
INTRODUCTION
Sézary syndrome (SS) is an aggressive, leukemic cutaneous T-cell lymphoma variant [1]. It is characterized by the presence of atypical, malignant Sézary cells (CD4+CD45RO+) in blood, lymph nodes and skin, with phenotype of central memory T-cells (TCM) [2], severe erythroderma, pruritus and lymphadenopathy. SS is a very rare disease (incidence rate 0.1/100 000) and consists of 3% of cutaneous T cell lymphoma (CTCL) [1, 3], but with an increasing incidence rate [4]. Median age at presentation is 60 years and despite treatment the prognosis for patients is bad [5]. Data from the largest cohort of advanced Mycosis fungoides (MF) and SS patients (1,275) revealed that the medium overall survival (OS) is 63 months, and, depending on the four risk factors (stage IV, age > 60 years, large-cell transformation, and increased lactate dehydrogenase), the 5-year survival differs markedly from 68% for low risk group, through 44% for intermediate risk to 28% for high risk group [6].
Molecular pathogenesis of SS is still unclear despite many studies on genetic alterations, gene expression and epigenetic regulations. Both cytogenetic and molecular studies revealed chromosomal instability with recurrent “hotspots” of chromosomal abnormalities like 17p (loss of TP53) [7], 10q (loss of PTEN and FAS) [8, 9], 8q (gain of MYC) [10, 11], 9p (loss of CDKN2A-CDKN2B) [12], 19p (loss of E2A) [11] and 6q (loss of A20) [13]. Transcriptome analysis revealed genes being overexpressed in SS like TOX [14] or downregulated like BIN1 [15] or STAT4 [16], with a demonstrated influence on cell biology. Recently, Boonk et al. [17] demonstrated that loss of CD26 and/ or CD7 in combination with altered expression of STAT4, TWIST1 and DNM3 or PLS3, could distinguish SS patients from benign erythroderma cases with 100% specificity. Analysis on whole genomes of CTCL patients [18–21] identified mutations affecting genes involved in important signaling pathways (JAK-STAT, T-cell receptor (TCR) signaling), cell cycle control and epigenetic regulation (chromatin modifying genes, histone and DNA methyltranferases).
The purpose of this study was to combine the whole genome and transcriptome NGS technology, in order to look globally at genetic alterations and their effect on the expression of the affected genes in SS patients. Our results not only underscore the genomic complexity of the Sézary cancer cell genome and make evident that key signaling pathways in SS are abrogated but also show an unpreceded large variety of novel gene rearrangements resulting in fusions transcripts and ectopically expressed genes.
RESULTS AND DISCUSSION
Copy number variations (CNVs) within regions of oncogenes and suppressor genes and new “candidate” genes
Whole genomes of tumor cells of nine Sézary syndrome patients were examined by NGS in combination with inspection of these data using Integrative Genomics Viewer (IGV, Broad Institute). This analysis revealed many large deletions and amplifications (Supplementary Table 1). Recurrent CNVs occurred in regions of already known and described oncogenes, like MYC (8q amplifications in 4/9 patients) and suppressor genes like TP53 (17p deletions in 6/9 patients) (Figure 1) and PTEN (10q deletions in 3 patients). MYC amplifications resulted in altered gene expression, especially in patient 5 with two extra copies of this gene (Reads per Kilobase per Million mapped reads - RPKM: Cn=10=59.54; P5=224.05). MYC amplifications are common in SS [10, 11], but also in many other cancer types. Another gene, TOX, in the 8q region was shown to be highly expressed in SS and involved in abnormal cell proliferation [14]. Furthermore, in contrary to MYC, TOX was stated as a marker in differential diagnosis between SS and erythrodermic dermatitis [22]. Due to this reports, TOX expression was checked in our nine SS patients (RPKM: Cn=10=5.1; SSn=9=63.0) and its overexpression was confirmed (Table 1). For three patients: P5 (RPKM=102), P8 (RPKM=155) and P9 (RPKM=88) this overexpression was the most significant, probably due to the presence of amplification in the 8q region. Strong upregulation of TOX in SS was confirmed by RQ-PCR on an additional cohort of SS patients. TOX showed the highest expression among the genes analyzed in SS, and was 21.88 times upregulated (p<0.0001) as compared to CD4+ controls (Figure 2).
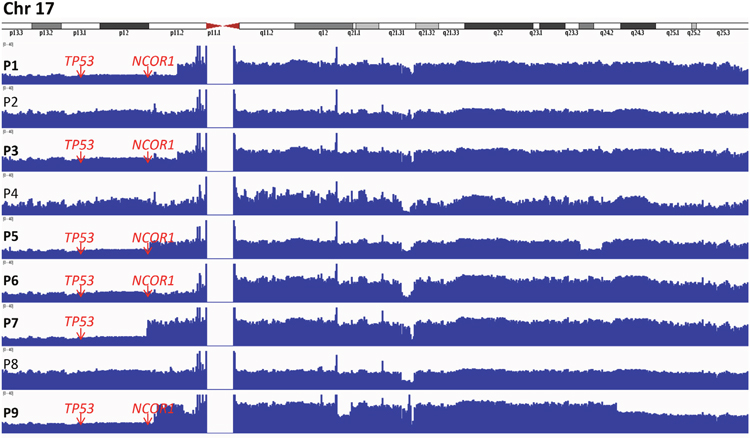
Figure 1: Integrative genomics viewer (IGV) visualization of alignments and coverage of the Illumina reads from the whole genome sequence analysis of 9 SS patients for chromosome 17. Whole genome analysis revealed deletions in the 17p region, involving TP53 (P1, P3, P5-7, P9) and NCOR1 (P1, P3, P5-6, P9). In patient P5 the break was located within NCOR1 sparing the first exon.
Table 1: Expression of genes involved in rearrangements in SS and control samples (C)
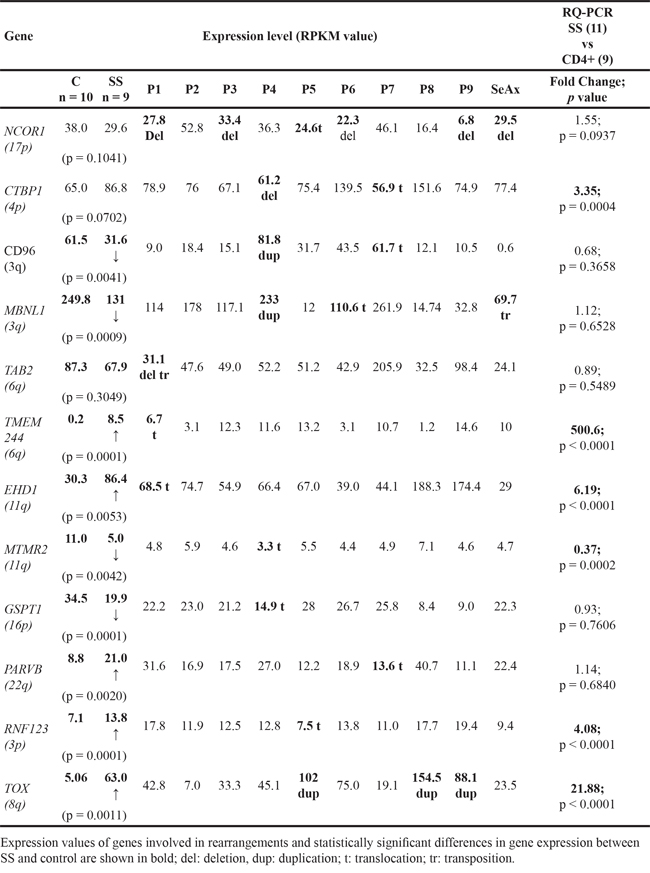
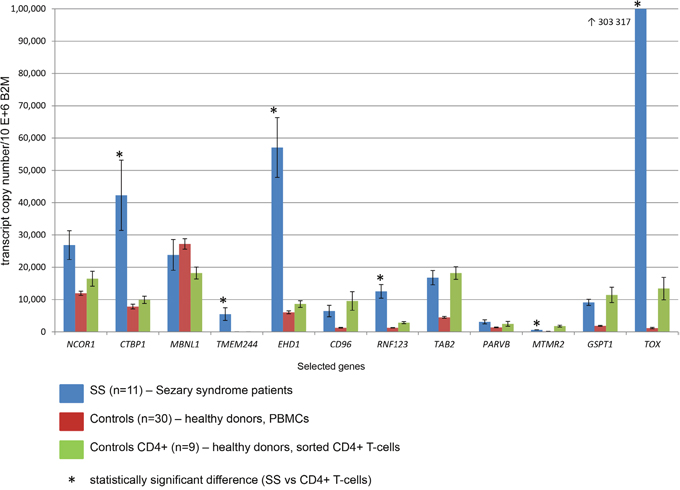
Figure 2: Expression of selected 12 genes measured by real-time quantitative PCR with specific TaqMan probes. Standard errors are reported with bars.
TP53 deletions and mutations were described to be frequent in SS, and as a result this gene was suggested to be a cancer driver gene in Sezary syndrome [7, 18–21]. In recent papers alterations in TP53 were detected in 24-92.5% of patients [18–20]. In this study monoallelic deletions of TP53 resulted in decreased expression of this gene in 5 patients (RPKM: Cn=10=22.73; P1=11.88; P3=10.71; P6=10.19; P7=11.62; P9=10.69), but in P5 increased expression was detected (RPKM=46.49). Mutations that may influence the function of TP53 were identified in P1 (TGT→TTT; C238F) and P5 (splicing site at the 5’ end of intron 7; GT→TT).
The 10q region is recurrently deleted in SS and the status of two genes in this region: PTEN, FAS have already been investigated. PTEN was described to be downregulated in SS, mostly by deletions [8]. In this study in 3 patients heterozygous PTEN deletions were found, with no significant influence on its expression (RPKM: Cn=10=20.37; P1=14.6; P5=17.43; P7=29.33). The second gene, FAS, was deleted in 5 SS patients, but its expression was decreased in all of them (RPKM: Cn=10=49.5; SSn=9=11.5). This is in agreement with previous studies showing that in majority of SS patients FAS expression was lost [9], not only because of recurrent deletions in this region, but also due to hypermethylation of CpG islands in the FAS promoter. Furthermore, inactivating FAS mutations are frequent in SS (10%-42.5%) [18, 19], confirming the putative role of different FAS alterations in malignant transformation and its possible role as a cancer driver. The 10p region is also frequently deleted in SS. Recent whole genome studies showed not only deletions, but also loss of function mutations (45.2%-65%) in Zinc Finger E-Box Binding Homeobox 1 (ZEB1) encoding an important T cell transcription factor [18–20]. In our study we identified deletions of ZEB1 in 5 patients, including one biallelic in P7, all of them having an impact on ZEB1 expression (RPKM: Cn=10=21.5; P1=11.7, P5=13.8; P6=14.2; P7=2.5; P9=7.7).
Interestingly, deletions in the 6q region, that were described to be quite common in SS (50% of patients) [13], were detected only in P1 in this study, and it didn’t include the tumor necrosis factor, alpha-induced protein 3 (TNFAIP3) gene. However, on the contrary to this previous study including pretreated patients, only tumor cells from newly diagnosed patients were studied in this one, suggesting TNFAIP3 deletions to be late events in the SS progression. Late occurrence of TNFAIP3 deletions is further supported by their absence in newly diagnosed Mycosis fungoides patients (unpublished data).
Deletions within 2p region resulted in a loss of DNA (cytosine-5-)-methyltransferase 3 alpha gene (DNMT3A) in four patients, biallelic in patient P8 and monoallelic in patients P1, P5 and P9, with a significant influence of its expression (RPKM: Cn=10=15.32; P1=7.9; P5=6.41; P8=0.1; P9=7.13). In other SS patients the expression of DNMT3A, for other reasons, was also lower compared to controls (RPKM: SSn=9=8.33). According to previous studies DNMT3A and other DNMT genes were usually overexpressed in cancer [23], leading to hypermethylation, gene silencing and possible malignant transformation. However, in aging cells and age-related diseases global hypomethylation of the genome is detected [24], suggesting the possible decreased activity of DNA methyltransferases. Alterations in genes like DNMT3A, DNMT3B, TET1, TET2 have recently been described to be very frequent in SS [18–21] and our results support a potential role of epigenetic modifiers in SS pathogenesis.
New candidate genes were investigated in commonly deleted regions. Three patients (P4, P5, P7) had monoallelic deletions of ubiquitin specific peptidase 28 gene (USP28) (11q) (RPKM: Cn=10=10.91; P4=3.56; P5=4.27; P7=2.46). Genomic studies revealed that both, deletions and overexpression of USP28 could be detected in human cancer [25]. USP28 is a modulator of tumor suppressor FBW7 [26], but in a dose-dependent manner [25]. Only the complete knockdown or upregulation of USP28 was proven to promote oncogenic transformation, due to FBW7 destabilization and accumulation of FBW7 substrates, like MYC [25]. On the contrary, partial deletions of USP28 stabilize FBW7 and drives FBW7 substrates degradation, promoting tumor suppression. Therefore, in our samples USP28 deletion is not likely to be the mechanism driving the malignant transformation, but rather the self-defense mechanism in those Sézary cells with the highest levels of MYC expression (RPKM: Cn=10=59.54; P4=136.46; P5=224.05; P7=144.81).
Two patients (P1, P5) had caspase activity and apoptosis inhibitor 1 gene (CAAP1) (9p) deleted, the second patient on both alleles (RPKM: Cn=10=12.92; P1=6.36; P5=0.93). Knockdown of CAAP1 was proven to induce apoptosis in a caspase-10 manner [27], which is not observed in SS samples with decreased (P1) or lost (P5) CAAP1 expression. One should keep in mind, that most likely p53 is non-functional in those two samples (due to LOH, see above), and since caspase-10 is activated in a p53-induced manner [27], this pathway could be blocked.
Genes involved in rearrangements have altered expression in SS patients
A variety of rearrangements (306; Supplementary Table 2) were detected in SS patients. Most of them were unique for each patient. We decided to focus on those changes that affect the same genes and their expression in more than one patient.
NCOR1 (nuclear receptor corepressor 1) is situated in the 17p region, which is one of the most commonly deleted regions in SS [20]. NCOR1 deletions were detected previously in SS patients (38/80) [20], in some cases they were associated with mutations leading to biallelic loss of function. In this study NCOR1 was fully deleted in four patients (P1, P3, P6, P9) (Figure 1) and in one patient (P5) most of the gene was deleted, except for exon 1, which was translocated to chromosome 15 and fused head-to-head with SV2B gene [chr15:91,813k/chr17:16,099 k]. CTBP1 (C-terminal binding protein 1) (4p) was fully deleted in P4, while in P7 it was involved in a complex rearrangement: translocation t(2;4), [chr4: 1,241k (CTBP1i1)/chr2: 9,994k (TAF1Bi4); chr2: 231,508k/chr4: 1,240k (CTBP1i1)], and t(4;11) [chr4: 1,240k/chr11: 76,788k (CAPN5i1)]. Both NCOR1 and CTBP1 have similar function in cells, they encode proteins involved in histone deacetylases (HDAC) complexes [28], which regulate HDACs activity. Despite the genomic alterations they seem to have higher mRNA expression in SS, yet only overexpression of CTBP1 was confirmed by PCR (Table 1; Figure 2). Destabilization of HDAC activity can lead to imbalance in gene expression, which is observed in human diseases [29], including SS [30]. Since HDAC inhibitors revolutionized the treatment of CTCL patients [31], the status of HDACs in the pathogenesis of SS should be studied in details, also in terms of HDACs’ interactions with proteins, regulating their activity.
CD96 and MBNL1 (muscleblind-like splicing regulator 1), both located in the amplified region in patient P4 (3q), were overexpressed in this sample (Table 2), on the contrary to other SS patients, where those genes were clearly downregulated. In another patient (P7) the 5’ end of the CD96 gene was deleted, the coding region, except for the last exon 15 (rearranged within chr 3), was involved in complex translocation between chr 2 and chr 22 [chr2: 239,042k/chr3: 111,338k (CD96i9)-111,368k (CD96i14)/chr22: 44,503k (PARVBi4)]. MBNL1 was involved in two gene fusions, in sample P6 and SeAx (see below). In P6 MBNL1 fused with MYB as a result of reciprocal translocation (3;6) associated with deletions [chr6: 135,518,448 (MYBi9)/chr3: 152,111,859 (MBNL1i2); chr3:151,811,183/chr6:135,633,875 (AHI1i26)] (Figure 3). This is yet another example of MYB involvement in rearrangements and deletions in SS, noticed by our team [32]. In SeAx MBNL1 was broken within intron 1, 5’ end was transpositioned in a close proximity to KIAA2018 gene [chr3: 151,998,982 (MBNL1i1)/chr3: 113,432,006] and created the fusion transcript, while 3’ end fused tail-to-tail with the part of MEDL12 gene [chr3: 152,000,045 (MBNL1i1)/chr3: 151,111,621 (MEDL12i36)]. MBNL1 mediates an important role in pre-mRNA alternative splicing regulation and depletion of Mbnl protein was proven to misregulate this process [33]. Because both fusions with MBNL1 gene downregulated its expression (Table 1), we decided to look up this gene in other SS patients. Although, RNAseq analysis clearly showed reduced expression in most SS patients compared to controls, it was not confirmed by RQ-PCR (Table 1; Figure 2).
Table 2: Fusion transcripts expressed in SS samples as a result of DNA rearrangements
Sample |
Rearrangement |
Breakpoints (DNA) |
Transcript (RNA) |
|---|---|---|---|
P1 |
t(11;19) |
chr11: 64,628,862/chr19: 39,235,710 |
EHD1(e1,2) – CAPN12(e3→9) |
P4 |
complex |
ch11:+95,529,671/ch11:+34,158,725-34,161,514/chr16:8 872 293 |
CEP57(e1)-NAT10(e22→e23+GTA)-ABAT(i14 chr16:8,872,293-8,872,087) |
P4 |
complex |
chr8:+29,928,614/chr17:-79,602,847–~79,603,500NPLOC4/ |
TMEM66(e1→e2)-BAIAP2(e7→e12?) |
P5 |
t(9;19) |
chr9: 137,760,563/chr19: 19,119,871 |
C9orf9(e1→e2)-SUGP2(e6→e12) |
P5 |
t(1;17) |
ch1:+26,171,755/ch17:+65,161,538 |
HELZ(e1→e15)-AUNIP(e2→e5) |
P6 |
t(1;3) |
chr3:-129,151,514/chr1:+198,647,866/ |
MBD4(e1→e6)-PTPRC(e3→e4→?) |
P6 |
t(1;3) |
chr1: 198,644,193/chr2:39,030,368-39,030,512/chr3: 194,062,664 |
PTPRC(e1→2)-CPN2(fr e2) |
P6 |
t(3;6) |
chr6:+135,518,448/chr3:+152,111,859 |
MYB(e1→e9)-MBNL1(e3→e10) |
P6 |
transpos |
chr3: 100,334,867/chr3:100,446,145 |
TFG(e1→e3)-GPR128(e2→e15) |
P8 |
Transposchr2 |
chr2:68,543,463/chr2:39,049,173–39,050,512/chr2: 69,247,504 |
ARMC6 (e1→8, fr e9 [→19,168,467])-DHX57 (e17→fr i17)[→39,049,173]-CNRIP(fr i2) [68,543,463→68,540,755…] |
P8 |
Transposchr2 |
chr2:71,014,668/chr2:39,563,447 |
MAP4K3(e1→6)-FIGLA(e3→5) |
P8 |
t(3;15;9) |
chr3: -53,370,567/chr15: -44,079,110-44,076,343SERF2/chr9: -34,664,314 |
Variant 1: DCP1A(e1,2,3)–CCl27(e2-3) |
P9 |
del chr2 |
chr2:27,743,165-27,911,890 |
Variant 1: AHDCI(e1)-WASF2(e5→e9) |
SeAx |
inv chr3 |
chr3:151,998,982/chr3:113,432,006 |
MBNL1(e1)-KIAA2018(e2→e7) |
SeAx |
t(1;16) |
chr16: 11, 002,416/chr1: 29,065,400-29,065,342/chr1: 29,065,438 |
Variant 1: CIITA(e1→e10)–YTHDF2(e4,5) |
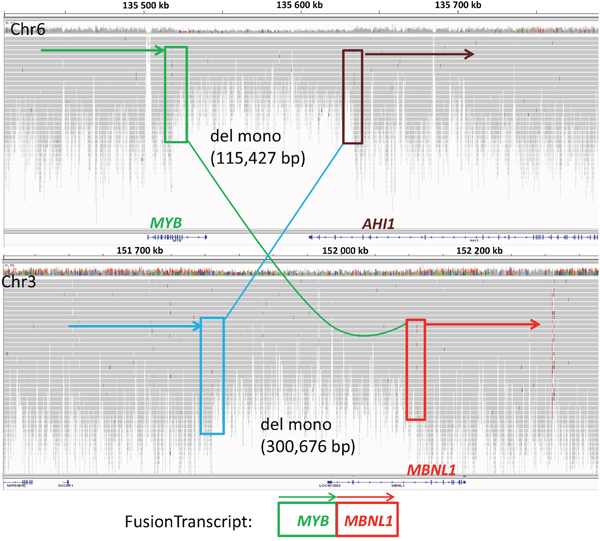
Figure 3: Integrative genomics viewer (IGV) visualization of alignments of the Illumina reads from the whole genome sequence analysis of SS patient (P6) for chromosome 6 (top) and 3 (bottom). Shown are two deletions, on chromosomes 6 and 3, associated with a reciprocal translocation t(3;6), in this SS patient. The 5’ part of MYB (green box) on chromosome 6 was fused to the 3’ part of MBNL1 (red box) on chromosome 3, resulting in a in frame MYB-MBNL1 fusion. An uncoding region of chromosome 3 (blue box) was rearranged to the 3’ part of AHI1 (brown box). Regions of chromosome 3 (containing the 3’ part of MYB and the 5’ part of MBNL1) and chromosome 6 (containing the 5’ part of MBNL1) were deleted during the translocation.
Several genes (TMEM244, RNF123, PARVB, EHD1, MTMR2, GSPT1, TAB2), involved in a rearrangement in only one patient, were selected based on their expression pattern in all SS patients as determined by RNAseq (Table 1). To confirm the pattern, expression of those genes was studied using TaqMan probes on a different cohort of SS patients and controls (Table 1; Figure 2). Four of those genes: TMEM244, EHD1, MTMR2, RNF123 had statistically significant difference in expression level compared to controls. TMEM244 (transmembrane protein 244) was only expressed in SS, no expression was detected in healthy T-cells. Even in P1, where one copy of the TMEM244 genewas involved in a complex rearrangement, it still had higher expression compared to controls (Table 2). The function of TMEM244 is unknown, however expression of other genes from the same family (TMEM176A, TMEM276B, TMEM16A) have been already linked to cancer [34, 35].
Novel fusion transcripts created as a result of rearrangements
Fifteen rearrangements detected in SS patients and SeAx resulted in an expression of new fusion transcripts (Table 2). Those transcripts, except for one (TFG-GPR128) [36], have never been detected before, either in SS, or any other malignancy. TFG-GPR128 was detected in patients with atypical myeloproliferative neoplasms, but also in a small percent of healthy individuals [36]. This is in agreement with other studies showing that chimeric RNA can be expressed in healthy individuals [37], especially when two genes involved reside closer on the genome. Moreover, even oncogenic fusion transcripts, like BCR-ABL, can be detected in normal cells [38, 39]. Knowing that, 30 healthy donors were checked for all fusion-driving rearrangements detected in SS patients in this study, but no fusion transcripts were detected.
Nine of identified fusions (Table 2) were in frame, with no premature stop codons. Theoretically, they could create fully length new proteins. However, for unknown reasons, fusion partners were not always fully expressed, like in EHD1-CAPN12, where exon 9 was the last to be expressed in a fusion transcript. Interestingly, sometimes the breakpoint was not within the partner gene, yet the fusion was still created. In DCP1A-CCL27, the breakpoint on DNA was within intron 3 of DCP1A, followed by a part of SERF2 gene (2, 767 bp), translocated approximately 1.6 kb upstream from CCL27 gene. Similarly, in MBNL1-KIAA2018, the first exon of MBNL1 gene was transpositioned 16, 500 kb upstream from KIAA2018, just to create the transcript with the 2nd exon. In this case the fusion occurred between parts of 5’UTR, so the partner gene KIAA2018 was intact, but the regulatory elements of MBNL1 led to its overexpression compared to other samples (RPKM: Cn=10=12.1; SSn=9=13.7; SeAx=53.1). In TMEM66-BAIAP2, both genes on DNA where separated by a 6 kb insertion, including the small part of NPLOC4 gene. The expression of BAIAP2 gene ended probably with exon 12, as there was another rearrangement starting within intron 12: t(17;21) [79, 082k;44, 412kPKNOX1i1].
PTPRC was involved in two fusions (Figure 4). The breakpoint was within intron 2, first two exons were translocated to chr 3 and forced the expression of the fragment of the second exon of CPN2 gene with 3’UTR. The second part of PTPRC gene was also translocated to chr 3, but under the influence of MBD4 gene. PTPRC encodes the Protein Tyrosine Phosphatase, Receptor Type C, which is highly expressed in lymphocytes where it is also known as the CD45 antigen [40, 41]. By interaction with the LCK proto-oncogene, belonging to the Src family tyrosine kinases, PTPRC/CD45 is involved in TCR signaling pathways regulating T-cell growth, differentiation and function, and contributes to the malignant transformation [42, 43]. There are many variants of this gene expressed in T-cells, each with unique roles for signal transduction and apoptosis [44–46], therefore destabilization of this molecule could lead to lymphomagenesis.
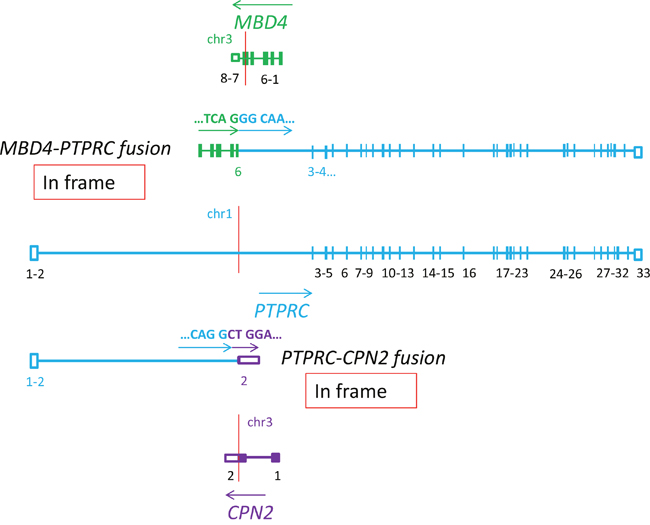
Figure 4: Schematic representation showing that the PTPRC gene, encoding the important T-cells antigen CD45, is involved in two in frame fusions: MBD4-PTPRC and PTPRC-CPN2 (P6).
Not in frame fusions (Table 2) had premature stop codons usually in the first exon after the breakpoint, probably leading to Nonsense-Mediated Decay (NMD) [47], which is a well-known mechanism preventing cells from accumulation of non-functional RNA and producing abnormal peptides.
Expression of chimeric RNA, with intronic elements have already been reported [37]. This whole transcriptome analysis revealed fusions with large intronic fragments: CEP57-NAT10-ABAT and ARMC6-DHX57-CNRIP. First exon of CEP57 gene was fused in frame with exon 22 of NAT10 gene (Table 2; Figure 5). Exon 23 was included in a fusion, but due to another breakpoint within the following intron, the expression was continued within intron 14 of ABAT gene (chr16). Twelve triplets encoding amino acids were present before the stop codon appeared, however, at this point it could not be determined whether it would be translated to a protein. In the second fusion, the transcription started with ARMC6 gene, but interestingly it stopped in the middle of the 3’UTR, just to go on within genes DHX57 and CNRIP. The implications of such expression remain unclear.
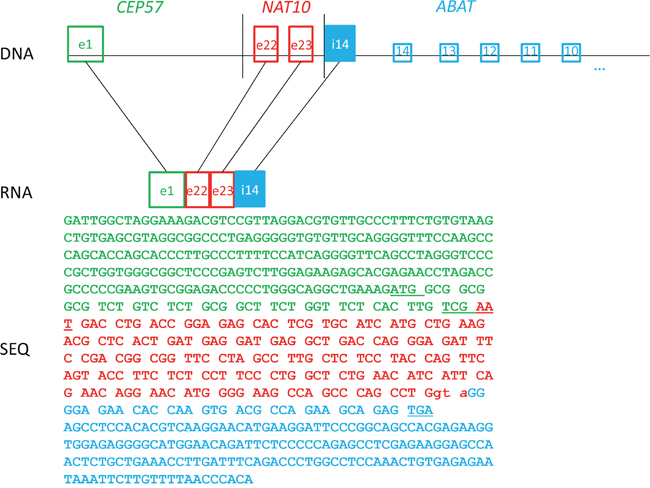
Figure 5: Schematic representation illustrating a complex rearrangement in patient P4. This rearrangement resulted in a fusion transcript between first exon of CEP57, two exons (22nd and 23d) of NAT10 with trinucleotide insertion GTA, and a fragment of intron 14 of the ABAT gene.
Ectopic expression of gene fragments involved in gene fusions
Five gene fusions resulted in ectopic expression of fragments of genes usually not expressed in T-cells: BAIAP2 (RPKM: P4=5; Cn=10=0.74; SSn=8=1.54), CPN2, (RPKM: P6=24.63; Cn=10=0.08; SSn=8=0.12), GPR128, (RPKM: P6=16.03; Cn=10=0.06; SSn=8=0.07), CAPN12, (RPKM: P1=11.36; Cn=10=0.60; SSn=8=0.76), FIGLA (RPKM: P8=1.37; Cn=10=0.07; SSn=8=0.10). Most of those genes, like FIGLA, have tissue specific expression. FIGLA is expressed in ovaries, as a central regulator of oocyte-specific genes that play roles in folliculogenesis, fertilization and early development [48]. In SS P8 FIGLA was ectopically expressed as a result of a fusion with MAP4K3, that have already been described as a fusion partner and a tumor suppressor in cancer [49, 50], and, in general, as an important player in a cell signal transduction.
Conclusion
In conclusion, with this NGS analysis we showed that the variety of genetic events in Sézary cells is enormous, though common denominators could not be identified. It appears that malignant transformation is a result of alterations on many levels: genomic (mutations, CNVs, rearrangements), transcriptomic (elevated expression or downregulation, fusion transcripts, ectopic expression) and epigenomic (methylation, HDACs activity). Although, it is still unclear, which changes are the drivers that caused the disease, and which are the passengers that occurred during the disease progression, this study strongly confirms the role of known tumor suppressors (TP53, FAS), epigenetic modifiers (NCOR1, DNMT3A) and dysregulated signaling pathways (PTPRC in TCR signaling) in SS pathogenesis. Due to the huge variety of alterations we hypothesize that in case of SS different pathways lead to the same clinical presentation. SS is a disease of elderly, so different molecular changes could accumulate during lifetime in skin homing TCM, that persist long term in normal skin following resolution of an immune response [2, 51]. In the future, it would be worth to analyze SS patients individually, to classify them, based on their molecular profile, to choose the best treatment options.
MATERIALS AND METHODS
Clinical samples
Nine SS blood samples were included in the study: P1 (23) (F, age: 80), P2 (19) (M, age: 65), P3 (33) (M, age: 54) from the Department of Dermatology, University of Medical Sciences, Poznan, Poland and P4 (WZ1) (F, age: 74), P5 (WZ4) (M, age: 70), P6 (WZ8) (F, age:71), P7 (WZ9) (M, age: 59), P8 (3-0030) (M, age: 73), P9 (3-0044) (F, age: 64) from the Department of Dermatology, Leiden University Medical Center, Netherlands and the SS derived cell line (SeAx). Patient clinical information is given in Supplementary Table 3. Blood samples were collected once the diagnosis of SS was confirmed. The use of human material was approved by the Local Ethics Committees of both universities, and performed in accordance with the Declaration of Helsinki. Samples were used to extract DNA and RNA, three of them were Ficoll purified mononuclear cells (PBMCs) (P1-P3) while others were sorted CD4+ T-cells (P4-P9). PBMCs were enriched for CD4+ T cells by depletion of non-CD4+ T cells using the CD4+ T Cell Isolation Kit I (Miltenyi Biotec, Bergisch Gladbach, Germany), resulting in a cell purity >95% as claimed by the manufacturer [52]. The purity of the samples was confirmed by quantitative NGS analysis of the T cell receptor alpha/delta locus (Supplementary Figure 1). In samples P4-P9 no germline TCRAD sequence was visible, indicating almost 100% purity of the samples, while in samples P1-P3 an admixture of non-malignant cells, without TCRAD rearrangements, was present.
Healthy individual control samples
For RNASeq 10 RNA samples from five healthy individuals (males: 24, 24, 28, 61 and 67 years old), isolated from sorted peripheral blood CD4+ or IL-2 activated CD4+ cells, were used as controls.
For RQ-PCR 9 RNA samples isolated from sorted peripheral blood CD4+ cells from healthy individuals (males: 21, 24 and 46 years old; females: 33, 33, 36, 49, 58 and 62 years old), and 30 RNA samples isolated from Ficoll purified mononuclear peripheral blood cells from healthy individuals (41-57 years old) were used as controls.
Paired-end next-generation sequencing
Paired-end 15x coverage whole genome NGS was performed by BGI-Hong Kong on the HiSeq2000 Illumina platform. At least two non-identical inserts with discordant 5’ and 3’ reads localized in different regions of the genome were considered to carry a rearrangement.
RNAseq
Paired–end RNASeq analysis was performed using two different NGS platforms: for 7 SS patients (P1-P7), SeAx and 10 control samples SOLID5500 platform was used (Life Technologies; mRNA fragments: 100-150 bp; reads: 75 bp/35 bp), while two other SS patients (P8, P9) were analyzed on HiSeq2000 Illumina platform (mRNA fragments: 150 bp reads: 100 bp).
NGS data analysis
Sequence analysis was based on GRCh37/hg19. Sequences, as bam files and bam.tdf files, were analyzed using Integrative Genomics Viewer 2.3 (IGV 2.3, Broad Institute). RPKM values were used to measure mRNA abundance. Lifescope software was used to find fusion transcripts: Paired-end reads that mapped to different genes were considered as evidence of fusion transcripts.
PCR, RT-PCR
Selected rearrangements and fusion transcripts were confirmed on DNA and cDNA using standard PCR and RT-PCR. Products were Sanger sequenced (Institute of Biochemistry and Biophysics PAS, Warsaw, Poland) to confirm the breakpoint. 30 DNA samples from healthy donors were used as controls.
Real-time quantitative PCR (RQ-PCR)
RQ-PCR was performed using the CFX96 Real-Time System (BioRad, Hercules, CA). The following genes were analyzed by commercially obtained TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA): NCOR1 (Hs01094540_m1), CTBP1 (Hs00972284_m1), TAB2 (Hs00248373_m1), CD96 (Hs00175524_m1), EHD1 (Hs00199030_m1), GSPT1 (Hs01093019_m1), MTMR2 (Hs01547438_m1), PARVB (Hs00203381_m1), RNF123 (Hs00222902_m1), TMEM244 (Hs02340633_m1), MBNL1 (Hs02569862_s1), USP28 (Hs00363603_m1). Beta-2 microglobuline (B2M) (Hs00984230_m1), not affected by alterations and suitable for CD4+ lymphocytes, [53] was used as a reference gene for sample normalization. cDNA samples from 11 SS patients, 30 Ficoll purified peripheral blood mononuclear cell samples (PMBCs) from healthy donors and 9 sorted CD4+ T-cell samples from healthy donors were analyzed. All samples were assayed in duplicate. Statistical analysis of the differences in the gene expression was performed using two-tailed Student’s t-test for unpaired data (GraphPad Software) and the 2-ΔΔCT Method [54].
ACKNOWLEDGMENTS
We would like to thank Robert Weissmann for excellent assistance in statistical analysis as well as Christian Sperling and Corinna Jensen for superior technical assistance.
FUNDING
This project was funded by the National Science Centre; decision No 2013/08/M/NZ2/00962 (KI, GKP), Foundation for Polish Science (KI), the German José Carreras Leukemia Foundation No R12/33 (CG, PG, CAS) and Dutch Cancer Society grant UL2013-6104 (CPT and MHV).
CONFLICTS OF INTEREST
The authors state no conflicts of interest.
REFERENCES
1. Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, Ralfkiaer E, Chimenti S, Diaz-Perez JL, Duncan LM, Grange F, Harris NL, Kempf W, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005; 105:3768-85.
2. Campbell JJ, Clark RA, Watanabe R, Kupper TS. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: a biologic rationale for their distinct clinical behaviors. Blood. 2010; 116:767-71.
3. Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009; 113:5064-73.
4. Wilson LD, Hinds GA, Yu JB. Age, race, sex, stage, and incidence of cutaneous lymphoma. Clin Lymphoma Myeloma Leuk. 2012; 12:291-6.
5. Li JY, Horwitz S, Moskowitz A, Myskowski PL, Pulitzer M, Querfeld C. Management of cutaneous T cell lymphoma: new and emerging targets and treatment options. Cancer Manag Res. 2012; 4:75-89.
6. Scarisbrick JJ, Prince HM, Vermeer MH, Quaglino P, Horwitz S, Porcu P, Stadler R, Wood GS, Beylot-Barry M, Pham-Ledard A, Foss F, Girardi M, Bagot M, et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sezary Syndrome: effect of specific prognostic markers on survival and development of a prognostic model. J Clin Oncol. 2015; 33:3766-73.
7. Lamprecht B, Kreher S, Mobs M, Sterry W, Dorken B, Janz M, Assaf C, Mathas S. The tumour suppressor p53 is frequently nonfunctional in Sezary syndrome. Br J Dermatol. 2012; 167:240-6.
8. Cristofoletti C, Picchio MC, Lazzeri C, Tocco V, Pagani E, Bresin A, Mancini B, Passarelli F, Facchiano A, Scala E, Lombardo GA, Cantonetti M, Caprini E, et al. Comprehensive analysis of PTEN status in Sezary syndrome. Blood. 2013; 122:3511-20.
9. Jones CL, Wain EM, Chu CC, Tosi I, Foster R, McKenzie RC, Whittaker SJ, Mitchell TJ. Downregulation of Fas gene expression in Sezary syndrome is associated with promoter hypermethylation. J Invest Dermatol. 2010; 130:1116-25.
10. Vermeer MH, van Doorn R, Dijkman R, Mao X, Whittaker S, van Voorst Vader PC, Gerritsen MJ, Geerts ML, Gellrich S, Soderberg O, Leuchowius KJ, Landegren U, Out-Luiting JJ, et al. Novel and highly recurrent chromosomal alterations in Sezary syndrome. Cancer Res. 2008; 68:2689-98.
11. Steininger A, Mobs M, Ullmann R, Kochert K, Kreher S, Lamprecht B, Anagnostopoulos I, Hummel M, Richter J, Beyer M, Janz M, Klemke CD, Stein H, et al. Genomic loss of the putative tumor suppressor gene E2A in human lymphoma. J Exp Med. 2011; 208:1585-93.
12. Laharanne E, Chevret E, Idrissi Y, Gentil C, Longy M, Ferrer J, Dubus P, Jouary T, Vergier B, Beylot-Barry M, Merlio JP. CDKN2A-CDKN2B deletion defines an aggressive subset of cutaneous T-cell lymphoma. Mod Pathol. 2010; 23:547-58.
13. Braun FC, Grabarczyk P, Mobs M, Braun FK, Eberle J, Beyer M, Sterry W, Busse F, Schroder J, Delin M, Przybylski GK, Schmidt CA. Tumor suppressor TNFAIP3 (A20) is frequently deleted in Sezary syndrome. Leukemia. 2011; 25:1494-501.
14. Huang Y, Su MW, Jiang X, Zhou Y. Evidence of an oncogenic role of aberrant TOX activation in cutaneous T-cell lymphoma. Blood. 2015; 125:1435-43.
15. Esmailzadeh S, Huang Y, Su MW, Zhou Y, Jiang X. BIN1 tumor suppressor regulates Fas/Fas ligand-mediated apoptosis through c-FLIP in cutaneous T-cell lymphoma. Leukemia. 2015; 29:1402-13.
16. Litvinov IV, Cordeiro B, Fredholm S, Odum N, Zargham H, Huang Y, Zhou Y, Pehr K, Kupper TS, Woetmann A, Sasseville D. Analysis of STAT4 expression in cutaneous T-cell lymphoma (CTCL) patients and patient-derived cell lines. Cell Cycle. 2014; 13:2975-82.
17. Boonk SE, Zoutman WH, Marie-Cardine A, van der Fits L, Out-Luiting JJ, Mitchell TJ, Tosi I, Morris SL, Moriarty B, Booken N, Felcht M, Quaglino P, Ponti R, et al. Evaluation of immunophenotypic and molecular biomarkers for Sezary syndrome using standard operating procedures: multicenter study of 59 cases. J Invest Dermatol. 2016; 136:1364-72.
18. Wang L, Ni X, Covington KR, Yang BY, Shiu J, Zhang X, Xi L, Meng Q, Langridge T, Drummond J, Donehower LA, Doddapaneni H, Muzny DM, et al. Genomic profiling of Sezary syndrome identifies alterations of key T cell signaling and differentiation genes. Nat Genet. 2015; 47:1426-34.
19. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, Bjornson RD, Maman Y, Wang T, Tordoff J, Carlson K, Overton JD, Liu KJ, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015; 47:1011-9.
20. Kiel MJ, Sahasrabuddhe AA, Rolland DC, Velusamy T, Chung F, Schaller M, Bailey NG, Betz BL, Miranda RN, Porcu P, Byrd JC, Jeffrey Medeiros L, Kunkel SL, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK-STAT pathway in Sezary syndrome. Nat Commun. 2015; 6:8470.
21. da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, Vermeer MH, Rabadan R, Ferrando A, Palomero T. The mutational landscape of cutaneous T cell lymphoma and Sezary syndrome. Nat Genet. 2015; 47:1465-70.
22. Boonk SE, Cetinozman F, Vermeer MH, Jansen PM, Willemze R. Differential expression of TOX by skin-infiltrating T cells in Sezary syndrome and erythrodermic dermatitis. J Cutan Pathol. 2015; 42:604-9.
23. Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014; 4:80.
24. Rodriguez-Rodero S, Fernandez-Morera JL, Fernandez AF, Menendez-Torre E, Fraga MF. Epigenetic regulation of aging. Discov Med. 2010; 10:225-33.
25. Schulein-Volk C, Wolf E, Zhu J, Xu W, Taranets L, Hellmann A, Janicke LA, Diefenbacher ME, Behrens A, Eilers M, Popov N. Dual regulation of Fbw7 function and oncogenic transformation by Usp28. Cell Rep. 2014; 9:1099-109.
26. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008; 8:83-93.
27. Zhang Y, Johansson E, Miller ML, Janicke RU, Ferguson DJ, Plas D, Meller J, Anderson MW. Identification of a conserved anti-apoptotic protein that modulates the mitochondrial apoptosis pathway. PLoS One. 2011; 6:e25284.
28. Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. J Biomed Biotechnol. 2011; 2011:129383.
29. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. 2013; 7:47-60.
30. Lee CS, Ungewickell A, Bhaduri A, Qu K, Webster DE, Armstrong R, Weng WK, Aros CJ, Mah A, Chen RO, Lin M, Sundram U, Chang HY, et al. Transcriptome sequencing in Sezary syndrome identifies Sezary cell and mycosis fungoides-associated lncRNAs and novel transcripts. Blood. 2012; 120:3288-97.
31. Prince HM, Dickinson M. Romidepsin for cutaneous T-cell lymphoma. Clin Cancer Res. 2012; 18:3509-15.
32. Izykowska K, Zawada M, Nowicka K, Grabarczyk P, Braun FC, Delin M, Mobs M, Beyer M, Sterry W, Schmidt CA, Przybylski GK. Identification of multiple complex rearrangements associated with deletions in the 6q23-27 region in Sezary syndrome. J Invest Dermatol. 2013; 133:2617-25.
33. Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA, Swanson MS. Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA-mediated disease. Mol Cell. 2014; 56:311-22.
34. Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013; 110:E1026-34.
35. Cuajungco MP, Podevin W, Valluri VK, Bui Q, Nguyen VH, Taylor K. Abnormal accumulation of human transmembrane (TMEM)-176A and 176B proteins is associated with cancer pathology. Acta Histochem. 2012; 114:705-12.
36. Chase A, Ernst T, Fiebig A, Collins A, Grand F, Erben P, Reiter A, Schreiber S, Cross NC. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica. 2010; 95:20-6.
37. Greger L, Su J, Rung J, Ferreira PG, Lappalainen T, Dermitzakis ET, Brazma A. Tandem RNA chimeras contribute to transcriptome diversity in human population and are associated with intronic genetic variants. PLoS One. 2014; 9:e104567.
38. Boquett JA, Alves JR, de Oliveira CE. Analysis of BCR/ABL transcripts in healthy individuals. Genet Mol Res. 2013; 12:4967-71.
39. Ismail SI, Naffa RG, Yousef AM, Ghanim MT. Incidence of bcrabl fusion transcripts in healthy individuals. Mol Med Rep. 2014; 9:1271-6.
40. Huntington ND, Tarlinton DM. CD45: direct and indirect government of immune regulation. Immunol Lett. 2004; 94:167-74.
41. Clark MC, Baum LG. T cells modulate glycans on CD43 and CD45 during development and activation, signal regulation, and survival. Ann N Y Acad Sci. 2012; 1253:58-67.
42. Ventimiglia LN, Alonso MA. The role of membrane rafts in Lck transport, regulation and signalling in T-cells. Biochem J. 2013; 454:169-79.
43. Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004; 23:7990-8000.
44. Tchilian EZ, Beverley PC. Altered CD45 expression and disease. Trends Immunol. 2006; 27:146-53.
45. Ogilvy S, Louis-Dit-Sully C, Cooper J, Cassady RL, Alexander DR, Holmes N. Either of the CD45RB and CD45RO isoforms are effective in restoring T cell, but not B cell, development and function in CD45-null mice. J Immunol. 2003; 171:1792-800.
46. Zheng X, Li AS, Zheng H, Zhao D, Guan D, Zou H. Different associations of CD45 isoforms with STAT3, PKC and ERK regulate IL-6-induced proliferation in myeloma. PLoS One. 2015; 10:e0119780.
47. Miller JN, Pearce DA. Nonsense-mediated decay in genetic disease: friend or foe? Mutat Res Rev Mutat Res. 2014; 762:52-64.
48. Joshi S, Davies H, Sims LP, Levy SE, Dean J. Ovarian gene expression in the absence of FIGLA, an oocyte-specific transcription factor. BMC Dev Biol. 2007; 7:67.
49. Carvalho D, Mackay A, Bjerke L, Grundy RG, Lopes C, Reis RM, Jones C. The prognostic role of intragenic copy number breakpoints and identification of novel fusion genes in paediatric high grade glioma. Acta Neuropathol Commun. 2014; 2:23.
50. Lam D, Dickens D, Reid EB, Loh SH, Moisoi N, Martins LM. MAP4K3 modulates cell death via the post-transcriptional regulation of BH3-only proteins. Proc Natl Acad Sci U S A. 2009; 106:11978-83.
51. Clark RA. Skin-resident T cells: the ups and downs of on site immunity. J Invest Dermatol. 2010; 130:362-70.
52. Boonk SE, Zoutman WH, Marie-Cardine A, van der Fits L, Out-Luiting JJ, Mitchell TJ, Tosi I, Morris SL, Moriarty B, Booken N, Felcht M, Quaglino P, Ponti R, et al. Evaluation of immunophenotypic and molecular biomarkers for Sezary syndrome using standard operating procedures: a multicenter study of 59 patients. J Invest Dermatol. 2016; 136:1364-72.
53. Wang T, Liang ZA, Sandford AJ, Xiong XY, Yang YY, Ji YL, He JQ. Selection of suitable housekeeping genes for real-time quantitative PCR in CD4(+) lymphocytes from asthmatics with or without depression. PLoS One. 2012; 7:e48367.
54. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-8.