression in glioblastomas Promoter of microtubule assembly and neuronal development |
|
CRMP1 |
Decreased in glioblastomas expressing EGFRvIII Invasion suppressor gene in lung cancer Associated with microtubule-containing structures in mitotic cells |
|
ALYREF |
Transport of fully processed mature mRNA out of the nucleus NUP107 complex (consisting of ALYREF, SNRPE, NUP107 and NUP50) is altered in 19% of glioblastomas |
Our network implies that these analyzed proteins are involved in alterations in normal cell metabolism using signaling pathways, including the WNT and ATM pathways, as well as Aurora B, Sema3A, and E-cadherin signaling. Overexpression of Aurora B mRNA and protein has been detected in a number of cancers [40–42], and tumorigenic changes have also been observed for the WNT and ATM pathways [43–45]. This suggests that GBM does not evolve through a single pathway, but through numerous pathways, which will be related to angiogenesis, and cell invasion, signal transduction, and metabolism [46, 47]. This complex nature of GBM thus implies that successful therapies will need to target different biological processes, to parallel these diverse pathways that GBM uses to ensure growth [48].
CONCLUSIONS
With combined use of nanobodies, proteomics and bioinformatics, we identified a set of proteins with altered expression in GBM samples. The analysis of the genes of interest using RNA sequencing data from TCGA suggested the possible use of some of them as biomarkers for glioma class differentiation, with DPYSL2 demonstrated as a glioma-specific biomarker. The use of CRMP1, NAP1L1, NUCL, ACTB, and VIM as potential class predictors also needs to be validated in biological fluids. Our bioinformatic results were verified using a small scale confirmatory study. Validation of our findings on a dataset that contains a greater number of glioma samples and reference samples should now be initiated. Further studying the significance of these biomarkers for glioma formation, progression and prognosis would help development of new treatment strategies, which may be beneficial for GBM patients.
MATERIALS AND METHODS
Ethics statement
The present study was approved by the National Medical Ethics Committee of the Republic of Slovenia (Numbers: 92/06/12, 89/04/13 and 95/09/15). Written informed consent was signed by the patients prior to their surgery. Reference samples were obtained during autopsies, following the legal regulations valid for the Republic of Slovenia. All of the samples used in this study are anonymous.
Preparation of protein extracts
Protein extracts used for phage particle enrichment and during panning were obtained from two commercially available GBM stem-like cell lines: NCH644 and NCH421K cells (Cell Line Service, Eppelheim, Germany) [49, 50]. Cells were grown in complete medium, as neurobasal medium (Gibco) plus 20 μM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, 2% B-27 supplement, 20 ng/mL basic fibroblast growth factor, 20 ng/mL epidermal growth factor, and 1 U/mL heparin.
Brain tissues originated from the hippocampus and subventricular and periventricular zones of 10 patients, were dissected out during autopsies and used as reference samples. Tissue samples were sealed in containers, labeled, snap frozen, and kept at -80 °C until they were used for protein extraction.
Glioblastoma samples from 13 patients (nine male, four female; aged 25-81 years), and lower grade glioma samples (WHO grade II and III) from 10 patients (six male, four female; aged 28-53) were dissected out during surgery, sealed in sterile containers, labeled, snap frozen, and kept at -80 °C until they were used for protein extraction. Data is presented in Table 2.
Two commercially available kits were used to separate the membrane and cytosolic protein fractions from all of the samples: ProteoExtract Transmembrane Protein Extraction kit (Novagen), and ProteoExtract Native Membrane Protein Extraction kit (Calbiochem). Protein concentrations were determined using the Bradford method [51].
Bio-selection and ELISA screening
An existing nanobody library was used in the present study. An adult alpaca was immunized with whole GBM cells, as indicated above and described previously [35]. The nanobody library was constructed by the Nanobody Service Facility (Vrije Universiteit Brussel, Brussels, Belgium), according to their established protocols [52–54].
Phage enrichment (i.e., bio-selection) was performed on solid-phase coated cytosolic proteins isolated from NCH644 and NCH421K GBM stem-like cell lines. Protein for coating of the GBM and reference samples was at 200 μg/mL (100 μL/well). Three rounds of phage enrichment were performed, following a published protocol [53]. Briefly, phages were incubated with the protein samples coated on plastic microtiter plates, extensively washed and target-specific phages eluted with triethylamine. Eluted phages were used to infect fresh Escherichia coli cells for overnight proliferation in 2× Yeast Extract and Tryptone with 100 μg/mL ampicillin and 70 μg/mL kanamycin. The following morning, phage particles were precipitated with PEG-6000/ 2.5 M NaCl and used in the next round of panning. Periplasmic extract ELISA was used to screen for clones expressing nanobodies with specificity for GBM proteins [53], with protein for ELISA at 2 μg/mL (100 μL/well) for GBM and reference samples. Briefly, individual clones were inoculated in 1 mL Terrific Broth medium and the nanobody expression was induced by adding 10 μL 1 M isopropyl β-D-1-thiogalactopyranoside. The next day, periplasmic extracts were obtained by incubating the E. coli initially in a hypertonic Tris/ EDTA/ sucrose solution, and then in a hypotonic solution, following the addition of distilled H2O. These periplasmic extracts were applied in parallel to wells that were coated overnight with proteins from GBM and reference samples. To detect the antigen-specific nanobodies, 100 μL/well primary and secondary antibodies, mouse anti-hemagglutinin, and goat-anti-mouse IgG (whole molecule)–alkaline phosphatase conjugate (both Sigma Aldrich), respectively, were used. The ELISA signals were measured at 405 nm after applying 100 μL/well alkaline phosphatase conjugate (Sigma Aldrich). The nanobody genes for the ELISA positive clones were amplified using colony PCR (6 min at 95 °C, 45 s at 94 °C, 45 s at 55 °C, 45 s at 72 °C, 10 min at 72 °C) with the primers RP (5'→3' TCA CAC AGG AAA CAG CTA TGA C) and GIII (5'→3' CCA CAG ACA GCC CTC ATA G), and were sequenced at Macrogen (Amsterdam, The Netherlands). Those that had appropriate sequences were chosen for large-scale expression.
Recloning and nanobody production
The nanobody genes were recloned in the pHEN6c vector and transformed into E. coli WK6 cells prior to large-scale production. The nanobodies were produced as described previously [55, 56]. Briefly, clones were grown overnight in Luria–Bertani–Miller medium. These cultures were used to inoculate 1.5 L Terrific Broth medium for each clone. Nanobody production was induced by adding 1.5 mL 1 M isopropyl β-D-1-thiogalactopyranoside, and the cultures were further shaken overnight. The periplasmic extracts were incubated with Ni+-nitrilotriacetic acid agarose (Qiagen), overnight at 4 °C. Immobilized nanobodies were washed with three column volumes phosphate-buffered saline (PBS) and then eluted with one column volume freshly prepared PBS supplemented with 0.5 M imidazole. Nanobodies were further purified by size-exclusion chromatography.
Antigen identification
Protein lysates from NCH644 and NCH421K cells were freshly prepared as follows: ~60 ×106 GBM cells were centrifuged at 125x g for 5 min at room temperature, supernatant was removed, and the cells were resuspended in lysis buffer (PBS supplemented with 0.1% NP-40). Cells were sonicated for 1 min (30 ms ON, 30 ms OFF). The resulting suspension was centrifuged at 290x g for 5 min at 4 °C, and the resulting supernatant was used for the antigen capture (Figure 7). Nanobodies were immobilized on Ni-(tris(carboxymethyl)ethylene diamine) resin (Machery-Nagel), and then incubated on a rotating platform for 1 h at 4 °C with fresh protein lysate. Supernatant was then removed, and the samples were washed three times with equilibration buffer (50 mM NaH2PO4•2H2O, 300 mM NaCl) and transferred to new microcentrifuge tubes. Samples were then washed five times with PBS, and the protein was digested with 10 μL 5 ng/μL trypsin solution (0.5 μg porcine trypsin in 100 μL PBS), overnight at room temperature. The next day, supernatant was removed, samples were washed twice with PBS, and the antigens were separated from the nanobodies with 150 μL 6 M urea. Urea elutions and overnight digests were cleaned up using stop-and-go-extraction tips (Stage Tips) [57], and analyzed using liquid chromatography–tandem mass spectrometry with the Easy-nLC system connected to an electron-transfer dissociation ion trap (Amazon; Bruker). The chromatogaphy was developed using a 75-min discontinuous gradient from 0% to 80% methanol in 0.1% formic acid. The tandem mass spectrometry spectra were searched against the human database using the X!tandem and MASCOT search engines, allowing a 5% false-recovery rate.
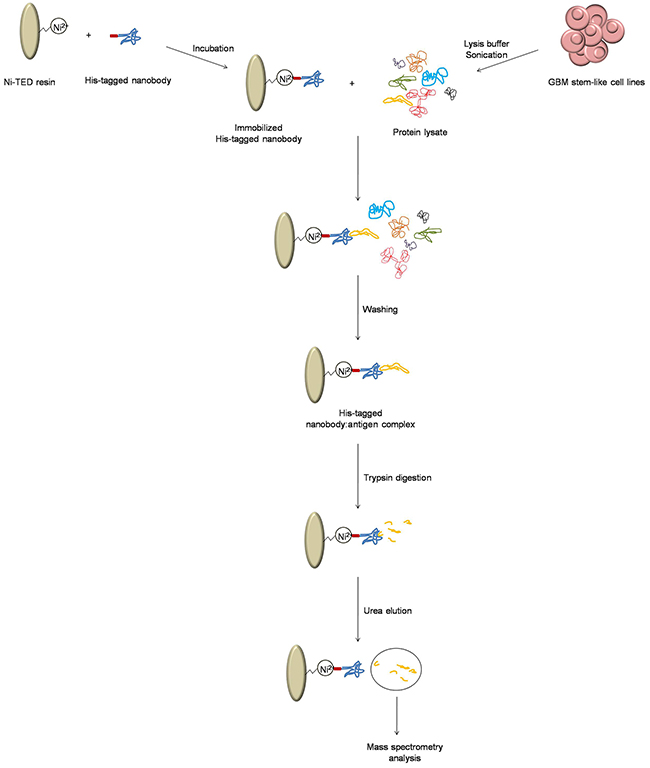
Figure 7: Schematic representation of the sample preparation for mass spectrometry. The process to obtain nanobody: antigen pairs, and antigen preparation for mass spectrometry analysis.
Western blotting
The vast majority of nanobodies bind to conformational epitopes, which are lost in denatured proteins [58]. This complicates the use of nanobodies as detection probes in Western blotting. Therefore, standard monoclonal antibodies (Sigma Aldrich) were used in the Western blots to confirm the differential occurrence of the target proteins. Protein extracts from glioma samples were divided into two groups: GBM, as 13 pooled tissue samples from patients with WHO Grade IV gliomas; and LGG, as 10 pooled tissue samples from patients with WHO Grade II and Grade III astrocytomas. Pooled protein extracts from 22 post-mortem brain samples were used as the reference samples. Here, ~20 μg to 30 μg pooled protein extracts were separated using 4% to 12% NuPAGE Bis-Tris Mini Gels (Invitrogen), and transferred to an Immobilion-P Transfer Membrane (Milipore). BlueStar Prestained Protein Ladder (Nippon Genetics) was used as the protein marker. Residual protein binding sites on the membranes were blocked with 5% PBS–milk for 1 h at room temperature. A mouse monoclonal anti-GAPDH antibody (Sigma Aldrich) was used for the loading control. The incubations with the primary antibodies were at 4 °C overnight, with shaking at 60 rpm. The incubations with the secondary anti-mouse IgG (whole molecule)–peroxidase antibody produced in goat (Sigma Aldrich) were for 1 h at 4 °C, with shaking at 60 rpm. The bands were revealed with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific), visualized with Fujifilm LAS-4000 (Tokyo), and analyzed with the Multi Gauge version 3.2 software. The numerical values of the antigen intensities were calculated as the ratio between the arbitrary units (AU) of the antigen and the loading control, as GAPDH .
To determine the specific antigen for Nb314 (Figure 8), this nanobody was immobilized on Ni-(tris(carboxymethyl)ethylene diamine) resin (Machery-Nagel) and incubated with fresh protein lysate (for preparation see “Antigen identification”) on a rotating platform for 1 h at 4 °C. After this incubation, the resin was allowed to settle and the supernatant (i.e., antigen-free protein lysate) was transferred to a new microcentrifuge tube. Total protein concentration was determined using the Bradford method. Here, ~20 μg to 30 μg protein extracts (antigen-free protein lysate, protein lysates from GBM, LGG and reference samples) were used for Western blotting. One membrane was probed with mouse monoclonal anti-DPYSL2 monoclonal antibody (Sigma Aldrich), and another with the MTHFD2 monoclonal antibody (M01), clone 4G7-2G3 (Abnova). Probing with primary and secondary antibodies, band detection and quantification were performed as described above.
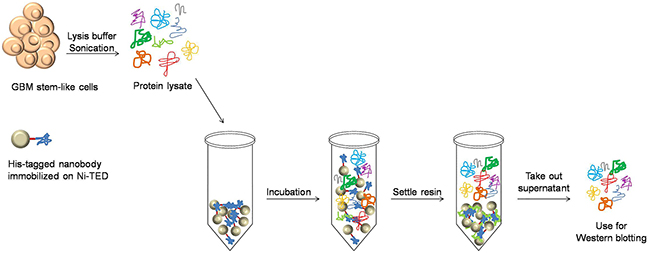
Figure 8: Protein sample preparation for determination of the Nb314 antigen. Preparation of antigen-free protein lysate used to determine the matching antigen for Nb314.
Bioinformatics
Possible interactions between the identified proteins were analyzed using the OncoFinder software [59] and the Reactome [60], Kyoto Encyclopedia of Genes and Genomes [61], and HumanCyc [(www.humancyc.org)] molecular interactions databases. The results from our previous study for TRIM28 and ACTB [35] were also included in the present analysis. Furthermore, the genes of interest were analyzed against TCGA RNA sequencing dataset which consists of 171 GBM, 530 LGG and five reference samples. This contains information about 19801 genes, including the genes of interest in the present study (i.e., CRMP1, DPYSL2, NAP1L1, NUCL, TRIM28, VIM, and ACTB), with the sole exception of ALYREF. The genes were checked for specific overexpression or underexpression among the data available from TCGA, using “cancer-to-normal” ratios calculated with OncoFinder. The AUC values for all of these genes were calculated as “GBM versus LGG”, “GBM versus LGG versus reference” and “glioma versus reference”. The RNA sequencing data were normalized using the DeSeq2 package, and the Firehose software was used to retrieve data.
Quantitative reverse transcription polymerase chain reaction
qPCR was used to determine changes in mRNA expression in the samples. mRNA was extracted from 13 GBM, 10 LGG and 22 paired reference samples. Extractions were performed with TRI Reagent (Sigma Aldrich), according to the manufacturer instructions. mRNA concentrations were estimated using a NanoDrop ND-1000 (NanoDrop Technologies, USA), and mRNA purities were determined using A260/A280 and A260/A230 ratios. The RNA integrity numbers (RINs) were determined using a bioanalyzer (Agilent 2100; Agilent Technologies, USA). Twelve samples with insufficient RNA quality (two GBM, one LGG, nine reference samples, with RINs from 2.2-2.9) were excluded from further analysis. Then, 2 μg of each mRNA sample was used for cDNA reverse transcription. The samples were treated with recombinant RNase-free DNase I (Roche) to remove possible DNA traces (15 min 30 °C, 10 min 75 °C) and transcribed with Transcriptor Universal cDNA Master (Roche), as follows: 5 min at 25 °C, 10 min at 55 °C, and 5 min at 85 °C.
The qPCR was performed using a Roche LightCycler 480 platform. The total reaction volume was 5 μL, and consisted of: 0.75 μL cDNA, 2.5 μL 2× LightCycler 480 SYBR Green I Master (Roche), 0.3 μL of each of the 2.5 mM primers, and 1.15 μL distilled H2O. The reactions were performed in triplicate using the following thermal cycling: pre-incubation 10 s at 95 °C; cycling, 20 s at 60 °C, 20 s at 72 °C, for 45 cycles; melting curve, 5 s at 95 °C, 1 min at 65 °C; continuous at 97 °C; and cooling, 30 s at 4 °C. From the five candidate reference genes (i.e. TBP, HPRT1, RPL13A, GAPDH, CYC1) chosen from the literature [62–64], RPL13A and CYC1 were selected for gene normalization using the NormFinder algorithm [65]. The primers for the reference genes (Table 4) were chosen from the literature [64], while the primers for the genes of interest (Table 5) were obtained from the PrimerBank PCR primer database for quantitative gene expression analysis (https://pga.mgh.harvard.edu/primerbank/) [66]. Relative quantification was performed as previously described [67]. For samples following Gaussian distribution differential expression analyzed as “GBM versus LGG versus reference” was calculated using one-way ANOVA and Holm-Sidak's corrections for multiple comparisons. For samples not following Gaussian distribution differential expression was calculated using Kruskal-Wallis test. In both cases P ≤0.05 was considered statistically significant (*, P ≤0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001).
Table 4: Primers for candidate reference genes
Gene |
Primer sequence (5'→3') |
Amplicon |
Gene name |
Function |
|---|---|---|---|---|
TBP |
F: CAG CAT CAC TGT TTC TTG GCG T |
232 |
TATA-box binding protein |
General transcription factor |
HPRT1 |
F: CAG CCC TGG CGT CGT GAT TAG T |
226 |
Hypoxanthine-guanine phosphoribosyltransferase |
Metabolic salvage of purines |
RPL13A |
F: CCT GGA GGA GAA GAG GAA AGA GA |
126 |
60s ribosomal protein L13a |
Component of 60S |
GAPDH |
F: TCG CCA GCC GAG CCA CAT C |
222 |
Glyceraldehyde-3-phosphate |
Glycolysis enzyme |
CYC1 |
F: GAG GTG GAG GTT CAA GAC GG |
160 |
Cytochrome C-1 |
Mitochondrial electron transport |
F, forward; R, reverse
Table 5: Primers for the genes of interest
Gene |
Primer sequence (5'®3') |
Amplicon length (bp) |
|---|---|---|
NUCL |
F: GAA CCG ACT ACG GCT TTC AAT |
93 |
ACTB |
F: CCA ACC GCG AGA AGA TGA |
97 |
VIM |
F: TGC CGT TGA AGC TGC TAA CTA |
248 |
NAP1L1 |
F: AAA GCA CGT CAG CTA ACT GTT |
146 |
TUFM |
F: AAA GAA GGG AGA CGA GTG TGA |
80 |
DPYSL2 |
F: GTG ACT ACT CTC TGC ATG TGG A |
87 |
CRMP1 |
F: AGT GAC CGA CTC CTC ATC AAA |
119 |
ALYREF |
F: ACA TTC AGC TTG TCA CGT CAC |
77 |
TRIM28 |
F: TGA GAC CTG TGT AGA GGC G |
93 |
F, forward; R, reverse
Statistical analysis
To confirm the bioinformatic findings, relative mRNA expression levels of the genes of interest were analyzed as “glioma versus reference” and “GBM versus LGG” in GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA). For samples following Gaussian distribution analysis was performed using unpaired two-tailed Student's t-test. Mann-Whitney test was used where samples did not follow Gaussian distribution. In both cases P ≤0.05 was considered statistically significant (*, P ≤0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001).
ACKNOWLEDGMENTS AND FUNDING
The authors would like to thank the Nanobody Service Facility (Vrije Universiteit Brussel, Brussels, Belgium) for the library construction, and Helena Klavžar (Centre for Functional Genomics and Bio-Chips, Faculty of Medicine, University of Ljubljana, Ljubljana, Slovenia) for technical support. This study was supported by the P1-0104 and P1-0390 Research Programme Grants from the Slovenian Scientific Agency (ARRS) to RK. SM acknowledges Strategic Research Programme financing from the Vrije Universiteit Brussel. IP and AB were supported by a EuroTransBio 'Omicsglioma' grant from the Foundation for Assistance to Small Innovative Enterprises, Russia. The funders had no roles in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest to disclose.
REFERENCES
1. Veliz I, Loo Y, Castillo O, Karachaliou N, Nigro O, Rosell R. Advances and challenges in the molecular biology and treatment of glioblastoma-is there any hope for the future? Ann Transl Med. 2015; 3: 7. doi: 10.3978/j.issn.2305-5839.2014.10.06.
2. Anderson MD, Hamza MA, Hess KR, Puduvalli VK. Implications of bevacizumab discontinuation in adults with recurrent glioblastoma. Neuro Oncol. 2014; 16:823-8. doi: 10.1093/neuonc/nou021.
3. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370: 699-708. doi: 10.1056/NEJMoa1308573.
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005; 352: 987-96. doi: 10.1056/NEJMoa043330.
5. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015; 129: 829-48. doi: 10.1007/s00401-015-1432-1.
6. Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013; 310: 1842-50. doi: 10.1001/jama.2013.280319.
7. Seymour T, Nowak A, Kakulas F. Targeting Aggressive Cancer Stem Cells in Glioblastoma. Front Oncol. 2015; 5: 159. doi: 10.3389/fonc.2015.00159.
8. van Woensel M, Wauthoz N, Rosiere R, Amighi K, Mathieu V, Lefranc F, van Gool SW, de Vleeschouwer S. Formulations for Intranasal Delivery of Pharmacological Agents to Combat Brain Disease: A New Opportunity to Tackle GBM? Cancers (Basel). 2013; 5: 1020-48. doi: 10.3390/cancers5031020.
9. Agrawal NS, Miller R Jr, Lal R, Mahanti H, Dixon-Mah YN, DeCandio ML, Vandergrift WA 3rd, Varma AK, Patel SJ, Banik NL, Lindhorst SM, Giglio P, Das A. Current Studies of Immunotherapy on Glioblastoma. J Neurol Neurosurg. 2014; 1.
10. Gruber ML, Buster WP. Temozolomide in combination with irinotecan for treatment of recurrent malignant glioma. Am J Clin Oncol. 2004; 27: 33-8.
11. Bloch O, Lim M, Sughrue ME, Komotar RJ, Abrahams JM, O’Rourke DM, D’Ambrosio A, Bruce JN, Parsa AT. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin Cancer Res. 2017 Feb 13. doi: 10.1158/1078-0432.CCR-16-1369. [Epub ahead of print].
12. Ma C, Zhou W, Yan Z, Qu M, Bu X. beta-Elemene treatment of glioblastoma: a single-center retrospective study. Onco Targets Ther. 2016; 9: 7521-6. doi: 10.2147/OTT.S120854.
13. da Fonseca CO, Simao M, Lins IR, Caetano RO, Futuro D, Quirico-Santos T. Efficacy of monoterpene perillyl alcohol upon survival rate of patients with recurrent glioblastoma. J Cancer Res Clin Oncol. 2011; 137: 287-93. doi: 10.1007/s00432-010-0873-0.
14. Hernandez-Pedro NY, Rangel-Lopez E, Vargas Felix G, Pineda B, Sotelo J. An update in the use of antibodies to treat glioblastoma multiforme. Autoimmune Dis. 2013; 2013: 716813. doi: 10.1155/2013/716813.
15. Kaufmann JK, Chiocca EA. Glioma virus therapies between bench and bedside. Neuro Oncol. 2014; 16: 334-51. doi: 10.1093/neuonc/not310.
16. Lesterhuis WJ, Haanen JB, Punt CJ. Cancer immunotherapy--revisited. Nat Rev Drug Discov. 2011; 10: 591-600. doi: 10.1038/nrd3500.
17. Hatanpaa KJ, Burma S, Zhao D, Habib AA. Epidermal growth factor receptor in glioma: signal transduction, neuropathology, imaging, and radioresistance. Neoplasia. 2010; 12: 675-84.
18. Gan HK, Kaye AH, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009; 16: 748-54. doi: 10.1016/j.jocn.2008.12.005.
19. Li L, Chakraborty S, Yang CR, Hatanpaa KJ, Cipher DJ, Puliyappadamba VT, Rehman A, Jiwani AJ, Mickey B, Madden C, Raisanen J, Burma S, Saha D, et al. An EGFR wild type-EGFRvIII-HB-EGF feed-forward loop regulates the activation of EGFRvIII. Oncogene. 2014; 33: 4253-64. doi: 10.1038/onc.2013.400.
20. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006; 66: 9339-44. doi: 10.1158/0008-5472.CAN-06-3126.
21. Kondo Y, Katsushima K, Ohka F, Natsume A, Shinjo K. Epigenetic dysregulation in glioma. Cancer Sci. 2014; 105: 363-9.
22. Tamura K, Aoyagi M, Wakimoto H, Ando N, Nariai T, Yamamoto M, Ohno K. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. J Neurosurg. 2010; 113: 310-8. doi: 10.3171/2010.2.JNS091607.
23. Moren L, Bergenheim AT, Ghasimi S, Brannstrom T, Johansson M, Antti H. Metabolomic Screening of Tumor Tissue and Serum in Glioma Patients Reveals Diagnostic and Prognostic Information. Metabolites. 2015; 5: 502-20. doi: 10.3390/metabo5030502.
24. Shen F, Zhang Y, Yao Y, Hua W, Zhang HS, Wu JS, Zhong P, Zhou LF. Proteomic analysis of cerebrospinal fluid: toward the identification of biomarkers for gliomas. Neurosurg Rev. 2014; 37: 367-80. doi: 10.1007/s10143-014-0539-5.
25. Syed P, Gupta S, Choudhary S, Pandala NG, Atak A, Richharia A, Kp M, Zhu H, Epari S, Noronha SB, Moiyadi A, Srivastava S. Autoantibody Profiling of Glioma Serum Samples to Identify Biomarkers Using Human Proteome Arrays. Sci Rep. 2015; 5: 13895. doi: 10.1038/srep13895.
26. Gao X, Mi Y, Ma Y, Jin W. LEF1 regulates glioblastoma cell proliferation, migration, invasion, and cancer stem-like cell self-renewal. Tumour Biol. 2014; 35: 11505-11. doi: 10.1007/s13277-014-2466-z.
27. Kijanka M, Dorresteijn B, Oliveira S, van Bergen en Henegouwen PM. Nanobody-based cancer therapy of solid tumors. Nanomedicine (Lond). 2015; 10: 161-74. doi: 10.2217/nnm.14.178.
28. Muyldermans S. Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013; 82: 775-97. doi: 10.1146/annurev-biochem-063011-092449.
29. Muyldermans S. Single domain camel antibodies: current status. J Biotechnol. 2001; 74: 277-302.
30. Azzazy HM, Highsmith WE Jr. Phage display technology: clinical applications and recent innovations. Clin Biochem. 2002; 35: 425-45.
31. De Groeve K, Deschacht N, De Koninck C, Caveliers V, Lahoutte T, Devoogdt N, Muyldermans S, De Baetselier P, Raes G. Nanobodies as tools for in vivo imaging of specific immune cell types. J Nucl Med. 2010; 51: 782-9. doi: 10.2967/jnumed.109.070078.
32. Muyldermans S, Cambillau C, Wyns L. Recognition of antigens by single-domain antibody fragments: the superfluous luxury of paired domains. Trends Biochem Sci. 2001; 26: 230-5.
33. Hovanessian AG, Soundaramourty C, El Khoury D, Nondier I, Svab J, Krust B. Surface expressed nucleolin is constantly induced in tumor cells to mediate calcium-dependent ligand internalization. PLoS One. 2010; 5: e15787. doi: 10.1371/journal.pone.0015787.
34. Borisov NM, Terekhanova NV, Aliper AM, Venkova LS, Smirnov PY, Roumiantsev S, Korzinkin MB, Zhavoronkov AA, Buzdin AA. Signaling pathways activation profiles make better markers of cancer than expression of individual genes. Oncotarget. 2014; 5: 10198-205. doi: 10.18632/oncotarget.2548.
35. Jovcevska I, Zupanec N, Kocevar N, Cesselli D, Podergajs N, Stokin CL, Myers MP, Muyldermans S, Ghassabeh GH, Motaln H, Ruaro ME, Bourkoula E, Turnsek TL, et al. TRIM28 and beta-Actin Identified via Nanobody-Based Reverse Proteomics Approach as Possible Human Glioblastoma Biomarkers. PLoS One. 2014; 9: e113688. doi: 10.1371/journal.pone.0113688.
36. Berger CM, Gaume X, Bouvet P. The roles of nucleolin subcellular localization in cancer. Biochimie. 2015; 113: 78-85. doi: 10.1016/j.biochi.2015.03.023.
37. Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010; 5: e8918. doi: 10.1371/journal.pone.0008918.
38. Tan F, Thiele CJ, Li Z. Collapsin response mediator proteins: Potential diagnostic and prognostic biomarkers in cancers (Review). Oncol Lett. 2014; 7: 1333-40. doi: 10.3892/ol.2014.1909.
39. Trog D, Yeghiazaryan K, Schild HH, Golubnitschaja O. Up-regulation of vimentin expression in low-density malignant glioma cells as immediate and late effects under irradiation and temozolomide treatment. Amino Acids. 2008; 34: 539-45. doi: 10.1007/s00726-007-0007-4.
40. Tsuno T, Natsume A, Katsumata S, Mizuno M, Fujita M, Osawa H, Nakahara N, Wakabayashi T, Satoh Y, Inagaki M, Yoshida J. Inhibition of Aurora-B function increases formation of multinucleated cells in p53 gene deficient cells and enhances anti-tumor effect of temozolomide in human glioma cells. J Neurooncol. 2007; 83: 249-58. doi: 10.1007/s11060-007-9335-1.
41. Borges KS, Castro-Gamero AM, Moreno DA, da Silva Silveira V, Brassesco MS, de Paula Queiroz RG, de Oliveira HF, Carlotti CG Jr, Scrideli CA, Tone LG. Inhibition of Aurora kinases enhances chemosensitivity to temozolomide and causes radiosensitization in glioblastoma cells. J Cancer Res Clin Oncol. 2012; 138: 405-14. doi: 10.1007/s00432-011-1111-0.
42. Zeng WF, Navaratne K, Prayson RA, Weil RJ. Aurora B expression correlates with aggressive behaviour in glioblastoma multiforme. J Clin Pathol. 2007; 60: 218-21. doi: 10.1136/jcp.2006.036806.
43. Blake SM, Stricker SH, Halavach H, Poetsch AR, Cresswell G, Kelly G, Kanu N, Marino S, Luscombe NM, Pollard SM, Behrens A. Inactivation of the ATMIN/ATM pathway protects against glioblastoma formation. Elife. 2016; 5. doi: 10.7554/eLife.08711.
44. Kierulf-Vieira KS, Sandberg CJ, Grieg Z, Gunther CC, Langmoen IA, Vik-Mo EO. Wnt inhibition is dysregulated in gliomas and its re-establishment inhibits proliferation and tumor sphere formation. Exp Cell Res. 2016; 340: 53-61. doi: 10.1016/j.yexcr.2015.12.010.
45. Lee Y, Lee JK, Ahn SH, Lee J, Nam DH. WNT signaling in glioblastoma and therapeutic opportunities. Lab Invest. 2016; 96: 137-50. doi: 10.1038/labinvest.2015.140.
46. Bastien JI, McNeill KA, Fine HA. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer. 2015; 121: 502-16. doi: 10.1002/cncr.28968.
47. Milinkovic V, Bankovic J, Rakic M, Stankovic T, Skender-Gazibara M, Ruzdijic S, Tanic N. Identification of novel genetic alterations in samples of malignant glioma patients. PLoS One. 2013; 8: e82108. doi: 10.1371/journal.pone.0082108.
48. Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Emerging targets for glioblastoma stem cell therapy. Journal of Biomedical Research. 2016; 30: 19-31. doi: 10.7555/jbr.30.20150100.
49. Campos B, Zeng L, Daotrong PH, Eckstein V, Unterberg A, Mairbaurl H, Herold-Mende C. Expression and regulation of AC133 and CD133 in glioblastoma. Glia. 2011; 59: 1974-86. doi: 10.1002/glia.21239.
50. Podergajs N, Brekka N, Radlwimmer B, Herold-Mende C, Talasila KM, Tiemann K, Rajcevic U, Lah TT, Bjerkvig R, Miletic H. Expansive growth of two glioblastoma stem-like cell lines is mediated by bFGF and not by EGF. Radiol Oncol. 2013; 47: 330-7. doi: 10.2478/raon-2013-0063.
51. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72: 248-54.
52. Bourkoula E, Mangoni D, Ius T, Pucer A, Isola M, Musiello D, Marzinotto S, Toffoletto B, Sorrentino M, Palma A, Caponnetto F, Gregoraci G, Vindigni M, et al. Glioma-associated stem cells: A novel class of tumor-supporting cells able to predict prognosis of human low-grade gliomas. Stem Cells. 2013; 32:1239-53. doi: 10.1002/stem.1605.
53. GhassabehG, Saerens D, Muyldermans S. Isolation of Antigen-Specific Nanobodies. In: Kontermann R and Dübel S, editors. Antibody Engineering. Heidelberg: Springer Berlin Heidelberg; 2010. pp. 251-66.
54. Pardon E, Laeremans T, Triest S, Rasmussen SG, Wohlkonig A, Ruf A, Muyldermans S, Hol WG, Kobilka BK, Steyaert J. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc. 2014; 9: 674-93. doi: 10.1038/nprot.2014.039.
55. Chames P, editor. Antibody Engineering, Methods and Protocols, 2nd edition. Marseille, France: Humana Press; 2012. pp 734.
56. Vincke C, Gutierrez C, Wernery U, Devoogdt N, Hassanzadeh-Ghassabeh G, Muyldermans S. Generation of single domain antibody fragments derived from camelids and generation of manifold constructs. Methods Mol Biol. 2012; 907: 145-76. doi: 10.1007/978-1-61779-974-7_8.
57. Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007; 2: 1896-906. doi: 10.1038/nprot.2007.261.
58. Oliveira S, Heukers R, Sornkom J, Kok RJ, van Bergen En Henegouwen PM. Targeting tumors with nanobodies for cancer imaging and therapy. J Control Release. 2013; 172: 607-17. doi: 10.1016/j.jconrel.2013.08.298.
59. Buzdin AA, Zhavoronkov AA, Korzinkin MB, Venkova LS, Zenin AA, Smirnov PY, Borisov NM. Oncofinder, a new method for the analysis of intracellular signaling pathway activation using transcriptomic data. Front Genet. 2014; 5: 55. doi: 10.3389/fgene.2014.00055.
60. Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, Caudy M, Garapati P, Gillespie M, Kamdar MR, Jassal B, Jupe S, Matthews L, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014; 42: D472-7. doi: 10.1093/nar/gkt1102.
61. Nakaya A, Katayama T, Itoh M, Hiranuka K, Kawashima S, Moriya Y, Okuda S, Tanaka M, Tokimatsu T, Yamanishi Y, Yoshizawa AC, Kanehisa M, Goto S. KEGG OC: a large-scale automatic construction of taxonomy-based ortholog clusters. Nucleic Acids Res. 2013; 41: D353-7. doi: 10.1093/nar/gks1239.
62. Valente V, Teixeira SA, Neder L, Okamoto OK, Oba-Shinjo SM, Marie SK, Scrideli CA, Paco-Larson ML, Carlotti CG Jr. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC Mol Biol. 2009; 10: 17. doi: 10.1186/1471-2199-10-17.
63. Wang Q, Ishikawa T, Michiue T, Zhu BL, Guan DW, Maeda H. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int J Legal Med. 2012; 126: 943-52. doi: 10.1007/s00414-012-0774-7.
64. Grube S, Gottig T, Freitag D, Ewald C, Kalff R, Walter J. Selection of suitable reference genes for expression analysis in human glioma using RT-qPCR. J Neurooncol. 2015; 123: 35-42. doi: 10.1007/s11060-015-1772-7.
65. Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004; 64: 5245-50. doi: 10.1158/0008-5472.CAN-04-0496.
66. Wang X, Spandidos A, Wang H, Seed B. PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2012; 40: D1144-9. doi: 10.1093/nar/gkr1013.
67. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3: RESEARCH0034.
68. Christian S, Pilch J, Akerman ME, Porkka K, Laakkonen P, Ruoslahti E. Nucleolin expressed at the cell surface is a marker of endothelial cells in angiogenic blood vessels. J Cell Biol. 2003; 163: 871-8. doi: 10.1083/jcb.200304132.
69. Koutsioumpa M, Papadimitriou E. Cell surface nucleolin as a target for anti-cancer therapies. Recent Pat Anticancer Drug Discov. 2014; 9: 137-52.
70. Watanabe T, Hirano K, Takahashi A, Yamaguchi K, Beppu M, Fujiki H, Suganuma M. Nucleolin on the cell surface as a new molecular target for gastric cancer treatment. Biol Pharm Bull. 2010; 33: 796-803.
71. Collet B, Guitton N, Saikali S, Avril T, Pineau C, Hamlat A, Mosser J, Quillien V. Differential analysis of glioblastoma multiforme proteome by a 2D-DIGE approach. Proteome Sci. 2011; 9: 16. doi: 10.1186/1477-5956-9-16.
72. Kidd ME, Shumaker DK, Ridge KM. The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol. 2014; 50: 1-6. doi: 10.1165/rcmb.2013-0314TR.
73. Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010; 24: 1838-51. doi: 10.1096/fj.09-151639.
74. Attia M, Forster A, Rachez C, Freemont P, Avner P, Rogner UC. Interaction between nucleosome assembly protein 1-like family members. J Mol Biol. 2011; 407: 647-60. doi: 10.1016/j.jmb.2011.02.016.
75. Mello JA, Almouzni G. The ins and outs of nucleosome assembly. Curr Opin Genet Dev. 2001; 11: 136-41.
76. Mosammaparast N, Ewart CS, Pemberton LF. A role for nucleosome assembly protein 1 in the nuclear transport of histones H2A and H2B. EMBO J. 2002; 21: 6527-38.
77. Schimmack S, Taylor A, Lawrence B, Alaimo D, Schmitz-Winnenthal H, Buchler MW, Modlin IM, Kidd M. A mechanistic role for the chromatin modulator, NAP1L1, in pancreatic neuroendocrine neoplasm proliferation and metastases. Epigenetics Chromatin. 2014; 7: 15. doi: 10.1186/1756-8935-7-15.
78. Kidd M, Modlin IM, Mane SM, Camp RL, Eick G, Latich I. The role of genetic markers--NAP1L1, MAGE-D2, and MTA1--in defining small-intestinal carcinoid neoplasia. Ann Surg Oncol. 2006; 13: 253-62. doi: 10.1245/ASO.2006.12.011.
79. He K, Guo X, Liu Y, Li J, Hu Y, Wang D, Song J. TUFM downregulation induces epithelial-mesenchymal transition and invasion in lung cancer cells via a mechanism involving AMPK-GSK3beta signaling. Cell Mol Life Sci. 2016; 73:2105-21. doi: 10.1007/s00018-015-2122-9.
80. Lei Y, Kansy BA, Li J, Cong L, Liu Y, Trivedi S, Wen H, Ting JP, Ouyang H, Ferris RL. EGFR-targeted mAb therapy modulates autophagy in head and neck squamous cell carcinoma through NLRX1-TUFM protein complex. Oncogene. 2016; 35:4698-707. doi: 10.1038/onc.2016.11.
81. Shi H, Hood KA, Hayes MT, Stubbs RS. Proteomic analysis of advanced colorectal cancer by laser capture microdissection and two-dimensional difference gel electrophoresis. J Proteomics. 2011; 75: 339-51. doi: 10.1016/j.jprot.2011.07.025.
82. Deighton RF, Le Bihan T, Martin SF, Barrios-Llerena ME, Gerth AM, Kerr LE, McCulloch J, Whittle IR. The proteomic response in glioblastoma in young patients. J Neurooncol. 2014; 119: 79-89. doi: 10.1007/s11060-014-1474-6.
83. Garcia JL, Perez-Caro M, Gomez-Moreta JA, Gonzalez F, Ortiz J, Blanco O, Sancho M, Hernandez-Rivas JM, Gonzalez-Sarmiento R, Sanchez-Martin M. Molecular analysis of ex-vivo CD133+ GBM cells revealed a common invasive and angiogenic profile but different proliferative signatures among high grade gliomas. BMC Cancer. 2010; 10: 454. doi: 10.1186/1471-2407-10-454.
84. Mukherjee J, DeSouza LV, Micallef J, Karim Z, Croul S, Siu KW, Guha A. Loss of collapsin response mediator Protein1, as detected by iTRAQ analysis, promotes invasion of human gliomas expressing mutant EGFRvIII. Cancer Res. 2009; 69: 8545-54. doi: 10.1158/0008-5472.CAN-09-1778.
85. Shih JY, Yang SC, Hong TM, Yuan A, Chen JJ, Yu CJ, Chang YL, Lee YC, Peck K, Wu CW, Yang PC. Collapsin response mediator protein-1 and the invasion and metastasis of cancer cells. J Natl Cancer Inst. 2001; 93: 1392-400.
86. Viphakone N, Cumberbatch MG, Livingstone MJ, Heath PR, Dickman MJ, Catto JW, Wilson SA. Luzp4 defines a new mRNA export pathway in cancer cells. Nucleic Acids Res. 2015; 43: 2353-66. doi: 10.1093/nar/gkv070.
87. Boyd JC. Mathematical tools for demonstrating the clinical usefulness of biochemical markers. Scand J Clin Lab Invest Suppl. 1997; 227: 46-63.