
INTRODUCTION
Recently in vitro and in vivo data suggest a crucial role of extracellular tau in the initiation/progression of AD [1, 2, 3]. Accumulating evidence shows that the clearance of extracellular pathological tau forms, which represents the rationale for the promising results of the ongoing tau-based immunotherapy, is an actual AD therapeutic alternative to the not-encouraging Aβ-based pharmacological and immunological approaches [4, 5]. Although full length tau is found in cerebral spinal fluid (CSF) from both normal humans [6] and rodents [7, 8], a heterogeneous population of fragments of protein -including the NH2terminal and/or proline-rich domain- is for the most part detected in CSF from AD subjects [9, 10, 11, 12] and in conditioned media from AD patient-derived induced pluripotent stem cells (iPSC) cortical neurons [13]. Interestingly, the largest part of tau secreted from different in vitro neuronal models has been recently confirmed to be actually C-terminally truncated, soluble, unaggregated and released both by vital and dead neurons in a way independent of cell damage [14]. Pathological hyperphosphorylation and caspase-3 cleavage of tau (Asp421) taking place in diseased subjects promote its aggregation and also favor the in vitro secretion [15]. Besides, the N-terminal projection domain of human tau -which interacts with the plasma membrane [16] and mediates neurotoxicity mainly at synapses [17]- is required for its secretion to the extracellular space in in situ tauopathy model [18]. Exosomes-associated NH2derived tau fragments are also detected in CSF from AD patients and a different pattern of NH2-tau fragments in CSF may reflect disease-specific neurodegenerative processes [19]. Accordingly, passive immunization with different tau antibodies directed against the extreme and mid-region in N-terminal end of protein turns out to be the most beneficial in reducing pathological tau hyperphosphorylation and in improving cognition of aged (16-months-old) 3XTg AD mice [20], thus fostering the targeting of these selective epitopes as an actual opportunity for the cure of AD and other tauopathies. Relevantly, a comprehensive mass spectrometry (MS)-based screening carried out on CSF samples from patients affected from AD and other tauopathies has confirmed that the first NH2 26-44 aminoacids of human tau are epitopes potentially targetable for promising immunotherapeutic interventions, being largely represented into proteomic profile of diseased secreted proteins [21]. However, the identification of the molecular nature of extracellular noxious tau species is of central importance for developing an effective tau-based immunotherapy and, in this framework, the biological effects of AD-relevant NH2-truncated human tau forms outside of neurons still remain to be determined. In addition, although a potential extracellular physiopathological role of full length tau has been highlighted, consistent with the recent findings that secreted tau is per se toxic on synaptic function [22, 23], no study has yet investigated whether one of the biologically relevant NH2-truncated forms of protein -which are largely detected in diseased CSF and released in vivo from AD nerve endings [9, 10, 11]- could affect the neurotransmission at the pre-synaptic level.
To this point, we previously reported that higher levels of the 20-22kDa NH2-truncated form of human tau - a neurotoxic fragment of the full length protein (htau40) mapping between 26 and 230 aminoacids of the longest human tau isoform- are largely detectable in presynaptic terminals [24, 25, 26] as well in peripheral CSFs from living patients affected by AD and not-AD human neurodegenerative diseases associated to dementias [27]. Importantly, following potassium-induced depolarization, this specific C-terminal truncated peptide is preferentially released from AD presynaptic nerve endings in comparison with age-matched healthy controls [28], thus providing a rationale for investigating its potential action outside of neurons. Therefore, given the physiopathological role of extracellular tau in the regulation of inter-neuronal signaling at synapses [29, 30, 31, 32] and the prevailing presence of N-terminal/middle region-containing truncated forms of protein ranging from 20-25kDa in AD-derived biological fluids [9, 10, 11, 13], we attempted to elucidate the biological effects induced by short- and long-term exposure of hippocampal isolated synaptosomes and mature primary neurons to subtoxic doses of extracellular human NH2htau 26-44 fragment, which is the minimal biologically active moiety of longer secreted 20-22kDa parental peptide [25, 33, 34].
RESULTS
NH2htau 26-44 (aka NH2htau) is highly flexible and mainly monomeric in solution
Although tau is an intrinsically disordered protein characterized by dynamic ensemble of interconverting conformations, recent detailed NMR spectroscopy analysis carried out on the full-length longest human tau 1-441isoform (htau40) has revealed that the first N-terminal 50 residues of protein favor a compact conformation displaying an intrinsic propensity to local secondary structure(s), as demonstrated by the presence of strong contacts within the residue stretch 1-20 and from this region to residues 30-50 [35]. In addition, the N-terminal residues Leu-Thr transiently form α-helical structure promoting a long-range intramolecular interactions [36, 37]. In view of these considerations, we set about investigating the conformational changes that chemically-synthesized (purity up to 99%) human NH2 26-44 (aka NH2htau) and its reverse control sequence (aka reverse) undergo in aqueous solution by circular dichroism (CD) spectroscopy, which allows to estimate the secondary structural composition of proteins in different experimental environments. As shown in Figure 1a, NH2htau displayed within the wide range of explored pH (pH 4-11) a spectrum typical for an unfolded peptide characterized by a strong negative ellipticity below 200nm which is suggestive of a random coil unfolded conformation. No evident sign of structured peptide chains was observed in CD spectra recorded in experimental conditions similar to those occurring near the lipid bilayers (i.e.membrane-mimicking media) such as in presence of increasing detergent SDS concentrations (Figure 1b) or anionic-zwitterionic phospholipid large unilamellar vesicles (LUV) composed by POPC/POPS in a 7/3 ratio at pH7.4 (Figure 1c). Only in presence of high percentage of TFE (more than 60%), a solvent which has a dielectric constant lower than water and is widely used to mimic the hydrophobic environments of membranes, a slight propensity towards a coil-helix transition could be discernable in spectral profiles (Figure 1d). The shape and magnitude of CD curves from its reverse counterpart were not dissimilar from those of NH2htau (data not shown), just resembling the traditionally accepted “random coil” spectra, although conformational changes appeared more pronounced starting with lower TFE percentages (40%).
Next, in order to evaluate the capability of both tau peptides dissolved in aqueous buffer to form amyloid-like fibrils, we carried out Thioflavin T (ThT)-binding fluorescence assay. The kinetics of fiber growth in solution were monitored and time traces from NH2htau in the absence (red curve) and presence of LUV composed by total lipid brain extract (TLBE, green curve) were calculated. TLBE bilayers contain a physiologically important mix of lipid components -cholesterol, gangliosides, sphingolipids, isoprenoids in addition to both acidic and neutral phospholipids- making these bilayers a biologically-relevant model surface. As shown (Figure 1e), unlike the classical sigmoidal kinetic profile characterizing the ongoing Aβ1-40 fibrillogenesis which resulted in progressive 12-14 fold increase in ThT fluorescence intensity (black curve), no time-dependent emission signal was measured for NH2htau up to 48h. Relevantly, ThT measurements did not change over time showing the same signal recorded at time zero even after longer incubation, indicating that NH2htau was not prone to aggregate and was highly stable in solution. Likewise, reverse control counterpart did not show any ability to form amyloid fibrils in solution when tested both in the absence (blue curve) and presence of TLBE-LUV membrane model (cyan curve).
Taken together the above data demonstrate that both NH2htau and its reverse control counterpart have intrinsically unstructured disordered nature (unfolded random coil) because they neither undergo any significant structural transition in different chemical and/or membrane-mimicking environments nor form aggregation fibers over time, although CD and ThT measurements do not completely exclude the possibility that the analyzed peptides might form amorphous and/or partially structured aggregates. Consistently with its monodisperse disordered structure in aqueous solution, SDS-PAGE Western blotting analysis with ad hoc devised 12A12 monoclonal antibody directed against the extreme N-terminal 26-36 aa of human tau protein (Figure 1f-1g), validated that the nature of NH2htau was mainly monomeric with a corresponding single band that migrated more slowly than expected in a calculated mass of 2.0 kDa, in line with previous studies of disease-related disordered proteins [16, 38]. In support of the finding that the prevailing molecular form of NH2htau under our experimental conditions appeared to be unaggregated, we found out that the overall proportion of soluble peptide in oligomers turned out to be low as its monomeric concentration increased. A lowly-populated fraction of aggregates of higher order oligomerization migrating around 35-50kDa was faintly detected only after prolonged exposition of high-performing PVDF transfer membranes, as shown in Supplementary Figure 1.
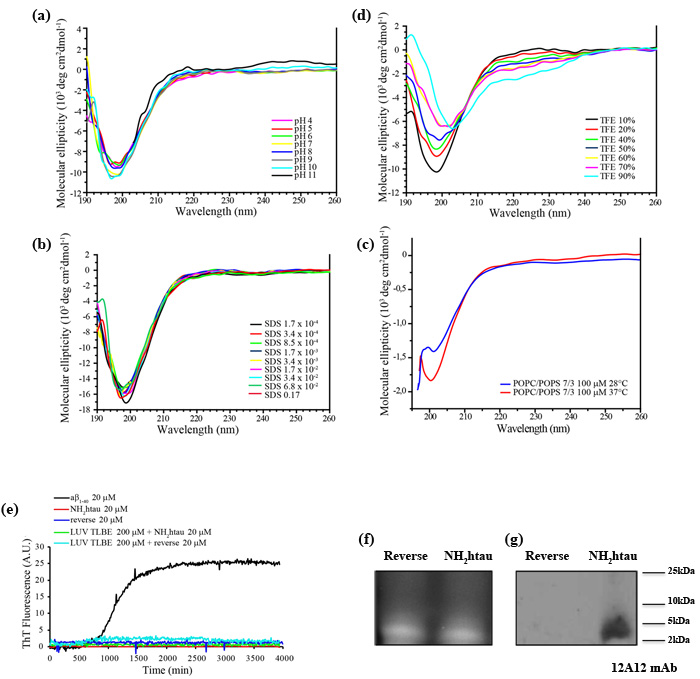
Figure 1: NH2htau 26-44 (i.e.NH2htau) shows an intrinsically disordered structure and is mainly monomeric in water environment. a.-b.-c.-d. CD spectra of NH2htau26-44 (10µM ) at different pH(A), at different concentrations of SDS(B), in presence of large unilamellar vesicles (LUV) composed by POPC/POPS 100 µM in a 7/3 ratio at pH7.4 at increasing temperatures (c) and at different percentage of TFE(d) are shown. Notice that NH2htau shows in aqueous solution a negative peak typical for an unfolded protein with a minimum at 200nm, indicating the general absence of local major conformational changes. e. Kinetics of fiber formation was measured by ThT binding assay and ThT fluorescence of Aβ1-40 20 µM (black curve), NH2htau 20 µM in the absence (red curve) and presence of lipid LUV TLBE 200 µM (green curve), reverse control peptide (i.e reverse) 20 µM in the absence (blue curve) and presence of lipid LUV TLBE 200 µM (cyan curve) are reported. All samples were prepared in 10 mM phosphate buffer, pH 7.4, 100 mM NaCl. Time traces reported are the average of three experiments. Kinetic curves for Aβ1-40 are classically sigmoidal-shaped indicating an ongoing aggregation growth process while no noticeable differences in ThT emission intensity over time are contextually detected for NH2htau and reverse peptides both in solution and in presence of lipid membrane mimetics. f.-g. NH2htau and its reverse sequence were dissolved as described in material and methods and 150pmol of both peptides were analyzed on 15% SDS-PAGE in reducing conditions. After reversible Sypro Ruby protein staining of gel(f), Western blotting analysis was performed by probing with specific 12A12 monoclonal antibody directed against the extreme N-terminal 26-36 aa of human tau protein.Cropped representative WB is shown (g).
Extracellular-added NH2htau stably interacts with authentic neuronal plasma membranes under physiological conditions and perturbs artificial biomembrane-like environments in a time-dependent manner
Despite being a highly soluble protein, full-length tau has a strong propensity to interact with lipid membranes [39, 40, 41]. Indeed, although highly charged, it selectively inserts into synthetic anionic bilayer -likely owing to the attractive electrostatic interactions between the positively charged C-terminal microtubule (MT)-binding extremity and membrane lipids- inducing structural compaction [42, 43]. Besides, the negatively charged amino-terminal projection domain of tau has been shown to physiologically interact with more detergent-resistant microdomains (lipid rafts) of the plasma membrane in a phosphorylation-dependent manner [16, 44, 45, 46]. In view of these considerations, at first we set about testing the propensity of extracellular-added NH2htau to associate with biological plasma membranes in a native, physiological and cellular environment by means of morphological and biochemical experimental approaches. To this aim, live imaging experiments were first carried out on mature hippocampal primary neurons (15 DIV) after exposure to FITC-conjugated NH2htau (1µM) for short incubation times (5’-10’) and the binding of fluorescent peptide to neuronal cell membranes was revealed under non-permeabilizing conditions by TRITC-cholera toxin subunits B which is known to specifically target the lipid raft microdomains. As shown in Figure 2a-2h, NH2htau-exposed cultures exhibited a disperse punctate labeling which outlined the cell periphery and consistently colocalized with membrane-bound cholera toxin, indicating that this peptide was able to bind the neuronal plasma membrane near to these detergent-resistant microdomains. This specific, dot-like pattern of immunoreactivity appeared even from shorter incubation times (5’) and was distributed both in close proximity to and away from somatic compartment (arrowheads and arrows point to non-synaptic and synaptic areas, respectively). Notably, not all structures were double-labeled for components of both lipid rafts and NH2htau (asterisks) and, following incubation for short period of time and under not-saturable conditions, only a few rafts-positive axons as well as synaptic boutons were decorated with fluorescent NH2htau. To better investigate the specific association of NH2htau to the synaptic terminal ends, additional colocalization studies were carried out by double confocal analysis of isolated synaptosomal preparations marked with FM4-64 -a red fluorescent dye used to image functional synaptic terminals- in the presence of FITC-conjugated NH2htau (1µM). As shown in Figure 2i-2j, the punctate distribution of NH2htau overlapped with staining of the FM4-64 -positive synaptosomes (arrowheads) supporting thus the finding that this extracellular-added peptide was able to accumulate on the plasma membrane of these subcellular compartments in exposed-neurons.
Finally, biochemical experiments of subcellular fractionation aimed to isolate the membrane-containing portions from NH2htau-, reverse-treated and untreated primary hippocampal cultures followed by Western blotting analysis with our 12A12 tau antibody (26-36 aa) (Figure 1f-1g), confirmed that the NH2htau was copurified with N-Methyl-D-aspartate (NMDA) Receptor subunit NR1-enriched fraction (Figure 2k) when extracellular-added to neurons. Control experiments were carried out to check the purity of preparation which turned out to be free of contaminations from other, non-membranous compartments (Supplementary Figure 2). Altogether, these three different experimental approaches demonstrate that the NH2htau fragment is able to interact with genuine neuronal membrane and synaptic boutouns only after few minutes of exposure.
Next, in order to investigate whether membrane-bound NH2htau is also able to perturb lipid bilayer over time, we turned to differential scanning calorimetry (DSC) which is a highly sensitive non-perturbing technique useful for a thermodynamic and kinetic characterization of lipid/protein interactions in biomimetic models [47, 48]. The potential of DSC experiments to provide useful information concerning peptide/membrane interactions is based on the fact that the heat capacity (Cp) profiles of the main transitions of mixed lipid/peptide systems can elucidate not only the effects of the peptide on the membrane but also its topological arrangement when inserted into the lipid matrix. In fact, as the enthalpy variations observed during the main lipid transitions are mainly due to the efficiency in the packing of the hydrocarbon tail, it’s possible to correlate the peptide-induced reduction of the transition enthalpy of the bilayer to the extent of the interactions between guest molecules and the core of lipid membranes. Moreover, the main transition temperature is more sensitive to interactions involving the lipid headgroups, increasing when the membrane surface is involved in the interaction with the guest peptide [49, 50, 51]. To study the dynamic interaction of both NH2htau and its reverse peptide with lipid bilayer, we took advantage of the same experimental setting used to analyze another intrinsically disordered protein such as α-synuclein, which is also physiologically absorbed by raft microdomains onto the neuronal membrane surface and affects the thermotropic phase behavior of anionic lipid vesicles [52, 53]. Mixed acid-zwitterionic LUV composed by DMPC/DMPS (ratio 7:3) which mimic the composition of mammalian biomembranes [54]were thus used in our studies. This artificial biomembrane model undergoes spontaneous lipid phase segregation which can be observed by DSC thermograms. In particular, calorimetric heating scans displayed two endothermic peaks characterized by separate melting temperature (Tm) of transition centred at 27.2+/-0.1 °C and 28.7+/-0.1 °C which may be ascribed to DMPC- and DMPS-rich lipid domains, respectively (Figure 2l-2m, black curves). Gaussians deconvolution of DSC thermograms was then obtained (Table 1) to calculate the enthalpy change (ΔH) values corresponding to the two melting peaks of these distinct lipid domains (21.58+/-2.15 kJ mol-1 and 8.88+/-0.9 kJ mol-1 for the DMPC- and the DMPS-rich lipid domains). The stability of the lipid bilayers over time was also tested at 25°C up to 72h and no significant change in the thermogram and thermodynamic parameters in DSC runs was detected (data not shown). Interestingly as shown in Figure 2l, the incorporation of the NH2htau into the DMPC/DMPS bilayers caused significant and time-dependent perturbations in their thermotropic phase behavior suggesting that this peptide was able not only to promptly associate to lipid membrane model but also to gradually induce profound structural changes in their packing. Indeed, immediately after the addition of 20 µM NH2htau (Figure 2l red curve) to suspension, the phase segregation was more evident and interaction of NH2htau with the surface of the artificial membranes was clearly distinguished by the rise in the thermal transition temperature with partitioning of two discrete domains centred at 27.2+/-0.1°C and 29.2+/-0.1 °C, respectively. Besides, we did not observe any decrease in ∆H of the two domains at t = 0 (Table 1), indicating that this extreme N-terminal tau peptide at first interacted with lipid artificial membranes on the outside, stabilizing the more rigid DMPS-based regions. Similar results were obtained after 24 hours incubation of artificial biomembrane mimetics with NH2htau (Figure 2l, blue curve and Table 1), as evidenced by slight changes in both enthalpy and temperature of the main transition in comparison to corresponding values at t = 0. However, a weak interaction between NH2htau peptide and the deep hydrophobic core of membrane appeared to be clearly discernable merely after 48 hours incubation (Figure 2l, green curve), as evidenced by diminution in both parameters of thermal transition temperature (from 29.4+/-0.1°C to 29.2+/-0.2°C) and ∆H (from 9.24+/-0.9 kJ mol-1 to 8.64+/-0.8 kJ mol-1) of DMPS-rich domains when compared to t = 24h. A more pronounced reduction in total ∆H (from 28.95+/- 2.9 kJ mol-1 to 23.49+/- 2.3 kJ mol-1) was finally detected after prolonged incubation times (72 hours, Figure 2l cyan curve) in connection with a strong drop in the ∆H of the DMPC-rich domain (from 20.31+/-2.0 kJ mol-1 to 16.94+/-1.7 kJ mol-1), as calculated by deconvolution analysis of the peaks (Table 1). Taken together these findings clearly indicate that: (i) this extreme N-terminal tau peptide enriches artificial membranes in more rigid DMPS-based regions at t = 0 and only later (t = 72h) shifts towards the more fluid, not-raft DMPC-based regions; (ii) following its immediate adsorption on the surface of lipid membrane model, the deep interaction of NH2htau with the hydrocarbon region takes place only after longer incubation, suggesting that this peptide is more likely to be internalized after 48-72h exposure. Differently from NH2htau, its reverse counterpart showed a limited effect on the thermotropic phase behavior of DMPC/DMPS membranes displaying a poor tendency to interact with model membranes and to penetrate the lipid bilayer. Indeed, in parallel set of experiments we detected that the only interaction of this control peptide was with the DMPS-rich region on the surface of the membrane -as evidenced by the increase in thermal transition temperature of the second peak (from 29.2 +/- 0.1 to 29.3+/- 0.1) - but that no concomitant decrease of the ∆H (Table 1) occurred up to 72h , suggesting the absence of any its obvious dynamic and stable interaction with the hydrophobic core of model membranes (Figure 2m). To the point, it’s noteworthy that small differences in the spatial arrangement of aminoacids within a peptide sequence have been actually proved to cause big changes in the interaction of peptide with lipid membrane by affecting in particular the peptide orientation and/or the depth of membrane insertion. Therefore considering that: (i) the guest-induced decrease of ΔH is mainly ascribable to the insertion of the externally-added molecule into the hydrocarbon core of the bilayer and (ii) the NH2htau incorporation into the DMPC/DMPS membrane shows an increase in the gel-liquid crystal transition temperature of the bilayer with a decreased enthalpy change, it may be concluded that- following its immediate adsorption on the surface- only the NH2htau has the capability to stably and deeply interact with the lipid bilayer in a time-dependent manner.
Collectively, these morphological and biochemical studies of live-imaging and subcellular fractionation along with biophysical DSC analyses on DMPC/DMPS lipid vesicles, demonstrate that NH2htau: (i) is able to interact within few minutes at least in part with lipid components located in both synaptic and non-synaptic areas of authentic neuronal plasma membranes; (ii) dynamically and spontaneously interacts with artificial membrane model, enriching earlier (up to 24h) the lipid biomimetic environments in the more rigid DMPS-based domains and being more likely internalized only after prolonged incubation times (48-72h) in correlation with its shift towards the more fluid, not-raft DMPC-based regions.
Table 1: Thermodinamic parameters of samples containing LUV DMPC/DMPS 7/3 ratio and NH2htau or its reverse over time.
Sample |
T1 (°C) |
∆H1(kJ mol-1) |
T2 (°C) |
∆H2(kJ mol-1) |
∆Htot(kJ mol-1) |
DMPC/DMPS 7/3 200 µM |
27.2 ± 0. 1 |
21.58 ± 2.15 |
28.7 ± 0. 1 |
8.88 ± 0. 9 |
30.46 ± 3. 0 |
DMPC/DMPS 7/3 200 µM NH2htau 20 µM t=0 h |
27.2 ± 0. 1 |
22.80 ± 2.3 |
29.2 ± 0. 1 |
9.68 ± 1. 0 |
32.48 ± 3. 2 |
DMPC/DMPS 7/3 200 µM NH2htau 20 µM t=24 h |
27.2 ± 0. 1 |
23.05 ± 2.3 |
29.4 ± 0. 1 |
9.24 ± 0. 9 |
32.29 ± 3.2 |
DMPC/DMPS 7/3 200 µM NH2htau 20 µM t=48 h |
27.2 ± 0. 1 |
20.31 ± 2.0 |
29.2 ± 0. 1 |
8.64 ± 0. 8 |
28.95 ± 2.9 |
DMPC/DMPS 7/3 200 µM NH2htau 20 µM t=72 h |
27.2 ± 0. 1 |
16.94 ± 1.7 |
29.2 ± 0. 1 |
6.55 ± 0. 6 |
23.49 ± 2.3 |
DMPC/DMPS 7/3 200 µM reverse 20 µM t=0 h |
27.2 ± 0. 1 |
20.78 ± 2.1 |
29.2 ± 0. 1 |
8.07 ± 0. 8 |
28.85 ± 2.8 |
DMPC/DMPS 7/3 200 µM reverse 20 µM t=24 h |
27.2 ± 0. 1 |
21.63 ± 2.2 |
29.3 ± 0. 1 |
9.25 ± 0. 9 |
30.88 ± 3.1 |
DMPC/DMPS 7/3 200 µM reverse 20 µM t=48 h |
27.2 ± 0. 1 |
22.14 ± 2.2 |
29.3 ± 0. 1 |
9.4 ± 0. 9 |
31.54 ± 3. 1 |
DMPC/DMPS 7/3 200 µM reverse 20 µM t=72 h |
27.2 ± 0. 1 |
23.30 ± 2.3 |
29.3 ± 0. 1 |
8.54 ± 0. 8 |
31.84 ± 3.2 |
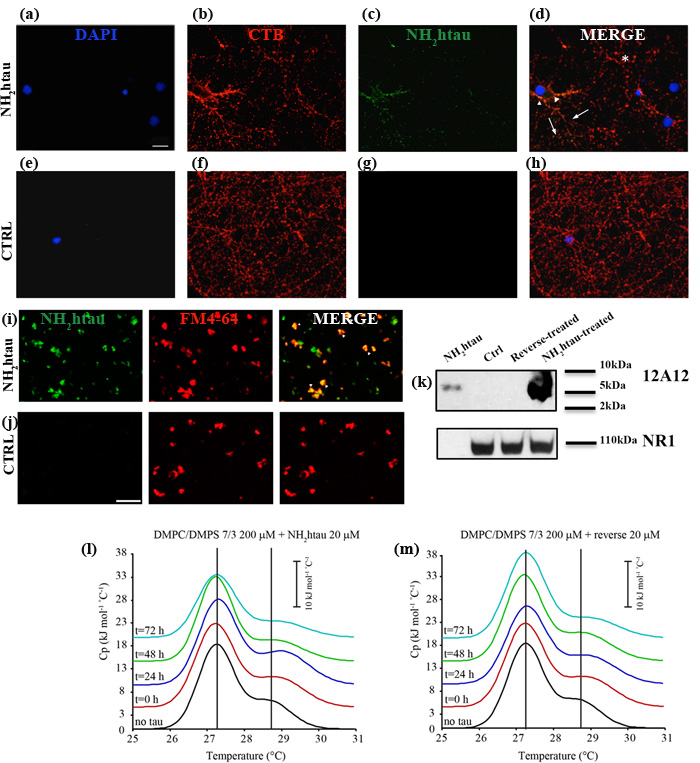
Figure 2: NH2htau is adsorbed by native neuronal plasma membranes under in vitro physiological conditions and deeply perturbs membrane-like lipid bilayers in a time-dependent manner. a.-b.-c.-d.-e.-f.-g.-h. Fluorescence analysis (a-b-c-d) under non-permeabilizing conditions on hippocampal neurons (DIV15) in the presence or absence of the FITC-conjugated NH2htau. Live primary cultures were 30’exposed to FITC-conjugated NH2htau (1µM) (green channel) and then stained with TRITC-conjugated cholera toxin subunit b (red channel). Nuclei were stained with DAPI (blue channel). Merge image shows the composition of two fluorescence channels. Untreated cultures were used as negative control (e-f-g-h). Notice that NH2htau displays a diffuse, dot-like pattern distributed both in close proximity to and away from somatic compartment (arrowheads and arrows point to non-synaptic and synaptic areas, respectively). Structures which are positive for components of lipid rafts but not for NH2htau (asterisks) are also present. Scale bar:5 µm. i.-j. Fluorescence analysis of purified hippocampal synaptosomal fractions in the presence or absence of the FITC-conjugated NH2htau. Isolated synaptosomes were dual-labeled (i) by staining with FM4-64 (50µM for 1’+ 30mMKCl for 90sec) (red channel) and with FITC-conjugated NH2htau (1µM) for 10’(green channel). Untreated synaptosomes were used as negative control (j). Overlay image (yellow, arrowheads) indicates colocalization of NH2htau and FM 4-64 in numerous granular spots. Few ring-like morphological structures (usually 1–3 µm in diameter) resembling spherical synaptosomes are labeled by NH2htau but not by membrane-bound FM4-64 dye .Scale bar:10 µm. k. Western blotting analysis (n=2) of isolated membrane-containing fractions from primary hippocampal neurons (15 DIV) exposed for 1h to NH2htau and its reverse (5µM). Untreated cultures and synthetic NH2 26-44 human tau peptide were used as controls. Immunoblotting was performed with 12A12 monoclonal antibody (26-36 aa) and with NR1 subunit antibody, to check the preparation purity. Cropped representative WB are shown. l.-m. DSC thermograms of LUV DMPC/DMPS 7/3 freshly prepared (black curve), LUV DMPC/DMPS 7/3 + NH2htau or reverse 20 µM each t = 0 (red curve), LUV DMPC/DMPS 7/3 + NH2htau or reverse 20 µM t = 24h (blue curve), LUV DMPC/DMPS 7/3 + NH2htau or reverse 20 µM t = 48h (green curve), LUV DMPC/DMPS 7/3 + NH2htau or reverse 20 µM each t = 72h (cyan curve). Values from deconvolution analysis of the peak profiles are reported in Table 1.
Sublethal doses of extracellular NH2htau induce prompt perturbation in K+-evoked calcium dynamics on isolated hippocampal synatopsomes
Properly controlled Ca2+ homeostasis is crucial for physiological maintenance of neuronal integrity as well as for survival and synaptic plasticity by controlling the release of neurotransmitter, membrane excitability and network activity. Consequently, perturbations in Ca2+ signaling have been proposed as one of the earliest age-related event underlying the synaptic pathology occurring in the AD and leading eventually to neurodegeneration [55, 56]. Having established that neuronal plasma membrane is a plausible site of action for extracellular N-terminally truncated tau species -which are more likely to be produced inside neurons by intracellular cleavage of full length-protein and then released into the extracellular space [13, 14]- we investigated the effects induced by acute application of low concentration of extracellular NH2htau and its reverse peptide on both the basal and the KCl-evoked Ca2+ dynamics turning to quantitative imaging with highly-specific Ca2+-sensitive fluorescent indicator. Besides, since changes of intracellular calcium homeostasis in nerve terminals directly trigger the discharge of synaptic vesicles and glutamate release evoked by K+-depolarization [57, 58, 59], we performed our assays on isolated hippocampal pinched-off nerve terminals, so-called synaptosomes, which are targeted by extracellular NH2htau (Figure 2) and have been proved to be particularly useful for biochemical studies of presynaptic stimulus-secretion coupling [60]. To this aim, purified synaptosomal preparations were preloaded with Fluo3 dye and then 5 min exposed to NH2htau or its reverse counterpart at 1 µM each, a concentration which does not cause any significant in vitro neuronal death up to 72h -as we previously assessed by MTT assay and Western blotting detection of active cleaved caspase-3 (Supplementary Figure 3)- and more closely mimics the in vivo pathological range of release-competent (i.e. soluble and microtubule unbound) amount of intracellular tau [61, 62, 63, 64]. Upon chemically-induced depolarization with 30mM KCl, each Ca2+ transient for three different experimental groups (saline-treated ctrl, NH2htau- and reverse-treated) was measured and the intensity and kinetics of the intrasynaptosomal Ca2+ changes produced by extracellular high [K+] were then visualized as a characteristic rapid rise to peak followed by a descending and apparent plateau phase, consistently with previous results [65]. Quantitative averaged data of fluorescence intensity (peak amplitude, ∆F) and of kinetic parameters (peak to baseline; baseline to peak; overall duration of signal) are shown in Figure 3. Following K+-stimulation (Figure 3a-3b-3c-3j), synaptic fractions acutely exposed to NH2htau displayed a pronounced potentiation (+35% and 36%, compared to both reverse- or untreated controls) in evoked increase of free-Ca2+ fluorescence intensity (∆F NH2htau: pixel intensity 72, 07% +/-12, 33 SD, +/- 2, 4 SEM n = 58, one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001) in comparison to both reverse- or untreated controls. On the contrary (Figure 3d-3e-3f-3g-3h-3i-3j), application of the reverse peptide failed to produce any significant change in magnitude of K+-induced Ca2+ peak height (-1%) (∆F reverse: pixel intensity, 53, 17% +/-14, 21 SD, +/- 1, 8 SEM n = 66, one-way repeated-measures ANOVA followed by Bonferroni post-hoc test p >0, 05) which did not significantly differ from untreated controls (∆F ctrl: pixel intensity, 52, 61% +/-10, 68 SD, +/- 1, 3 SEM n = 98). Further, as shown by kinetic analyses reported in Figure 3k, all the kinetic parameters displayed a significative difference when the NH2htau group was compared to both reverse and untreated controls. Indeed, the rise in synaptic Ca2+ signal (baseline to peak: NH2htau 2, 2 s +/-0, 42 SD, +/-0, 08 SEM; ctrl 2, 02 s +/-0, 21 SD, +/- 0, 02 SEM; reverse 2, 03 s +/-0, 34 SD, +/-0, 03 SEM) was significantly different among the three different experimental groups (+10% NH2htau vs ctrl and reverse, one-way repeated-measures ANOVA followed by Bonferroni post-hoc test **p <0, 01; -0, 4% ctrl vs reverse p >0, 05). The fall of synaptic Ca2+ transient (-22 % vs ctrl; -26 % vs reverse) as well as its time course (-21 % vs ctrl; -24 % vs reverse) in nerve endings preparations exposed to NH2htau (NH2htau: peak to baseline 44, 75 s +/-16, 25 SD, +/-2, 7 SEM one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001; duration 47 s +/-15, 53 SD, +/-2, 8 SEM n = 58 one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001) were significantly reduced compared to both reverse-and untreated controls. Conversely, reverse peptide (reverse: peak to baseline 60, 54 s +/-19, 62 SD, +/-3, 4 SEM; duration 62, 57 s +/-20, 15 SD, +/-3, 4 SEM n = 66 ) did not provoke any significant change in kinetic parameters (peak to baseline -4% vs ctrl one-way repeated-measures ANOVA followed by Bonferroni post-hoc test p >0, 05; duration -4% vs ctrl one-way repeated-measures ANOVA followed by Bonferroni post-hoc test p >0, 05) in comparison to untreated control (ctrl: peak to baseline 57, 59 s +/-14, 89 SD, +/-1, 7 SEM; duration 59, 61 s +/-14, 21 SD, +/- 1, 7 SEM). Importantly, no significant global change in Ca2+ amplitude over baseline was detected in parallel assays when hippocampal synaptosomes (up to 30’; data not shown) and mature (15 DIV) primary neuronal cultures as well (up to 1h; data not shown) were exposed to extracellular NH2htau and reverse peptide both used at the same experimental concentration (1 µM) but in the absence of KCl stimulus (resting conditions). To this point, we did not measure any significant variation in Ca2+ signal throughout the recording times ruling out thus the possibility that its external application could lead to other calcium-affecting mechanisms previously described for preaggregated and unaggregated full-length tau species, including ionic pore formation and membrane leakage [66, 67], activation of specific cell surface receptors [62] or inhibition of plasma-membrane Ca2+-ATPase (PMCA) [68].
Taken together, our data from Ca2+ functional imaging demonstrates that:(i) acute treatment with low, sublethal doses of extracellular-applied NH2htau is not able to modify the resting Ca2+ level but affects both the magnitude and the kinetic parameters of ionic intracellular signaling evoked by chemically-induced depolarization of the synaptic neuronal compartments; (ii), the amplitude of K+-evoked Ca2+ transient is greater in synaptosomes when exposed to NH2htau but declines to a stable plateau more quickly (i.e higher and shorter signal) than in the other two experimental groups (reverse- and saline-treated controls); (iii) this effect is specific because its reverse sequence is actually inactive in perturbing either basal or stimulated Ca2+ homeostasis when compared to saline-exposed untreated controls.
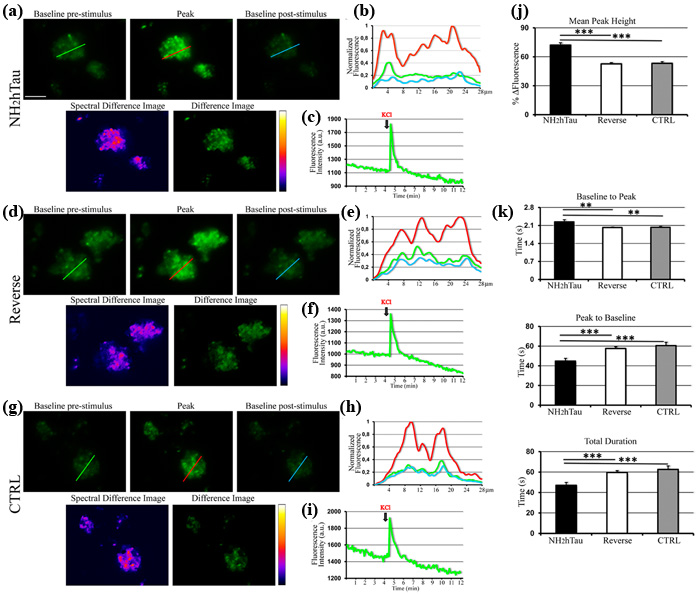
Figure 3: Acute administration of sub-toxic doses of NH2htau induces perturbation in regulation of K+-evoked intracellular calcium dynamics in isolated hippocampal synaptosomes. Changes in K+-evoked Ca2+ levels induced by NH2htau and its reverse counterpart were assessed in Fluo-3 loaded hippocampal synaptosomes exposed to NH2htau (1µM), its reverse peptide (1µM), and saline (CTRL) 5 min before KCl (30mM) stimulation. a.-d.-g. Upper row: wide field images of treated synaptosomes. Baseline pre-stimulus is the image immediately before the fluorescence increase towards the peak. Peak is the image showing the highest fluorescence intensity. Baseline post-stimulus is the image immediately after that the post-stimulus baseline is established. Lower row: false color image (fire palette; Spectral Difference Image) showing the fluorescence intensity of the difference image. Brighter spots correspond to functional presynaptic terminals. Difference image is the subtraction of the baseline pre-stimulus image from the peak one. b.-e.-h. Normalized traces of fluorescence intensity spatial profiles derived from the line shown in a, d, and g. Green line refers to the baseline pre-stimulus image. Red line refers to the peak image. Cyan line refers to the baseline post-stimulus image. Note the relative fluorescence height intensity of the peak image (red line) which appears spatially distributed to partially fused multiple sites. c.-f.-i. Representative traces of fluorescence time courses derived from the three experimental groups. KCl black arrows indicate the time of the stimulus addition. j. Comparison, among the three experimental groups, of the average percentages of the fluorescence intensity difference between the peak and the baseline pre-stimulus values. Note the statistically significant increase of calculated value from NH2htau in comparison to those from the reverse and CTRL groups (one-way ANOVA followed by Bonferroni post-hoc test ***p<0,0001 versus NH2htau). k. kinetic analysis of the fluo-3 intensity time-course: baseline to peak, i.e. time for the rate of signal rise; peak to baseline, i.e. time for the rate of signal decay; total duration, i.e. the overall time from baseline pre-stimulus to baseline post-stimulus. Data were derived from five independent experiments. In each experiment two coverslips for each experimental group were analyzed. Values are means of at least four independent recordings and statistically significant differences were calculated by one-way ANOVA followed by Bonferroni post-hoc test (**p<0,01; ***p<0,0001 versus NH2htau). Scale bar: 20µm.
Extracellular-added NH2htau acutely inhibits K+-induced FM1-43 destaining and presynaptic glutamate release in hippocampal synaptosomal preparations
Owing to potent relationship between Ca2+ and exocytosis, small changes in Ca2+ influx during a presynaptic potential action are highly effective in modulating the neurotransmitter output at synapses [69]. Glutamate is the primary excitatory neurotransmitter in the brain playing an important role in hippocampal-dependent learning and memory and deregulation of glutamatergic synaptic transmission is considered a primary step of synaptic failure occurring in the pathogenesis of AD [70]. In view that our results showing an early modulatory effect of NH2htau on K+-evoked regulation of intracellular Ca2+ (Figure 3) hinted at possible alterations in Ca2+-dependent mechanisms underlying synaptic activity, we evaluated the acute effects evoked by the extracellular addition of either NH2htau and its reverse sequence (1 µM each) on live dynamics of vesicles release from isolated nerve terminals (i.e. synaptosomes) of mature (15 DIV) hippocampal neurons by means of functional FM1-43 fluorescence imaging. FM1-43 styryl dye is more fluorescent when partitioned in membranes, so its release from synaptic vesicles can be measured as a monoexponential loss in signal which actually reflects the rate of exocytosis occurring at nerves terminals. Although this protocol is unable to discriminate glutamatergic from GABAergic synapses, its activity-dependent unloading is widely used to study the properties of presynaptic release on purified synaptosomal preparations, providing thus a direct measure of their in vitro secretory function [59, 71, 72, 73]. After 5 min exposure to NH2htau, its reverse sequence and saline, the decay of FM1-43 fluorescence evoked by synaptosomes stimulation with high-potassium (30 mM KCl) was measured and quantitative analysis of live imaging data from the three different experimental groups is shown in Figure 4a-4b-4c-4d-4e-4f-4g-4h. Results clearly indicate that short-term application of subtoxic doses of extracellular NH2htau caused a significant reduction of pre-synaptic FM1-43 destaining (NH2htau: 12, 42 %+/- 4, 7 n = 90, one-way repeated-measures ANOVA followed by Bonferroni post-hoc test **p < 0, 01) in individual hippocampal axonal terminals which displayed severe deficits in vesicles release dynamics when compared to both reverse- and untreated controls (-36, 56 % vs ctrl; -30, 94 % vs reverse). Conversely, reverse peptide did not provoke any appreciable change in activity-dependent dye unloading (reverse: 17, 98 % +/- 6, 1 n = 86) and reverse-treated nerve terminal preparations did not significantly differ (-8%) from untreated controls (ctrl: 19, 58% +/- 3, 2 n = 90, one-way repeated-measures ANOVA followed by Bonferroni post-hoc test p >0, 05). Of note, when hippocampal synaptosomes were exposed to extracellular NH2htau used at the same experimental concentration (1 µM) but in the absence of KCl stimulus (resting conditions, saline-exposed), we did not detect significant differences (Figure 4g) in comparison to an untreated and saline-exposed group ruling out thus the possibility that this extracellular-added peptide might have a destaining effect per se .
To further investigate the nature of potent in vitro inhibitory effect shown by NH2htau on presynaptic compartment of hippocampal neurons, high-sensitive radioactivity-based assays of glutamate release were also carried out according to standard procedure [74, 75]. To this aim, isolated nerve endings preloaded with [3H]D-aspartate ([3H]D-Asp) -which allows to measure the neurotransmitter exocytosis avoiding drawbacks due to its enzymatic degradation- were incubated with synthetic NH2htau or its reverse counterpart (1, 10, 100nM) and then superfused with mild depolarizing stimulus (12 mM KCl; equiosmolar replacement of Na+) in order to elicit the Ca2+-dependent, exocytotic-like release of the radioactive tracer [76, 77, 78]. As shown in Figure 4i, transient and short-term exposure (-90 s) of synaptosomal terminals to nanomolar concentrations of NH2htau caused a dramatic and statistically significant inhibition of 12 mM K+-evoked tritium overflow in a concentration-dependent manner, the maximum of inhibition (-38, 3%) being observed when the peptide was added at 100 nM (NH2htau: 1 +/- 0, 09 n = 11 ANOVA followed by Bonferroni post-hoc test ***p < 0, 0001 vs ctrl). On the contrary, reverse peptide even if used at the highest experimental concentration (100 nM) was completely inactive in modulating the exocytotic-like glutamate release (reverse: 1, 63 +/-0, 09 n = 11) and treated samples did not significantly differ from saline-exposed untreated controls (ctrl 1, 62 +/- 0, 06 n = 11).
No significant effect on spontaneous basal glutamate release was detected in isolated synaptosomes upon long-term incubation with NH2htau and its effect on K+ evoked neurotransmitter exocytosis was abrogated by incubation in Ca2+-free milieu or by pretreatment with membrane-permeant ion chelator 1, 2-bis-(2-aminophenoxy)ethane-N, N, N’, N’-tetraacetic acid tetra(acetoxymethyl) ester (BAPTA/AM)BAPTA, suggesting that the presynaptic action of NH2htau is strictly Ca2+- dependent (Figure 4i).
Importantly, these functional changes at synaptic terminals occurred within few minutes of exposure, in agreement with prompt and marked tendency of NH2htau to interact with biomimetic and cell-surface membranes as we previously detected by morphological, biochemical and biophysical studies (Figure 2). Besides, the extracellular-added NH2htau, but not its reverse counterpart, was able to alter the K+- stimulated functional properties in neurotransmitter secretion of hippocampal neurons in the absence of any appreciable cell injury because the loss in synaptic components along with the structural changes in neuritic architecture and the reduction in mitochondrial density were measured only later, after 48h incubation times (see below) .
Collectively these experiments, carried out on isolated hippocampal synaptosomes by Ca2+-indicator dye measures and neurotransmitter exocytosis assays, points to a direct causal link between depolarization-dependent defects in Ca2+ handling and early presynaptic modulatory effect of extracellular NH2htau on glutamatergic neurotransmission demonstrating that: (i) the inhibitory action on depolarization-stimulated glutamate exocytosis induced by short-term external application of low, sublethal doses of NH2htau is accompanied by concomitant alteration in local Ca2+ dynamics in nerve endings; (ii) the peak amplitude and kinetic parameters of Ca2+current evoked by K+ stimulation develop respectively at a greater rate but decay to a less long-lasting plateau in NH2htau-exposed in nerve endings compared with control ones (i.e higher and shorter signal), resulting in an overall decrease in neurotransmitter release ; (ii) these changes are specific for NH2htau because its reverse sequence does not afford any significant effect either on stimulated Ca2+ signaling or on glutamate exocytosis when compared to saline-exposed untreated controls.
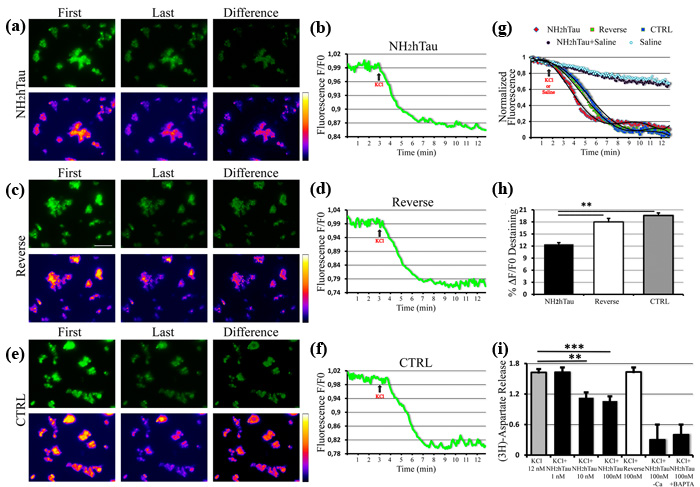
Figure 4: NH2htau acutely inhibits the K+-stimulated presynaptic vesicles release and glutamate exocytosis in purified synptosomal preparations. a.-b.-c.-d.-e.-f.-g.-h. K+-induced destaining of FM1-43 dye on isolated nerve terminals from mature (15 DIV) hippocampal neurons exposed to NH2htau (1µM), its reverse peptide (1µM), and saline (CTRL) 5 min before KCl (30mM) stimulus addition. a.-c.-e. Upper row: wide field images of treated synaptosomes. Lower row: false color vertically corresponding images (fire palette) which show the fluorescence intensity. Brighter spots correspond to functional presynaptic terminals. First is the image before KCl administration. Last is the image when the after-stimulus baseline is established. Difference is subtraction of the last image from the first one. Note that the yellow color is below the saturation level (i.e.white color). b.-d.-f. Representative traces of destaining time courses derived from the three experimental groups. g. Normalized, aligned and averaged fluorescence intensity traces derived from the three experimental groups (25 traces each) plus two saline(-KCl) additional controls. One, in the absence of both treatments and KCl stimulus, representing the fluorescence bleaching rate of our experimental setting. The other one, in the presence of NH2htau (1µM) and with saline added, showing that NH2htau alone is not able to induce significant destaining effects. Trend lines (black lines) superimposed to fluorescence intensity values of three experimental groups were calculated by polynomial fitting. h. Comparison of the average destaining percentage of the fluorescence intensity among the three experimental groups. In each experiment(n=5) two coverslips for each experimental group were analyzed. Values are means of at least three independent recordings and statistically significant differences were by one-way ANOVA followed by Bonferroni post-hoc test (**p<0,01versus NH2htau). Scale bar:15µm. i. The overall dose-effect of the NH2htau action (1-10-100nM) on glutamate release was evaluated by high-sensitive radioactive-based measure of depolarization-evoked release of [3H]D-Asp. Reverse sequence, used at the highest concentration(100nM) and saline-exposed untreated controls were also included. The K+-evoked tritium overlow is expressed as % of 12 mM KCl-evoked [3H]D-aspartate overflow versus saline-exposed untreated ctrl.Values are means of at least five independent experiments and data were considered statistically significant for p < 0.05 at least (**p < 0,01; ***p < 0,0001 versus saline-exposed untreated ctrl, one-way ANOVA followed by Bonferroni post-hoc test).
Deterioration in presynaptic terminals, neuritic degeneration, microtubule collapse and reduction of mitochondrial density are detected in living hippocampal neurons only after long-term exposure to sublethal doses of NH2htau
Structural alterations in synaptic terminals, instability of microtubules, dendritic retraction, mitochondrial abnormalities are all hallmarks of neuronal pathology playing a critical role in promoting cognitive dysfunction during the AD onset/progression and appearing prior to frank cell death [79]. Therefore, by Western blotting on total protein extracts and immunocytochemistry analyses, we investigated the expression level and subcellular distribution of different cellular markers of synaptic, cytoskeleton and mitochondrial compartments after administration of NH2htau (1-2µM) to hippocampal neuronal cultures for increasing incubation times (up to 48-72 h). Interestingly (Figure 5a-5b), we noticed only after 48h cultures treatment a significant and dose-dependent decline in selected proteins which are mainly located in the presynaptic compartment and are involved in local turnover of synaptic vesicles and/or Ca2+-triggered neurotransmitter release, such as α-synuclein, synapsin I, synaptosomal-associated protein 25 (SNAP-25), synaptophysin and vesicular glutamate transporter 1(vGLUT1). Importantly, no discernable change was detected at earlier incubation times (1-3-6-12-24h, data not shown) ruling out the possibility that the diminution in functional release of glutamate induced by the acute administration of NH2htau on isolated hippocampal synaptosomes -as we showed in Figure 4- could be ascribed to modifications in expression levels of these relevant presynaptic proteins which control the Ca2+-coupled neurotransmitter exocytosis at nerve endings. Others presynaptic proteins -such as synaptic vesicle protein 2 (SV2), dynamin, synaptotagmin- were unmodified, in line with the in vivo evidence that presynaptic proteins are not equally affected in AD brains [80, 81, 82]. Conversely, the expression level of two major postsynaptic proteins -N-Methyl-D-aspartate (NMDA) Receptor subunit NR1 and postsynaptic density protein 95 (PSD95) - did not change or even significantly increased, likely due to reactive/compensatory mechanisms reminiscent of those occurring in vivo during progression of AD pathology [83, 84]. Other postsynaptic proteins, such as NMDAR2A/B and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) Receptor subunit GluR1/5, were also unaffected throughout our experimental conditions (data not shown). No significant alterations in the intracellular amount of not-synaptic proteins located in different subcellular compartments- including specific markers of trans-Golgi network and endoplasmic reticulum such as golgin-97 and calnexin- were contextually found.
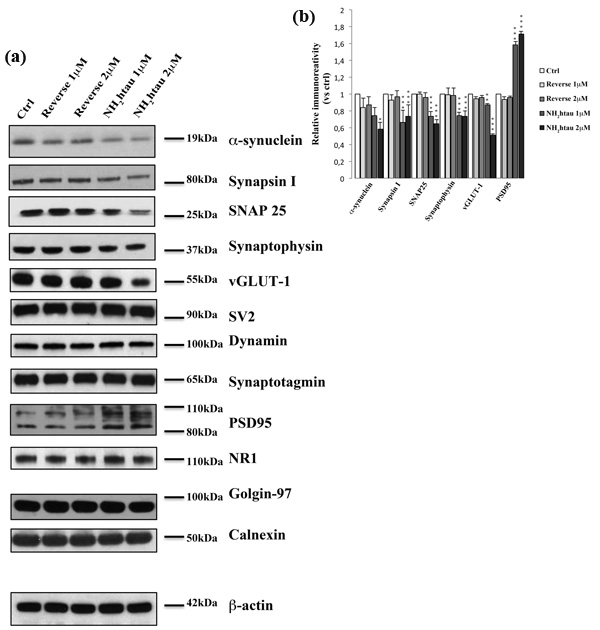
Figure 5: Long-term application of NH2htau induces a marked and selective loss of exocytotic presynaptic vesicles proteins in cultured hippocampal neurons. a.-b. Western blotting analysis (n=12) was carried out on equal amounts of total protein extract (40µg) from mature hippocampal primary neurons (DIV15) exposed for 48h to increasing subtoxic concentration (1-2µM) of NH2htau and its reverse control sequence. Immunoblots (a) were probed with antibodies against several presynaptic- (α-synuclein, synapsin I, synaptosomal-associated protein 25 SNAP-25, synaptophysin, vesicular glutamate transporter 1 vGLUT1 , synaptic vesicle protein 2 SV2, dynamin, synaptotagmin) and post-synaptic markers (N-Methyl-D-aspartate NMDA Receptor Subunit NR1, postsynaptic density protein 95 PSD95) and against not-synaptic proteins located in trans-Golgi network and endoplasmic reticulum (golgin-97 and calnexin). Cropped representative WB are shown. Densitometric quantification of immunoreactivity levels (b) was calculated by normalizing the values on the β-actin intensity and expressed as ratio respect to corresponding ctrl values.Values are means of at least nine independent experiments and statistically significant differences were calculated by one-way ANOVA followed by Bonferroni post-hoc test (*p < 0,05; **p<0,01; ***p<0,0001 versus untreated ctrl).
Furthermore morphological quantitative studies (Figure 6a-6b-6c-Supplementary Figure 4), in addition to confirming the loss of selective pre-synaptic proteins detected in above-mentioned biochemical analyses, revealed that a concomitant disruption of cytoskeleton took place in NH2htau-treated hippocampal cultures which displayed a simplification in network of neuronal processes along with an evident corruption/diminution in array of microtubule track (length and assembly). A pronounced decrease in immunoreactivity of MAP-2, a microtubule-associated protein largely used to trace the integrity of the neuritic network, was clearly evident in NH2htau-treated neuronal cultures when compared to control and reverse groups (one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001) in concomitance with a marked drop in the density of puncta positive for synaptophysin, a specific marker for presynaptic membrane vesicles (one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001) (Figure 6a, Supplementary Figure 4C-4D). The loss in MAP-2 staining turned out to be mostly localized to neurites of thin calibers (Figure 6A, arrows) which appeared distorted, fragmented and decorated with interspersed bead-like varicosities whereas the larger processes (Figure 6a, arrowheads) were still present and appeared uninjured. Moreover, after staining of neuron-specific βIII-tubulin, NH2htau-treated neuronal cultures showed in comparison to control and reverse groups an evident neuritic dystrophy (one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001) involving both high and low calibers processes (Figure 6b, arrows and arrowheads respectively, Supplementary Figure 4A). Destabilization and reduction in length of cellular microtubules occurred in concomitance to a net decrease in dotted labeling for α-synuclein(one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001, Supplementary Figure 4B), a presynaptic protein which is known to be also severely affected in several neurodegenerative tauopathies including AD [85]. Besides, a significant reduction in density of COX-1- positive mitochondria was also clearly appreciable in NH2htau-exposed neuronal cultures (one-way repeated-measures ANOVA followed by Bonferroni post-hoc test ***p <0, 0001, Supplementary Figure 4E-4F), just resembling the retrograde degeneration or “dying-back neuronal death” as detectable at prodromal AD stages [86, 87, 88]. In particular, mitochondria co-located (Figure 6c, asterisks in reverse-treated and control groups) or juxtaposed to synaptic sites (Figure 6c, arrowheads and arrows in opposition) appeared mainly affected in primary cultures upon long-term treatment with extracellular NH2htau. In line with previous findings reporting a causal relationship between tau malfunction, depletion of synaptic mitochondria and loss of synaptic markers [89], the impaired trafficking of these organelles which were likely no longer transported along compromised axonal projections towards terminal ends [90] occurred along with progressive and delayed in vitro presynaptic deterioration, as displayed by contextual decline in synapsin-I immunoreactivity. Importantly, in spite of these neuritic and synaptic degenerative alterations, only very few neurons displayed an evident nuclear heterochromatin, indicating that most of these pathological changes occurred before an overt neuronal death (Supplementary Figure 3). Finally, no changes in global cellular integrity/viability (Supplementary Figure 3) were found in untreated and reverse-treated controls used at the same experimental conditions (concentration and time incubation), although we detected a slight variability in the latter experimental group in agreement with previous findings reporting a quantifiable but not statistically-significant interfering response in hippocampal neurons exposed to other backward-reading sequence peptides such as the reverse Aβ peptide [91].
Taken together, these studies suggest that: (i) chronic exposure of mature hippocampal primary neurons to low and sublethal doses of extracellular NH2htau recapitulates important features of in vivo pre-symptomatic stages of AD neuropathology resembling the dying-back mechanisms of cell degeneration ; (ii) NH2htau -but not its reverse sequence- is able to specifically interfere with structural stability of presynaptic terminals only after chronic treatment of cultures (48-72h) in concomitance with pronounced perturbations in number of resident mitochondria and/or cytoskeleton organization.
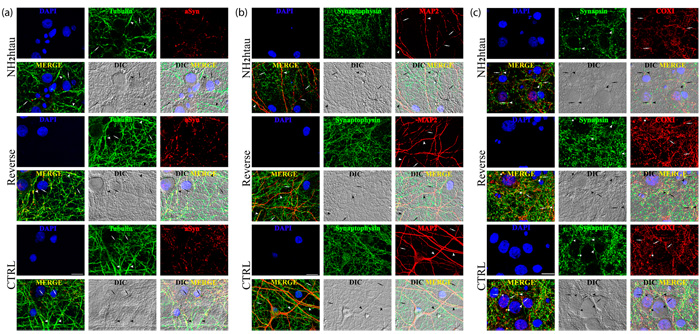
Figure 6: Distortion of the dendritic tree, microtubule breakdown and mitochondria loss occur in concomitance with decline of presynaptic proteins density in living hippocampal neurons chronically exposed to NH2htau. a.-b.-c. Confocal microscopy analysis of double immunofluorescence carried out on mature hippocampal primary neurons (DIV15) exposed for 48h to NH2htau and its reverse control sequence (1µM). Merge images show the overlay of the three fluorescence channels, Differential Interference Contrast (DIC; gray channel) enables the visualization of the neuritic network, DIC Merge is the composition of the three fluorescence and of the DIC channels. (a): presynaptic synaptophysin (green channel) and dendritic MAP-2 (red channel). Nuclei (blue) were stained with Hoechst 33258 (0.5 mg/ml). Arrowheads and arrows point to MAP2- positive neurites of larger and smaller caliber, respectively. (b): presynaptic α-synuclein (green channel) and neuron-specific cytoskeletal beta III tubulin (red channel). Arrowheads and arrows point to beta III tubulin-positive neurites of larger and smaller caliber, respectively. (c): presynaptic synapsin I (green channel) and mitochondrial marker COX I (red channel). Arrowheads point to synapsin I-labeled presynaptic spots and arrows point to COX I -positive mitochondrial structures. In the merge, DIC and DIC-Merge channels, arrowheads and arrows appear in opposition to give evidence to mitochondria resident at juxstaposed presynaptic sites. Asterisks mark typical punctuate structures immunoreactive for both synapsin I and COX I (yellow dots) representing mitochondria which are localized to presynaptic sites (i.e.synaptic mitochondria). Note the loss of double-stained synapsin I/COX I puncta and the decrease of juxstaposed presynaptic sites/mitochondria in the NH2htau-treated cultures. Images are representative of at least three independent experiments. Scale bar: A=20 µm ;B-C=10 µm.
NH2htau peptide shows a higher propensity to assume less compacted conformers than its reverse counterpart: conformational flexibility may account for their divergent in vitro biological effects
Proteolytic cleavage of tau alters its structure, functional capacity and propensity to aggregation [1]. Nevertheless, given their intrinsically flexible nature, tau and its truncated forms do not retain a fixed 3D structure but fluctuate among a large number of different configurations and, consequently, relative structural studies turn out to be problematic and cannot be performed by means of standard techniques, such as X-ray diffraction and electron microscopy [92, 93, 94, 95]. In this context, small angle X-ray scattering (SAXS) is one of the few biophysic approaches which is able to provide detailed structural information on this protein, allowing a quantitative characterization of its conformational polydispersity [93, 94, 95]. Accordingly, the SAXS method has been widely employed in recent years to study the overall structure of both full length tau and its truncated forms [95, 96, 97, 98, 99, 100, 101] and also to characterize other neuropathologically-relevant small peptides composed of only few tens of residues such as the neurotoxic Aβ1-42 (42 aminoacids) [102] and the chemically unfolded Angiotensin II (8 aminoacids) [103]. However, at variance with C-terminal fragments that have been extensively studied [95, 99, 104], very little is known about NH2-terminal fragments of tau protein whose structural characterization can provide a better understanding of their potent neurotoxic role [13, 33, 34, 105].
Here, we used SAXS to investigate the overall conformation of the NH2htau and its reverse sequence peptide. Figure 7a-7b displays the processed scattering curves I(s) of two measured tau constructs with both profiles appearing featureless, as expected for a flexible and unstructured peptide. Their flexible nature was also confirmed by the Kratky plot (s2I(s) as a function of s) which is traditionally employed to qualitatively identify disordered protein states (Figure 7c-7d, for NH2htau and its reverse respectively ) [92, 93, 95, 97, 98, 99, 106]. As expected, Kratky plots (Figure 7c-7d) were not bell-shaped with a clearly defined maximum. For both tau peptides, s2I(s) monotonously increased up to reach a plateau at high s values -a behavior which resembleed that of an ideal Gaussian chain [106]- further confirming their unstructured nature. The gyration radius RG of NH2htau and its reverse peptide were then estimated by using the Guinier approximation -according to which the scattering intensity has a Gaussian shape at small s (s<1.3/RG) [92, 93, 94, 95, 97, 98, 99, 106]- and the calculated Guinier plot (lnI(s) vs s2) is reported in Figure 7e-7f respectively. As shown by a linear fit of the data, we obtained RG = 1.32±0.02 nm for NH2htau26-44 and 1.23±0.02 nm for its reverse sequence peptide. Similar results were found by using the Debye’s approximation (data not shown) and the RG values are summarized in Table 2.
An important metric holding for unstructured proteins and peptides is given by the Flory equation that correlates RG with the length of the peptides [103]:
RG = R0Nν (eq. 1)
where N is the number of aminoacids, R0 is a constant that depends on the persistence length of the chain and ν = 1/2 for a Gaussian chain. For R0 = 2.54±0.01 nm and v = 0.598±0.028, Eq. 1 well describes the behavior of unfolded proteins and peptides ranging between 16 and 549 residues [103]. By using eq. 1, a theoretical value of RG = 1.18±0.03 nm could be estimated for both peptides and this value was consistent within one standard deviation with that we measured for the reverse sequence, again endorsing its flexible and disordered nature. Conversely, NH2htau showed a slightly higher RG than that expected for a Gaussian chain, being consistent with the theoretical values within three standard errors.
An independent experimental approach to probe the flexible nature of the two peptides was obtained by comparing the measured RG with the average hydrodynamic radius RH [107, 108]. In view of these considerations, we measured the hydrodynamic radius of NH2htau and its reverse sequence by dynamic light scattering (DLS), at the same temperature and concentration conditions used in SAXS experiment. In Figure 7 we report the number-weighted hydrodynamic radius distributions (PN (r) ) of NH2htau (Figure 7g) and its reverse sequence (Figure 7h) which were computed by averaging a few hundreds of independent PN(r) functions. As shown (Figure 7g-7h), distributions were monodisperse confirming that the two peptides were of high purity and did not contain significant amounts of aggregates. An average number-weighted hydrodynamic radius (RH )N = 0.95±0.05 nm (eq. 4) and (RH )N = 0.94±0.05 nm were measured for NH2htau and its reverse sequence, respectively. The stability of the two peptides was also assessed by monitoring (RH )N over a time of 12 hours (Figure 7g-7h lower panels). Interestingly and in agreement with data shown in Figure 1f, (RH )N remained constant during 12 hr incubation confirming thus that the large part of species in solution was in monomeric form.
A RG/RH ratio of 1.4±0.1 and 1.3±0.1 was obtained for NH2htau and its reverse sequence, respectively. As reported, these values were consistent with the value for an ideal Gaussian chain as expected for a highly denaturated protein or an intrinsically disordered protein in their Θ-state [107]. At the Θ-point*, chain-chain and chain-solvent interactions balanced each other such that the polymer was at a critical point, at which the thermodynamic phase boundaries disappeared. Collectively, these biophysical data further corroborate the highly flexible nature of the two peptides and are in close agreement with our CD experiments showing that the only structure detected in a wide pH range (pH 4-11) is random coil (Figure 1a).
To gain better insights into the overall structure of the two analyzed peptides, we fitted their experimental curves by using the Ensemble Optimization Methods (EOM) implemented in the software package ATSAS [92, 93, 94, 95], which has been widely used to characterize many intrinsically disordered proteins, including the full length tau and its truncated forms [95, 97, 98, 99] and the neurotoxic Aβ 1-42 peptide [102]. The EOM method -starting from the aminoacidic sequence- generates a pool of models spanning the entire chain’s conformational space and then it selects from that pool -by an iterative genetic algorithm- the most probable ensemble of conformations which best fit the experimental data. The RG distributions of the starting pool of conformations (gray shaded curve) as well the selected ensemble (open square curve) for both NH2htau and its reverse sequence peptide are reported in Figure 8A (a-b, respectively). As shown, the selected ensembles matched the experimental curves (Figure 7a-7b; continuous red lines). Notably, in both cases, the selected ensembles did not span the entire available conformational space but only two well-defined regions peaked around 1 and 1.5 nm, a behavior which was confirmed by generating four independent starting pools of different numerosity (Supplementary Figure 5). The selected ensembles clearly demonstrated that the reverse sequence peptide had a higher tendency to populate more compacted conformational states in comparison to NH2htau (Figure 8A a-b) and, relevantly, this propensity was consistent with the higher RG value of NH2htau obtained in Figure 7e-7f. A quantitative estimation of this trend can be provided by the A1, 5/A1 ratio, a parameter which was obtained by dividing the area of the peak at high RG by that of the peak at low RG value. To this point, we measured a value of A1, 5/A1 = 3.2±0.2 for NH2htau- and of A1, 5/A1 = 1.5±0.2 for its reverse counterpart (errors correspond to the weighted standard deviation calculated over the four different ensembles reported in Supplementary Figure 5). Importantly, the A1, 5/A1 ratio was found to be significantly different between the two analyzed peptides, further supporting the different sampling of the phase space which may underlie their different biological in vitro effect. Remarkably, it has been largely accepted that the pathogenic role of unstructured proteins and peptides cannot be related to misfolding of given fixed 3D structures, as in the case of globular protein, but it is more likely to be strictly correlated to the their different capability to fluctuate among different conformations which can be significantly altered in pathological conditions [43] .
Table 2: Peptides concentration as measured by using the Guinier’s approximation (first column); gyration radius RG (second column) and hydrodynamic radius (third column) as measured by SAXS and DLS; RG/RH ratio obtained by columns 2 and 3 (4th column); Calculated RG value for an ideal Gaussian Chain according to eq. 1 (5th coulmn).
Peptide |
Concentration mg/ml |
RG (nm) Guinier |
RH (nm) DLS |
RG /RH |
RG (nm) RG =R0Nν |
RG (nm) EOM |
A0.155/A0.97 |
NH2htau |
2.5±0.1 |
1.32±0.03 |
0.95±0.05 |
1.4±0.1 |
RG=1.18±0.06 |
1.31±0.05 |
3.1±0.2 |
Reverse |
2.4±0.1 |
1.23±0.03 |
0.94±0.05 |
1.3±0.1 |
RG=1.18±0.06 |
1.21±0.05 |
1.5±0.2 |
RG value computed by using the Ensable Optimization Method EOM (6th coulmn); A1.5/A1 values obtained by dividing the area of the peak at high RG by that of the peak at low RG value in Figure 8A.
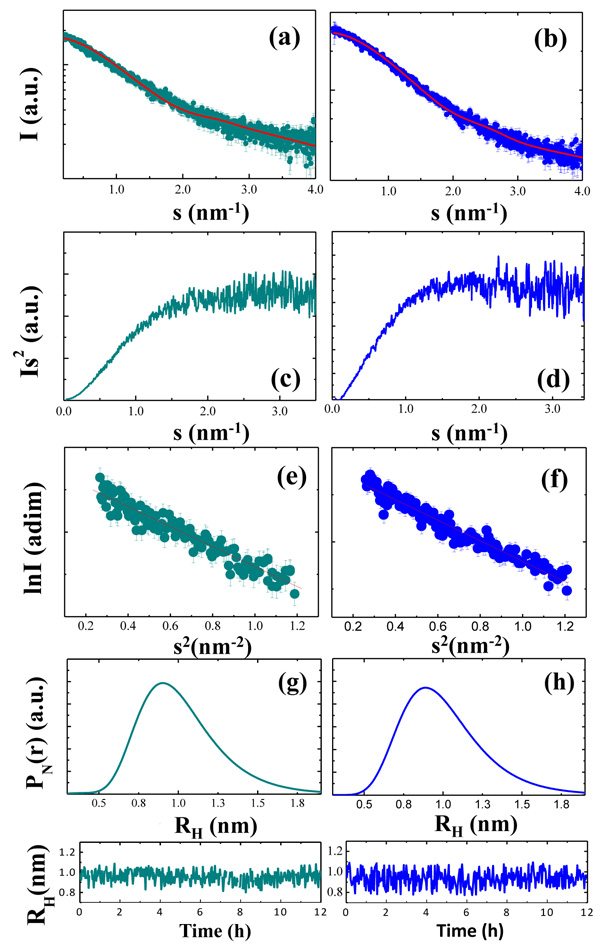
Figure 7: NH2htau is more extended than its reverse counterpart, hinting at different conformational flexibility. From top to bottom of the panel are reported: scattering profiles of NH2htau (a) and its reverse sequence (b) and the red continuous lines indicate the EOM fit of the experimental data; Kratky plots of the two peptides (c, d for NH2htau and its reverse control sequence, respectively); Guinier plot of the two peptides (e, f for NH2htau (a) and its reverse sequence, respectively); Number-weighted hydrodynamic radius distributions PN(r) (g, h for NH2htau and its reverse control sequence, respectively); time evolution of the average number-weighted hydrodynamic radius (lower panels of fig g-h for NH2htau and its reverse sequence, respectively).
Finally, Figure 8B displays four contact maps (Cα-Cα distance expressed in Ångström between all pairs of residues) of four highly probable conformations, namely: a highly probable NH2htau/reverse sequence conformation extracted from the peak at low RG values (Figure 8B a-c) and high RG values (Figure 8B b-d). A cut-off distance of 14 Å was used in these analyses. In Figure 8B a-c, we highlighted the occurrence of a long-range contact between the region entailing residues 15−19 and the region entailing residues 1−5. Furthermore, in the figure insets, a snapshot of the corresponding configuration is reported. It is worth stressing here that these conformers are not meant to provide a statistically significant description of the overall conformations of peptide. Conversely, they provide an idea of what might be the shape of a compacted and an extended conformer.
Altogether and in agreement with CD spectra and ThT fluorescence intensity measurements (Figure 1a-1b-1c-1d-1e), our SAXS analyses confirm that NH2htau clearly displays a disordered and monodispersed nature under the current experimental conditions. Furthermore, these results demonstrate that -in contrast to its reverse sequence counterpart- the NH2htau is more prone to adopt a specific subsets of “open” conformations - likely by exposing important surface residues essential for its functional in vivo interaction with specific biological partners- which might account for their selective and divergent biological effects. Finally, the present data will be in aid of future immunotherapeutic interventions [21] providing new structural information on the 18-residue domain located in N-terminal projection of human tau (26-44epitope) which is the minimal biologically active moiety of longer secreted 20-22kDa pathologic peptide [27, 34] .
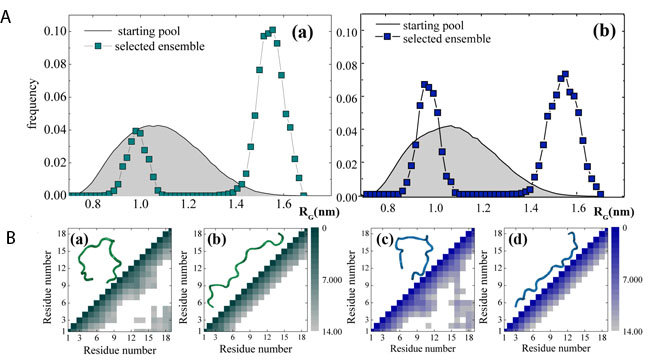
Figure 8: Ensembles of structures populated by NH2htau in aqueous solution differ from those by its reverse sequence. A. EOM starting pool of conformations (gray shaded curves) and selected ensembles (filled square) for NH2htau (a) and its reverse counterpart (b). B. contact maps (Cα–Cα distance between all pairs of residues) of four highly probable conformations, namely: a highly probable NH2htau/reverse sequence conformation extracted from the peak at low RG values (a-c) and high RG value (b-d). Distances between different residues are expressed in Å (a cut-off distance at 14 Å was used). In the figure insets, a snapshot of the corresponding configuration is reported.
Discussion
Truncation at N-terminal domain of tau plays an important role in both neurodegeneration and cognitive decline occurring in all brain human tauopathies, including the most prevalent Alzheimer’s disease (AD) [109]. Tau cleavage has been largely accepted to critically contribute to pathogenesis and progression of these devastating disorders not only by promoting misfolding /aggregation of protein but also by releasing soluble toxic fragments which induce neurodegeneration in a way independent of aggregation [110). Considered that: (i) a better understanding of early, asymptomatic and possible reversible states in progression of neuropathology of these illnesses could help to plan preventive and then more effective disease-modifying therapeutic strategies [111]; (ii) the extension of tau neuropathology more closely correlates with dementia status, being a better predictor of cognitive performance than Aβ deposition in any region of the brain [112]; (iii) immunotherapy-based in vivo Aβ reduction has been proved to have only a limited success [4, 5] it has been suggested that a greater clinical efficacy may be achieved by clearing extracellular, soluble toxic tau species in the earlier stages of the disease when cognitive impairments is still not evident [5]. In this regards, passive immunization targeting the N-terminally truncated forms of tau is being currently pursued in phase I clinical trials [113] based on the findings that multiple fragments containing NH2/mid-region of human protein -but not its C-terminally cleaved and full length isoform(s)- are mostly detected in CSF from AD patients [9, 10, 11, 12] and in conditioned media from AD patient-derived induced pluripotent stem cells (iPSC) cortical neurons [13, 14].
Here we show that low, sublethal doses of soluble and extracellular-added human NH2tau 26-44 fragment (i.e.NH2htau) - which is the minimal biologically active moiety of neurotoxic 20-22kDa parental peptide accumulating in vivo in AD presynaptic terminals and secreted into extracellular parenchyma [24, 25, 28, 33, 34]- are able to impact on normal synaptic function(s) by acutely interfering with depolarization-evoked glutamate release from purified hippocampal nerve terminals. Significant reduction in presynaptic neurotransmitter exocytosis is paralleled by local alteration in peak amplitude and kinetic parameters of Ca2+ transients which concomitantly take place in K+-stimulated isolated synaptosomal preparations. Biochemical and morphological changes, such as selective reduction in presynaptic proteins along with marked neuritic dystrophy and loss of synaptic mitochondria which are classically detected in the pre-symptomatic neuropathologic stages of human tauopathies, are found in in vitro neuronal cultures only after chronic incubation with extracellular NH2htau and prior to overt cell death. The specificity of these results is further supported by the lack of any significant biological effect contextually shown by application of reverse peptide which behaves as inactive control in all the above-summarized analyses, likely due to its poor conformational flexibility which makes it unable to dynamically perturb lipid membranes in contrast to NH2htau. This evidence is biologically relevant to human pathology and important in terms of translational outcome, in view the fact that a population of NH2-terminal truncated fragments of tau protein -including our peptide [21, 27, 28]- is mainly present in CSF from human AD patients and secreted from cryopreserved synaptosomes following depolarizing stimulus [9, 28]. Importantly, in support of a recent report elucidating the beneficial role sustained by the immunodepletion of N-terminal projection of tau (residues 6-18) in improving memory deficits in 3XTgAD mice [20], these results hopefully prospect that passive immunization with the our newly-developed 12A12 monoclonal antibody targeting the N-terminal sequence of human protein encompassing the 26-44 aminoacidic stretch could actually represent an effective therapeutic opportunity for AD and other tauopathies. In addition, the observation that the primary and earlier target of action of pathogenic extracellular NH2htau appears to be the functional release of neurotransmitter from nerve terminal ends -which is followed by delayed mechanisms of cell degeneration- are in agreement with previous observations reporting that synaptic impairment precedes the tardive cell loss in patients and in experimental models of tauopathies, including AD [114]. Finally, the findings that pathogenetic NH2-truncated tau is capable of negatively affecting the presynaptic glutamate release from isolated synaptosomes in Ca2+ -dependent manner are also in line with previous electrophysiological, behavioral, biochemical and morphological evidence showing that mutated htau -in addition to its classical postsynaptic actions [17] and prior to frank neuronal loss- is also able to induce an early synaptic dysfunction by means of presynaptic mechanisms including changes in Ca2+ homeostasis, altered probability of neurotransmitter release, ultrastructural abnormalities and reduced expression of presynaptic markers [115, 116, 117, 118, 119, 120, 121].
Extracellular NH2-truncated tau disrupts Ca2+- dependent glutamate release at presynaptic terminals: potential pathomechanism(s) and relevance to neurodegeneration
A causal association between intracellular accumulation of pathological tau species and dysregulation of calcium homeostasis in the brains of AD patients and in cellular and animal models has been already described [55, 56]. As shown by functional analyses carried out on purified synaptosomal preparations (Figures 3-4), in the present study we report that the secreted and pathologically AD-relevant NH2htau fragment from the extracellular milieu limits the depolarization-evoked glutamate release in a potent (at nanomolar concentrations) and rapid manner (within few minutes), likely acting on regulation of intracellular Ca2+ dynamics at the level of stimulus-exocytosis cascade. To this point, although several mechanisms should be taken into account, we favor the hypothesis of an indirect effect on intrinsic viscosity of lipid bilayers of neuronal plasma membrane leading to a plausible partitioning/diffusion of the (Cav)2.1 and (Cav)2.2 voltage-gated calcium channels (VDDC, P/Qtype and N-type respectively) which are known to be located into lipid-rafts and responsible for coupling of neuronal depolarization with neurotransmitter release at the level of presynaptic terminals [122]. In this regard, pathological tau has also been reported to accumulate within these cell-surface detergent-resistant microdomains of the plasma membrane in aging Tg2576 transgenic mice and AD brains [123] and the N-terminal projection domain of tau protein associates to lipid-rich rafts of plasma membrane in a phosphorylation-dependent manner [41]. Besides, our biochemical and biophysical results from kinetic studies of evoked Ca2+transient along with Western blotting and DSC analyses (Figures 2-3-4-5) further support the above observations demonstrating that: (i) the dose-dependent impairment of glutamate exocytosis induced by NH2htau attached to surface of isolated nerve endings occurs upstream of Ca2+ dynamics out of the action mechanism of neurotransmitter release induced by external high [KCl] which is known to involve only the depolarization-evoked activation of voltage-dependent Ca2+ channels [57, 59] and not of other ion-channels modulating the synaptic terminal excitability; (ii) treatment of neuronal cultures with NH2htau does not induce per se up to 48h any significant alteration in the protein expression level of some components of cytoskeleton and/or of release machinery which is relevantly involved in controlling the excitatory neurotransmission, excluding the possibility of a direct effect downstream of Ca2+ entry on synaptic vesicle trafficking/ exocytosis apparatus and on axonal transport; (iii) upon its prompt interaction with the surface of biomembrane-mimicking environments, the external application of NH2htau gives rise to a profound phase segregation of lipid bilayer leading to a selective enrichment in more rigid, raft-like microdomains which are largely accepted to be the cellular sites where functional assembly of SNARE (soluble N-ethyl-maleimide-sensitive fusion protein attachment protein receptor) components of vesicular exocytotic machinery with presynaptic P/Q calcium channels occurs [122]. In addition, our preliminary data (not shown) from microscopy visualization of membrane fluidity by means of 2-Dimethylamino-6-lauroylnaphthalene (Laurdan) fluorescence points to a significant perturbation in lipids packing upon short-term exposure of neuronal cultures to NH2htau but not to its reverse control sequence. Interestingly, it has been reported that α-synuclein associates with lipid-rafts microdomains [52] in a similar way of N-terminal end of tau [16] and that, when extracellular-applied in its monomeric form, is able to early perturb calcium homeostasis by affecting the portioning of several cell surface-associated proteins [124, 125]. Therefore consistent with our immunofluorescence stainings (Figure 2), it would be reasonable to hypothesize that secreted NH2htau might change the membrane fluidity/composition upon its prompt interaction with presynaptic boutons and alter thus the distribution and/or insertion of Ca2+ channels into sorting /signaling lipid platforms as well as the association of these channels with appropriate effector proteins, thus inhibiting their activities [126]. Of note, biophysical parameters (bending, fluidity and thickness) underlying the reciprocal spatial organization between lipids membranes and surface-associated proteins have been largely accepted to actively participate in presynaptic functions and to drive the neurotransmitter release in neurons via regulation of proteins trafficking and activity. In line with our DSC analyses, previous studies have also proved that one of the initial steps in neurotoxic biological effects evoked by rather low concentrations of Aβ stems from a “membrane-disordering effect” caused by alteration in acylchain layer and, thus, in fluidity of cell membranes [127]. Additional studies are required to test this possibility and further experiments are under current investigation to better address the presynaptic pathway(s) underlying the membrane-mediated actions of NH2htau in neurons. In relation to the presynaptic Ca2+-dependent coupling between stimulus and neurotransmitter secretion of interest is the fact that, as shown in our correlational studies of Ca2+ imaging and radioactivity-based measures of [3H]D-aspartate release, the peak of K+-evoked elevation in Ca2+ current turns out to be somewhat higher and to decline to a stable plateau more quickly in hippocampal isolated nerve terminals exposed to NH2htau than in other two experimental group (reverse- and untreated controls), resulting in overall decrease of glutamate release (compare Figure 3 to Figure 4). To this point, it’s worth stressing that the strength of glutamatergic synapses depends on both the peak and the kinetic of [Ca2+] transient (duration) near its sensors. It has been largely accepted that depolarization of synaptosomes by elevated KCl initiates a rapid “spike” of Ca2+ current within 1-2 s of stimulation followed by a more extensive slow plateau. The K+ -evoked secretion of glutamate neurotransmitter from synaptic terminals does not correlate with the increase in [Ca2+] bulk but occurs for the large part during its late recover to a plateau, undergoing a biphasic release which may reflect a dual localization of releasable vesicles at the active zone and in the cytoplasm [128]. Therefore, we argue that greater but shorter signal of activity-dependent Ca2+ transient could account for pathophysiological deficits induced by extracellular NH2htau on neurotransmission at the presynaptic level (Figure 3j-3k). Furthermore our findings on acute and potent inhibitory action exerted by pathogenetic NH2-truncated tau on glutamate release from isolated synaptosomes (Figure 4i) are also in line with the emerging concept that the unbalance in network activity occurring in vivo in prodromal AD subjects is more likely to reflect a decreased capacity of cerebral neurons to cope with existing glutamate level that becomes toxic at concentration that normally shows no harmful effect. Relevantly, magnetic resonance spectroscopy has revealed a significantly lower level of glutamate in AD brains compared to healthy controls and patients with mild cognitive impairment [129]. In line, soluble pathological tau species have been recently proved to be able to change the neuronal electrical properties in the rTg4510 (mutation P301L) mouse model of tauopathy, provoking a diminution in global neuronal activity with slower spontaneous oscillations and a reduced firing rates, prior to significant cell death or synapse loss [130].
Pathological molecular/structural determinants at N-terminal extremity of human tau protein: novel opportunities for tau-based immunotherapy in AD and other tauopathies
In addition to aberrant calcium homeostasis and imbalance in neurotransmitter release, selective and regional loss of presynaptic terminals, neurite retraction, breakdown of microtubules, prominent loss of mitochondria which specifically reside at nerve endings are also discernable in transgenic animal model of tauopathy and in AD patients by sensitizing neurons to “dying back” prior to overt cell death [30, 80, 81, 131, 132, 133]. Consistently, our in vitro biochemical data from Western blotting and immunofluorescence analyses (Figures 5-6-Supplementary Figure 4) show a dose-dependent diminution in selective markers involved in trafficking/priming of presynaptic vesicles along with a pronounced degeneration of neuronal processes and reduction in mitochondrial density only after 48h incubation time with low doses of NH2htau, in absence of any significant global change in cell viability (Supplementary Figure 3) which conversely appeared evident later on (72-96h). These findings are relevant because synaptotoxicity, axonopathy and mitochondrial abnormalities induced in vitro by chronic and sublethal exposure of mature hippocampal primary neurons to NH2htau recapitulate a few important in vivo biochemical and morphological changes that closely resemble the pre-symptomatic neuropathologic stages of human tauopathies. To the point and in line with our in vivo previous results [26], recent findings have endorsed a crucial role of tau dysfunction in affecting biology and density of mitochondria located at terminal ends (i.e. synaptic mitochondria) [89]. Proteomic analyses have also shown that presynaptic compartment, along with a significant impairment in dynamic stability of microtubules and reduction in number of synaptic vesicles, are preferentially deregulated in vivo into transgenic mice expressing another toxic human truncated tau fragment (aa 151-391) [134]. In addition, the A152T tau mutation -a tauopathies risk factor which lies within the N-terminal projection region of protein not involved in interaction with microtubules - has been recently shown to perturb presynaptic neurotransmission and mitochondrial distribution in C.elegans worms, in absence of any accumulation of insoluble intracellular aggregates [135]. Therefore and in a similar way of extracellular-added monomeric Aβ 1-42 whose continuous exposure (i.e. without replacing the culture media up to 72-96h) to cortical primary neurons is required to achieve maximal neurotoxicity due to its sequential binding to membranes, a threshold level of membrane-bound NH2htau could be needed to trigger a full-blown neuronal cell death. Alternatively, neurotoxicity initiated by binding of extracellular-added monomeric NH2htau to membranes could depend on the activation of long-term intracellular signaling transduction pathways which affect one or more second messengers controlling, directly or indirectly, the neuronal bioenergetics, cytoarchitecture and synapses integrity. Finally and in agreement with our DSC kinetic analyses (Figure 2) displaying that NH2htau is able to interact with the deep hydrocarbon region of lipid bilayer only after longer incubation times (48-72h), it’s also hypothesizable that -following its initial adsorption on the external side of plasma membrane- a residual amount of this peptide could be later internalized and even undergo an intracellular misfolding which, in turn, causes the delayed loss of synapses, neurite retraction and mitochondrial deficit. Consistent with these findings, monomeric exogenous tau has been demonstrated to be up-taken by neurons [136, 137] and aggregate into endosomes [136]. Interestingly, high intracellular levels of pathogenetic NH2htau are able to interfere with the mitochondrial biology leading to drop of energy (ATP) and eventually to neuronal death, as we previously reported [138]. Furthermore, quantitative mass spectrometry following co-immunoprecipitation with an antibody just encompassing the extreme N-terminal domain (27-40aa) of tau protein have also revealed that numerous pre-synaptic proteins involved in vesicle docking/fusion -together with a large number of mitochondrial markers- are selectively pulled-down from mice brains [139] .
Concerning a direct role of self-aggregation in affecting synaptic function, it’s also worth mentioning that the oligomerization process of full length or truncated tau species is not essential for internalization, secretion and propagation [13, 22, 62, 136, 140] and that truncation can cause degeneration independently of its aggregation [110]. Our observations from CD spectra, ThT-binding fluorescence intensity measurements and SAXS analysis confirm the unstructured/flexible and monodisperse nature of NH2htau in water environment under current experimental conditions along with EOM study unraveling its intrinsic propensity to adopt specific conformational ensembles which might account for its potent in vitro biological effects (Figures 1, 7, 8). Therefore, although we cannot completely rule out that NH2htau can undergo temporary structural changes [35] as well as membrane-induced conformational transitions [42, 43] facilitating its folding/clustering in amorphous and/or partially structured aggregates over longer periods of times, our findings clearly demonstrate that this N-terminal tau peptide adopts a pathological conformation being certainly in vitro toxic when extracellular-added to neurons in its prevailing monomeric form.
It’s worth pointing out that , in the present work we used concentrations for NH2htau ranging from 1nM to 1µM which are within the physiological range of unbound/free tau [61, 65] and within the range previously used by other authors in assessing the extracellular role of full length tau (62) or its C-terminal [141] and N-terminal [13] active fragments on cell lines and primary neurons respectively. Furthermore, it’s noteworthy that: (i) the activity-dependent secretion of full length tau may occur through synaptic transmission [142] and (ii) the pre-synaptic release of longer 20-22kDa tau fragment occurs following K+ depolarization of AD terminal ends [28]. Therefore, although the amount of extracellular NH2htau required to affect synaptic functions is higher than the diffusive levels of tau found in AD patient CSF or in the conditioned media from neuronal cultures and in interstitial fluids, it’s more likely to assume that the secretion of soluble tau species through a synaptic mechanism would necessarily generate local concentrations at the release site, and likely site of action, which are significantly higher than its circulating levels [13].
Regarding the observation that no significant effect on basal synaptic exocytosis was found out in neurons upon treatment with subtoxic doses of extracellular NH2htau, compelling evidence has proved that the stimuls-independent discharge at the level of the release machinery and the action potential-triggered neurotransmitter release: (i) rely on independent neuronal signal transduction pathways and on divergent presynaptic machineries and/or postsynaptic targets that may even operate in a spatially segregated manner; (ii) are not linked with respect to their relative activities because modulations of spontaneous release are not always accompanied by corresponding changes in evoked exocytosis. Therefore, following binding to plasma membrane and consequent alteration in the lipid bilayer fluidity as we detected by DSC (Figure 2), it’s conceivable that extracellular NH2htau might perturbate the localization and, then, the activation of one or more membrane-associated proteins which are selectivity associated to regulated neurosecretion. The specific effect induced by extracellular NH2htau on the movement/distribution of distinct membrane-associated proteins in lipid rafts and, then, on the differential regulation of evoked versus spontaneous neurotransmitter release, is possible to be dependent on the different strength of association of proteins with membrane bilayer, or on the differential role of lipid raft elements in the maintenance of proteins cluster and in the confinement of distinct proteins in their specific site of localization [125].
Finally it’s also noteworthy that, although the extracellular NH2tau 26-44 fragment (i.e.NH2htau) only comprises 19 amino acid sequence of over 400 amino acids included in full length tau sequence, it represents the minimal biologically active moiety of longer 20-22kDa parental peptide [27, 33, 34]. Therefore, given that (i) molecular characterization of identity of the extracellular toxic tau species is mandatory to design a best-targeted and more effective immunotherapy relying on the specific, epitope-directed and antibody-mediated depletion [4, 5]; (ii) the NH2 26-44aa is critical aminoacidic stretch representing the biologically active moiety of 20-22kDa AD-relevant secreted NH2-truncated tau forms [24, 27, 34], our findings can be particularly relevant for tau physiopathology in the field of AD and other tauopathies, helping to design more beneficial tau-directed and disease-modifying in vivo curative approaches. To this regard, it’s worth noting that passive immunotherapy with HJ 8.5 targeting the N-terminal projection domain of human tau (residues 25-30) has proved to be succesful in inhibiting the transcellular propagation of tau and improving cognitive deficits in P301S tau transgenic mice [143]. In addition, based on the encouraging in vivo results from tauopathy animal models [20, 143, 144], clinical trials with a set of monoclonal tau antibodies binding to and clearing the extracellular and/or intracellular pathological species including the extreme N-terminal region of human protein are currently in progress [5].
Concluding remarks
In summary, the present investigation not only confirms and extends the notion that extracellular tau is per se harmful for neurons [13, 23, 62] but also opens novel and potentially more effective therapeutic opportunities aimed at preventing the early impairment in synaptic plasticity and memory caused by one of the actually secreted [21, 27, 28] and, consequently, pathologically relevant N-terminal truncated species in human tauopathies.
MATERIALs AND METHODS
Chemicals and antibodies
Brain Total Lipid Extract (TLBE), 1, 2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1, 2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS) 1, 2-palmitoyl-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1, 2-palmitoyl-oleoyl-sn-glycero-3-phosphoserine (POPS) were purchased from Avanti Polar Inc. (Alabaster, AL). All N-fluorenylmethoxycarbonyl (Fmoc)-protected amino acids, Fmoc-NH-(PEG)11-COOH, 2-(1-H-benzotriazole-1-yl)-1, 1, 3, 3-tetramethyluronium tatrafluoroborate (TBTU), and NovaSyn TGR resin were obtained from Novabiochem (Switzerland); N, N-diisopropyl-ethylamine (DIEA), N-hydroxybenzotriazole (HOBt), DMF (peptide-synthesis-grade), triisopropylsilane (TIS), trifluoroacetic acid (TFA), Thioflavin T (ThT), Sodium dodecyl sulfate (SDS), 2, 2, 2-Trifluoroethanol (TFE), N, N-diisopropylethylamine (DIEA), N, N-dimethylformamide, piperidine, triisopropylsilane (TIS), trifluoroacetic acid (TFA) and all salts used for buffer preparation were purchased from Sigma-Aldrich (St.Louis, MO) with a purity of 99%. SynaptoRed C2 (FM4-64) 70021, SynaptoGreen C4 (FM1-43) 70022 were from Biotium (Hayward, CA); Calcium Indicator (Fluo-3AM) F1242 was from Thermo Fisher Scientific (Massachusetts, MA).
The following antibodies were used: β-III tubulin antibody rabbit ab18207 Abcam; β-III tubulin antibody mouse (clone 2G10) T8578 Sigma-Aldrich; β-actin mouse S3062 Sigma-Aldrich; Synapsin I antibody rabbit AB1543P Millipore Corporation; PSD95 antibody (clone 7E3-1B8) mouse MAB1598 Millipore; PSD95antibody (clone: 6G6-1C9) mouse ADI-VAM-PS002 EnzoLife Science; synaptophysin antibody mouse sc-17750 Santa Cruz; Calnexin antibody ( clone C-20) goat sc-6465 Santa Cruz ; MAP-2 antibody mouse MAB3418 Millipore; dynamin antibody mouse BD Transduction Laboraories 610246; GAPDH antibody (clone GAPDH-71.1) G8795Sigma-Aldrich ; SNAP25 antibody mouse (clone SMI81) Biolegend 836301; SV2A antibody (clone E-8) mouse sc-376234 Santa Cruz; α-synuclein antibody (clone 42) mouse 610786 BD Transduction Laboratories; α-synuclein antibody rabbit S3062 Sigma-Aldrich; synaptotagmin antibody (clone ASV48) mouse SYA-148 Stressgen; anti complex IV subunit I MTCO1 (clone 1D6E1A8) ab14705 MitoSciences; NMDAζ1 antibody (C-20) goat sc-1467 Santa Cruz; vGLUT1antibody rabbit 135 302 Synaptic System ; Anti- caspase 3cleaved (active) form antibody rabbit AB3623 Millipore; golgin 97 (E-16) antibody goat sc-74632 Santa Cruz.
Synthesis, purification, labeling and preparation of tau peptides
NH2 htau 26-44 and its reverse sequence control peptide were synthesized and purified using reverse-phase HPLC (D.B.A., Milan, Italy). Peptides mass and purity (>99%) were confirmed by reverse-phase HPLC and electrospray/ion trap mass spectrometry. A lyophilized powder of peptides was dissolved in at a stock concentration of 1 mM by brief stirring. Aliquots were stored at -80°C prior to use. The working solution was diluted with PBS or ultrapure water at a concentration of 50 μM at 4°C and used immediately. Peptide preparations were analyzed by Sypro Ruby protein staining and SDS-PAGE Western blot for each experiment.
NH2 htau 26-44 :NH2 QGGYTMHQDQEGDTD AGLK- COOH
Reverse sequence control peptide :NH2 KLGADTDGEQDQHM TYGGQ- COOH
FITC- conjugated NH2htau 26-44 was synthesized and purified (>99%) using reverse-phase HPLC (Biosynthesis, U.S.A.).
Generation of the N-terminal tau 12A12 antibody (26-36aa)
Affinity-purified mouse monoclonal antibody directed against the extreme N-terminal 26-36 aa of human tau protein (12A12) was produced, purified and characterized by Monoclonal Antibodies Core Facility (MACF) at EMBL- Monterotondo, Rome, Italy (Dott.Alan Sawyer).
12A12 was generated by immunizing mice with a peptide of amino acids 26-36 aa of hT40 (D25(NH2 QGGYTMHQDQ-COOH epitopes). The specificity of this mAb (IgG isotype) was verified by Western blot analysis (Figure 1f-1g) and enzyme-linked immunosorbent assay (ELISA) test (95% sensitivity and 90% specificity).
Preparation of artificial biomembrane model system
We used large unilamellar vesicles (LUVs) composed of TLBE or DMPC/DMPS (7/3 molar ratio). Model membranes were prepared as described elsewhere [47]. Briefly, aliquots of lipid stock solutions in chloroform were dried by using a stream of dry nitrogen gas and evaporated under high vacuum to dryness in a round-bottomed flask. To obtain multilamellar vesicles (MLVs), the resulting lipid film was hydrated with an appropriate amount of phosphate buffer (10 mM buffer, 100 mM NaCl, pH = 7.4) and dispersed by vigorous stirring in a water bath. LUVs were obtained by extruding MLVs through polycarbonate filters (pore size = 100 nm, Nuclepore, Pleasanton, CA) mounted in a mini-extruder (Avestin, Ottawa, ON, Canada) fitted with two 0.5 ml Hamilton gastight syringes (Hamilton, Reno, NV). Samples were typically subjected to 23 passes through two filters in tandem and as recommended elsewhere. An odd number of passages were performed to avoid contamination of the sample by vesicles that might not have passed through the filter.
Circular dichroism
The CD spectra were obtained at 25°C (if not differently specified) under a constant flow of N2 on a Jasco J-810 spectropolarimeter equipped with a Peltier thermoelectric type temperature control system. Experimental measurements were conducted in several condition, using 1 cm path length cuvettes. The CD measurements were carried out under a variety of experimental conditions, including different pH levels and membrane-mimicking enviroments (i.e negatively charged SDS, anionic-zwitterionic POPC/POPS LUV and water/TFE mixtures). The CD spectra were recorded in the UV region (190-260 nm) with peptide concentration of 10 µM. CD intensities are expressed as molecular ellipticity.
ThT assay
The kinetics of amyloid fiber formation were measured using the increase of fluorescence emission upon binding of commonly used amyloid specific dye, thioflavine T (ThT). Samples were prepared by adding 5 µL stock solution of tau26-44 or tau44-26 to 100 µL of 10 mM phosphate buffer solution pH 7.4, 100 mM NaCl or to 100 µL of a TLBE 7/3 LUVs solution, containing 60 µM ThT (final peptides concentration was 20.0 µM). Experiments were carried out in Corning 96 well non binding surface plates. Time traces were recorded using a Varioskan plate reader (λecc 440 nm, λem 482 nm) at 37 °C, shaking samples for 10 seconds before each read. Time traces are the average of three measurements.
Differential scanning calorimentry (DSC)
The interaction of NH2htau and reverse control peptide with model membranes was tested by performing DSC experiments. Samples were prepared by adding the proper amount of a stock solution (800 µM) of both tau peptides to 1 ml of LUV DMPC/DMPS 7/3 200 µM. The final peptides concentration was 20 µM. Five independent samples were prepared in order to perform experiments every 24 hours up to 72 hours . All the experiments were collected from 10 to 40 °C, 1 °C/min scanning rate at the pressure of 3 atm using a nanoDSC (TA instrument).
Animals
All protocols involving animals were performed in accordance with the guidelines established by the European Communities Council (Directive 2010/63/EU of 22 September 2010). Experiments involving animals were performed in accordance with the relevant approved guidelines and regulations accepted by the Italian Ministry of Health and approved by the Ethical Committee on animal experiments of EBRI “Rita Levi-Montalcini” Foundation (Rome, Italy).
Synaptosomes preparation
Mouse hippocampal purified synaptosomes were prepared by homogenizing tissue in 10 volumes of 0.32 M sucrose, buffered to pH 7.4 with Tris-(hydroxymethyl)-amino methane [Tris, final concentration (f.c.) 0.01 M] [75]. The homogenate was centrifuged at 1, 000 x g for 5 min and the supernatant was stratified on a discontinuous Percoll gradient (2%, 6%, 10% and 20% v/v in Tris-buffered sucrose) and centrifuged at 33, 500 x g for 5 min. The layer between 10% and 20% Percoll (synaptosomal fraction) was collected and washed by centrifugation. In a set of experiments, the tissue was homogenized in buffered sucrose containing 1 mM 1, 2-bis-(2-aminophenoxy) ethane-N, N, N’, N’, tetra-acetic acid (BAPTA), in order to entrap this agent into subsequently isolated synaptosomes [75]. The synaptosomal pellets were resuspended in a physiological solution with the following composition (mM): NaCl, 140; KCl, 3; MgSO4, 1.2; CaCl2, 1.2; NaH2PO4, 1.2; NaHCO3, 5; HEPES, 10; glucose, 10; pH 7.2-7.4. To ascertain whether fractionated preparations were really enriched in synaptic terminals and free of any contaminations from neuronal perikarya, Western blotting analysis was carried out to check the purity of samples by probing with antibodies against the presynaptic protein synapthophisin and cytosolic GAPDH, as previously reported [27].
Synaptosomal glutamate release
Synaptosomal glutamate release was estimated as previously reported [74, 75, 77] . In details, isolated hippocampal ending nerves were incubated for 45 min at 37°C in a rotary water bath in the presence of [3H]D-aspartate ([3H]D-ASP; a non-metabolizable analogue of glutamate used to label the synaptosomal glutamate-releasing pools used at final concentration 50 nM). Aliquots of synaptosomal suspension (5-10 μg protein) were equally distributed on microporous filters placed at the bottom of a set of parallel superfusion chambers maintained at 37 °C (Superfusion System, Ugo Basile, Comerio, Varese, Italy) [74]. Superfusion was then started with standard medium at a rate of 0.5 ml/min and continued for 48 min. After 39 min of superfusion to equilibrate the system, synaptosomes were transiently (90s) exposed to high K+ containing medium (12 mM, NaCl substituting for an equimolar concentration of KCl), and then fractions were collected according to the following scheme: two 3-min fractions (basal release), one before (t = 36-39 min) and one after (t = 45-48 min), a 6-min fraction (t = 39-45 min; evoked release). When indicated, synaptosomes were superfused with medium in which Ca2+ ions were omitted to prevent any Ca2+-dependent releasing activity.Collected fractions and superfused synaptosomes were measured for radioactivity. The amount of radioactivity released into each superfusate fraction was expressed as percentage of the total radioactivity. The K+-induced overflow was estimated by subtracting the neurotransmitter content the first and the third fractions collected (basal release, b1 and b3) from that in the 6-min fraction collected during and after the depolarization pulse (evoked release, b2). In a set of control experiments performed to evaluate the Ca2+-dependency of the induced [3H]d-ASP release, the superfusion medium was replaced, starting from t = 20 min, with a medium containing 0.1 mM Ca2+ and 500 μM EGTA. In some experiments, synaptosomes were incubated 30 min (15 min before and during [3H]d-Asp labelling) in the presence of 100 μM 1, 2-bis-(2-aminophenoxy)-ethane-N, N, N′, N′-tetraacetic acid, tetraacetoxymethyl ester (BAPTA-AM).
Vesicles exocytosis FM1-43 or FM4-64 assay
Depolarization-dependent FM1-43 or FM4-64 destaining was used to measure synaptic vesicle fusion events with the plasma membrane on intact nerve terminals (i.e., synaptosomes) [59, 71, 72, 73]. Isolated synaptosomal fractions from mature rat primary hippocampal neurons (15DIV) seeded on coverslips were incubated in physiological salt solution (PSS; NaCl 140 mM, glucose 11.5 mM, KCl 5.9 mM, MgCl2 1.4 mM, NaH2PO4 1.2 mM, NaHCO3 5 mM, CaCl2 1.8 mM, HEPES 10 mM) and then with FM1-43 (50 μM) for 2 min, followed by application of high KCl 30 mM to intrasynaptically load the fluorescent dye. After 1 min, synaptic preparations were washed twice to remove non-internalized FM1-43 and placed on an incubation chamber of a time-lapse system composed by an inverted fluorescence microscope (TiE; Nikon, Japan), equipped with an incubation chamber (Okolab), a cooled CCDcamera (Clara;Andor), a Perfect Focus System to avoid z-axis focus fluctuations and a Niss Elements imaging software (Nikon). After 1 min a baseline was established, synaptoneurosomes were incubated (t = 1) with either NH2htau peptide or its reverse sequence control (both 1µM) for additional 5 min and then (t = 6) stimulated by application of 30mM KCl which we previously stated in pilot experiments to be the minimum concentration able to induce robust responses in our samples. Video recordings were performed with a 40×oil objective (N.A 1.4) for at least 10 min and 14 bit images were captured by 200 ms exposure time every 2 s at room temperature (25°C). After fluorescence recordings, one differential interference contrast (DIC;Nomarski) image of the field was taken for visual inspection of global morphological features (data not shown). Video images were analyzed by using the Spot module of the ImarisSuite 7.6® software (Bitplane A.G., Zurich, Switzerland) by semiautomated application of 1.5 μm radius diameter mask on fluorescent puncta. The criteria for fluorescence puncta inclusion in the data analysis were the spherical shape ranging between 0.5-1.5 micron and the stimulation-dependent destaining response. Destaining time courses were generated by normalization of each fluorescence spot trace by using the formula Fi/F0 where F0 is the fluorescence before stimulus addition (average of 10 time points of the image t-stack) and Fi is the fluorescence at each time point. Quantification of FM1-43 exocytosis responses was accomplished by calculating the average percentage of fluorescence loss and a group of control experiments was also contextually run to test the specificity of dye loading and destaining, as following: (i) incubation in Ca2+-free Ethylene Glycol TetraAcetic acid (EGTA)-containing medium drastically reduced both the FM1-43 loading and inhibited the K+ stimulus-induced dye release; (ii) the FM1-43 photobleaching rate was measured in absence of the K+ stimulus. To verify whether NH2htau could induce per se synaptic release, we additionally performed a few control experiments using the same experimental setting conditions with the exception that the NH2htau was incubated with the synaptosomes and the KCl stimulus was substituted by saline addition.
Intracellular calcium imaging in cultured neurons and isolated synaptosomes
Relative changes in the cytosolic Ca2+ concentration were measured using the Ca2+ -sensitive fluorescent dye indicator Fluo-3 AM. Hippocampal primary neurons and isolated hippocampal ending nerves were incubated for 45 min in the dark at room temperature with 2 µM Fluo-3 AM dissolved in 0.02 % pluronic acid (both from Biotium, Hayward, CA) in physiological salt solution (PSS; NaCl 140 mM, glucose 11.5 mM, KCl 5.9 mM, MgCl2 1.4 mM, NaH2PO4 1.2 mM, NaHCO3 5 mM, CaCl2 1.8 mM, HEPES 10 mM). To allow for completion of Fluo-3 AM de-esterification process, cultures were washed and incubated in PSS alone for 10-15 min at 37 °C and then visualized and recorded by using a time-lapse system mounted on an inverted fluorescence microscope (TiE; Nikon, Japan), equipped with: (i) an incubation chamber (Okolab, Italy), (ii) a cooled CCD camera (Clara; Andor), (iii) filters for Differential Interference Contrast (DIC; Nomarski) acquisitions, (iv) Perfect Focus System to avoid z-axis focus fluctuations and a (v) Niss Elements imaging software (Nikon). Video recordings were performed with a 20X objective, for at least 20 min, and 12 bit images were captured by 200 ms exposure time every 2 s at room temperature (25°C). After 1 min a baseline was established, neurons were incubated (t = 1) with either NH2htau peptide or its reverse sequence control (1µM) for additional 5 min and then (t = 6) stimulated by 30 mM KCl addition, which we previously verified in pilot experiments to be the minimum concentration able to induce robust responses on our in vitro cultures. After fluorescence recordings, one DIC image of the fluorescence field was taken for visual inspection of morphological features. Fluorescence intensity changes were calculated as (F-F0)/F0, where F was the fluorescence intensity measured in each frame after stimulation and F0 the basal fluorescence level calculated by averaging the intensity values 20 sec before the peak. Video images and pixel intensities were evaluated afterwards, by using Imaris Suite 7.6® Track module (Bitplane A.G., Zurich, Switzerland) and Image J 1.4 (imagej.nih.gov/ij/) softwares by applying a 1.5 μm radius diameter mask on fluorescent puncta. However, some puncta which visually displayed a faint Fluo-3 staining were not detected by the Track module, owing to a low responses to KCl stimulation, and therefore were not included in the analysis. Inclusion criteria for data analysis were: (i) puncta labeling with Fluo-3, (ii) puncta showing a prompt Ca2+ rise and sustained Ca2+ response (which was more than 20% increase over resting baseline), (iii) puncta returning to fluorescence baseline level. For all calcium signals, amplitude, time to peak, peak to baseline and overall duration of the response were determined. In few control experiments performed to evaluate the source of Ca2+ increase, a medium containing 0.1 mM Ca2+ and 500 μM EGTA was used and no statistically significant fluorescence variation and/or typical curve shape was detected.
Neuronal culture and treatments
Hippocampal neurons were prepared from embryonic day 17-18 (E17/E18) embryos from timed pregnant Wistar rats (Charles River), as we previously reported [34]. In detail, the hippocampus was dissected out in Hanks’ balanced salt solution buffered with HEPES and dissociated via trypsin/EDTA treatment. Cells were plated at 1 x 106 cells on 3.5-cm dishes pre-coated with poly-D-lysine. After 2 days of culturing in neurobasal medium with B-27 supplement and glutamax, cytosine arabinofuranoside was added to reduce glial proliferation. Half of the medium was changed every 3-4 days and mature neuronal cultures were infected at 15 days in vitro, as reported in [34].
Assessment of neuronal viability
Cell viability was quantified by counting the number of intact nuclei, after lysing in detergent-containing solution and by the MTT tetrazolium salt assay, as reported in [26, 33, 34].
Immunofluorescence
Following treatment, neuronal cultures were washed twice with PBS and fixed in 4% (w/v) paraformaldehyde for 15 min at room temperature. Cells were permeabilized with 0.1% (v/v) Triton X-100/PBS pH 7.4 for 4 min at room temperature. Coverslips were saturated with 2% BSA and 10% normal goat serum (NGS) for 3h followed by incubation overnight at 4 °C in a humidified chamber with primary antibodies. Unbound antibody was removed by three washes and bound antibody was detected by incubation with donkey anti-rabbit Alexa 488 (1:300) and donkey anti-mouse rhodamine- conjugated (1:300) secondary antibodies (Invitrogen) at room temperature for 30 min. Nuclei were stained with nuclear marker 4, 6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma, St. Louis, MO, U.S.A.) 1:1000 in PBS for 5 min. Controls were performed by omitting either the primary or the secondary antibody. Images are representative of at least three independent experiments.
Live-fluorescence protocol
Morphological detection of cell-surface NH2htau was performed according to procedure aimed to detect direct interaction of extracellular-added Aβ 1-42 peptide to neuronal membrane lipids rafts with several modifications [145]. In brief, live neuronal cultures were washed twice with chilled PBS 1X , incubated for 10’ with 1µM FITC-conjugated NH2htau (Biosynthesis, USA) at 4°C and then labeled for lipid rafts by means of TRITC- conjugated cholera toxin subunit B (CT-B), according to manual instructions (Vybrant lipid raft labeling kit, Molecular Probes). After three washes with chilled PBS 1X, cells were fixed in 4% (w/v) paraformaldehyde for 15’ min at 4°C and coverslip slides were then mounted in medium containing the nuclear counterstain, DAPI. For morphological detection of synaptosomes-surface NH2htau, 1µM FITC-conjugated NH2htau was added 5 min to synaptosomal preparations and after two washes the FM4-64 assay(see above) was performed (50µM 1’+30mM KCl 90sec). Images are representative of at least three independent experiments.
Subcellular fractionation and isolation of membrane-containing fraction
Biochemical detection of cell-surface NH2htau was performed according to procedure aimed to detect direct interaction of extracellular-added Aβ 1-42 peptide to hippocampal membranes with several modifications [146]. In brief, mature hippocampal neurons were exposed to NH2htau or its reverse (5µM) at 4°C per 1h and then membrane-insoluble BS3 (Thermo Fisher) was used for crosslinking molecules on the cell surface, according to manual instructions. After that tau-treated and untreated control cultures underwent subcellular fractionation to obtain cytosol and membrane fractions. Neurons were scraped in 5mM EDTA in phosphate-buffered saline (PBS) e pelleted by centrifugation for 10’ at 2300 Xg at 4°C. Pellets (P1) were resuspended in 100µl of fractionation buffer (0.25M sucrose 1mM MgCl2 , 2mM EGTA, 25mM HEPES ph 7.4) and lysed by three cycles of freeze-thaw in liquid nitrogen. Lysated were then centrifugated at 100, 000X g for 30’ and the supernatant (cytosol fraction) was removed. The membrane-containing pellets (P2) were resuspended in 100µl of fractionation buffer and equivalent volume of loading dye. Equal amounts of proteins were analyzed by Western blotting with our 12A12 antibody directed against the extreme N-terminal 26-36 aa of human tau protein.
Protein cellular lysates preparation
Total proteins were extracted by scraping the cells in an SDS-reducing sample buffer or lysis in ice-cold RIPA buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 1% Triton, 2 mM EDTA, 0, 1% SDS plus proteases inhibitor cocktail (Sigma Aldrich, P8340) and phosphatase inhibitor cocktail (Sigma Aldrich, P5726/P2850) and centrifuged at 4 °C for 15 min at 1000 X g. The supernatant was then collected and boiled for 5 min.
Western blot analysis and densitometry
Equal amounts of protein were subjected to SDS-PAGE 7.5-15% linear gradient or Bis- Tris gel 4-12% (NuPage, Invitrogen). After electroblotting onto a nitrocellulose membrane (Hybond-C Amersham Biosciences, Piscataway, NJ) the filters were blocked in TBS containing 10% non-fat dried milk for 1h at room temperature or overnight at 4°C. Proteins were visualized using appropriate primary antibodies. All primary antibodies were diluted in TBS and incubated with the nitrocellulose blot overnight at 4°C. Incubation with secondary peroxidase coupled anti-mouse, anti-rabbit or anti-goat antibodies was performed by using the ECL system (Amersham, Arlington Heights, IL, U.S.A.) In a few experiments, multiple normalizations of the same filter on different loading controls, such as β-actin and GAPDH (glyceraldehyde 3-phosphate dehydrogenase), were carried. Final figures were assembled by using Adobe Photoshop 6 and Adobe Illustrator 10 and quantitative analysis of acquired images was performed by using ImageJ (http://imagej.nih.gov/ij/).
DLS experiment and data analysis
High-purity tau peptides were suspended in PBS solution (10× PBS: 1.3 M NaCl, 0.07 M Na2HPO4 and 0.03 M NaH2PO4, pH 7.4) at the nominal concentration of 2.5 mg/ml. Subsequently, the solution was centrifuged for 10 min at 10 000g and the supernatant filtered to eliminate aggregates. DLS measurements were performed by using a Zetasizer Nano ZS (Malvern, Herrenberg, Germany) equipped with a 633-nm He-Ne laser and operating at an angle of 173°. Solvent-resistant micro cuvettes (ZEN0040, Malvern, Herrenberg, Germany) have been used for experiments with a sample volume of 40 µL.
Dynamic light-scattering (DLS) measures the intensity autocorrelation function ![]() , where τ is the lag time and the bracket represents the ensemble average. The G2(τ) can be related to the field autocorrelation function g1(τ) through the Siegert relation G2(τ) = 1+βg12(τ), where β is an instrumental constant equal to 1 in our setup. The mathematical form of g1(τ) depends on the physical properties of the system investigated. For a monodisperse solution of noninteracting particles, a single exponential function with decay time τ is obtained. For a polydisperse sample, g1 (τ) is no longer a single exponential. In this case, the distribution of decay rates on g1(τ) can be taken into account by introducing a weighting function,
, where τ is the lag time and the bracket represents the ensemble average. The G2(τ) can be related to the field autocorrelation function g1(τ) through the Siegert relation G2(τ) = 1+βg12(τ), where β is an instrumental constant equal to 1 in our setup. The mathematical form of g1(τ) depends on the physical properties of the system investigated. For a monodisperse solution of noninteracting particles, a single exponential function with decay time τ is obtained. For a polydisperse sample, g1 (τ) is no longer a single exponential. In this case, the distribution of decay rates on g1(τ) can be taken into account by introducing a weighting function,
![]() eq. 2
eq. 2
where PI (r)dr is the intensity-weighted radius distribution function, describing the distribution of the fraction, in the interval dr, of the intensity scattered by a particle of hydrodynamic radius r and decay rates , ![]() with η is the water viscosity and k the Boltzmann constant [92, 93].The value PI(r) can be obtained by means of a regularized Laplace inversion of the intensity autocorrelation function. In this case the intensity-weighted radius distribution is obtained by a direct numerical inversion of the DLS data. The numerical inversion is automatically performed by the Malvern instrument software.
with η is the water viscosity and k the Boltzmann constant [92, 93].The value PI(r) can be obtained by means of a regularized Laplace inversion of the intensity autocorrelation function. In this case the intensity-weighted radius distribution is obtained by a direct numerical inversion of the DLS data. The numerical inversion is automatically performed by the Malvern instrument software.
The number-weighted radius distribution PN(r) can be recovered by taking into account the form factor P(qr) and the mass M, according to the following equation [93, 94],
![]() eq.3
eq.3
The recovery of PN (r) from PI (r) is automatically performed by the Malvern software package. Once the PN (r) and the PI (r) are known, it is thus easy to determine the number-weighted average hydrodynamic radius (RH )N:
![]() eq.4
eq.4
SAXS experiment and data analysis
High-purity tau peptides were dissolved in PBS solution (10× PBS: 1.3 M NaCl, 0.07 M Na2HPO4 and 0.03 M NaH2PO4, pH 7.4). Peptides concentration was carefully determined through the Guinier approximation and reported in tab. 1. Subsequently, the solution was centrifuged for 10 min at 10 000g and the supernatant was filtered to eliminate aggregates. SAXS measurements were acquired on the BioSAXS beamline (BM-29) at ESRF (Grenoble, France), equipped with a 2D detector (Pilatus 1M, Dectris). The sample to detector distance for normal operation is 2.5 m, which allows a momentum transfer of s = (4π sin θ/λ) in the range from 0.05 to 5.8 nm−1. A volume of 50 μL of solution was placed in a 1.8 mm diameter quartz capillary (mounted in vacuum) with a few tens of micrometer wall thickness, using an automated sample loader. The potential effect of radiation damage was evaluated performing a 10 s exposure at constant temperature without observing any radiation damage. An exposure time of 2 s at each temperature was used to avoid possible radiation damage and experimental measures were carried out at room temperature. Solvent scattering was evaluated- before and after measuring the samples - in the same capillary sample holder to allow for background scattering subtraction.
Peptides sizes were estimated by their radius of gyration RG obtained with two different methods: the Guinier approximation and the Debay model. The first model states that for very small angles (s < 1/RG) the scattered intensity I(s) is described by the following formula:
![]() eq. 5
eq. 5
where I(0) is the total scattered intensity at s = 0.
Given eq. 5, the scattering intensity plotted as ln I(s) vs. s2 (Guinier’s Plot) is a linear function for a particle of any shape. The radius of gyration of both peptides was determined by a linear fit of the Guinier plot. However, since the presence of aggregates and oligomers in solution can lead to an overestimation of RG, we also used the Debye’s approximation according to which, for a Gaussian chain, the scattered intensity I(s) of the RG of the chain can be expressed by the equation:
![]() eq. 6
eq. 6
This approximation holds for a larger momentum transfer ranges with the respect to the Guinier approximation and therefore is less affected by the presence of oligomers and aggregates [95]. We investigated the peptides flexibility by means of the Kratky plot (s2I(s) as a function of s) that is routinely employed to qualitatively identify disordered states that are extremely frequent in the full length human tau and in almost all its fragments exception made for 306VQIVYK311 and 275VQIINK281 , two hexapeptides with a high propensity to form beta-structures [98, 99].
Given the highly flexible nature of tau and of almost its fragments, an explicit description of the structural ensemble visited by both peptides was given by means of the EOM software package [95]. Different pools of representative backbone models with different numerosities were generated for each fragment, namely 10.000, 50.000 and 100.000. The theoretical scattering intensities corresponding to these models were calculated by means of the program CRYSOL. These intensities were then used by the genetic algorithm GAJOE to select from the initial pool an ensemble of conformers providing, on the average, the best fit of the experimental SAXS data. The following GAJOE parameters were set: number of generations, 1.000; number of ensembles, 50; number of curves per ensemble, 20; number of mutations per ensemble, 10; number of crossings per generation, 20. A total of five independent pools were used for fitting each SAXS curve.
Statistical analysis
Experiments were carried out in triplicates and repeated at least three times. Data were expressed as means ± S.D. (n = 4). Statistically significant differences were calculated by unpaired two-tailed t-Student’s test and one-way ANOVA followed by Bonferroni post-hoc test (*p < 0, 05; **p <0, 01; ***p <0, 0001) as indicated in the figure legends. Data from Western blot analysis and immunofluorescence studies were representative of at least three separate experiments.
Author contributions
F.F, C.V., C.G, O.G., I.F., F.M., S.M.F.M., S.A. performed morphological, biochemical and biophysical experiments; C.M.T. prepared primary neuronal cultures; L. V. contributed to 12A12 production; P.A., M.D., P.G., A.A., A.B., C.P., contributed through discussions; De S., P.M., provided analytical tools: G.A., designed the research, conceived and supervised all the experiments, wrote the manuscript.
Acknowledgments
We wish to thank Dott. Francesco Della Valle for critical suggestions and technical help in several biochemical experiments . We are are also grateful to Dott.ssa Francesca Malerba and Dott.Bruno Ercole Ercoli for their technical help in 12A12 antibody purification .
Conflicts of interest
The authors declare that they have no actual or potential conflicts of interest and that these data are not published elsewhere. In addition all authors approve the study described in this report.
Funding
This research was supported by PRIN 2010-2011 (prot. 2010M2JARJ-003) to G.A and by Magnetic Diagnostic Assay for neurodegenerative diseases H2020-ICT-2016-2017” SSI - Smart System Integration” Proposal 732678 SEP 210349930. The funders had no role in study design, data collection and analysis, decision to publish or preparation of manuscript.
REFERENCES
1.Hanger DP, Lau DH, Phillips EC, Bondulich MK, Guo T, Woodward BW, Pooler AM, Noble W. Intracellular and extracellular roles for tau in neurodegenerative disease. J Alzheimers Dis. 2014; 40:S37-45.
2. Mohamed NV, Herrou T, Plouffe V, Piperno N, Leclerc N. Spreading of tau pathology in Alzheimer’s disease by cell-to-cell transmission. Eur J Neurosci. 2013; 37:1939-1948.
3. Clavaguera F, Grueninger F, Tolnay M. Intercellular transfer of tau aggregates and spreading of tau pathology: Implications for therapeutic strategies. Neuropharmacology. 2014; 76:9-15.
4. Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008; 15:157-168.
5. Pedersen JT, Sigurdsson EM. Tau immunotherapy for Alzheimer’s disease. Trends Mol Med. 2015; 21:394-402.
6. Blennow K, Zetterberg H. The past and the future of Alzheimer’s disease CSF biomarkers-a journey toward validated biochemical tests covering the whole spectrum of molecular events. Front Neurosci. 2015; 9:345.
7. Barten DM, Cadelina GW, Hoque N, DeCarr LB, Guss VL, Yang L, Sankaranarayanan S, Wes PD, Flynn ME, Meredith JE, Ahlijanian MK, Albright CF. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J Alzheimers Dis. 2011; 24:127-141.
8. Mably AJ, Kanmert D, McDonald JM, Liu W, Caldarone BJ, Lemere CA, O’Nuallain B, Kosik KS, Walsh DM. Tau immunization: a cautionary tale? Neurobiol Aging. 2015; 36:1316-1332.
9. Meredith JE Jr, Sankaranarayanan S, Guss V, Lanzetti AJ, Berisha F, Neely RJ, Slemmon JR, Portelius E, Zetterberg H, Blennow K, Soares H, Ahlijanian M, Albright CF. Characterization of novel CSF Tau and ptau biomarkers for Alzheimer’s disease. PLoS One. 2013; 8:e76523.
10. Johnson GV, Seubert P, Cox TM, Motter R, Brown JP, Galasko D. The tau protein in human cerebrospinal fluid in Alzheimer’s disease consists of proteolytically derived fragments. J Neurochem. 1997; 68:430-433.
11. Portelius E, Hansson SF, Tran AJ, Zetterberg H, Grognet P, Vanmechelen E, Höglund K, Brinkmalm G, Westman-Brinkmalm A, Nordhoff E, Blennow K, Gobom J. Characterization of tau in cerebrospinal fluid using mass spectrometry. J Proteome Res. 2008; 7:2114-2120.
12. Wagshal D, Sankaranarayanan S, Guss V, Hall T, Berisha F, Lobach I, Karydas A, Voltarelli L, Scherling C, Heuer H, Tartaglia MC, Miller Z, Coppola G, et al. Divergent CSF τ alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J Neuro Neurosurg Psychiatry. 2015; 86:244-250.
13. Bright J, Hussain S, Dang V, Wright S, Cooper B, Byun T, Ramos C, Singh A, Parry G, Stagliano N, Griswold-Prenner I. Human secreted tau increases amyloid-beta production. Neurobiol Aging. 2015; 36:693-709.
14. Kanmert D, Cantlon A, Muratore CR, Jin M, O’Malley TT, Lee G, Young-Pearse TL, Selkoe DJ, Walsh DM. C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death. J Neurosci. 2015; 35:10851-10865.
15. Plouffe V, Mohamed NV, Rivest-McGraw J, Bertrand J, Lauzon M, Leclerc N. Hyperphosphorylation and cleavage at D421 enhance tau secretion. PLoS One. 2012; 7:e36873.
16. Brandt R, Léger J, Lee G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J Cell Biol. 1995; 131:1327-1340.
17. Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010; 142:387-397.
18. Kim W, Lee S, Jung C, Ahmed A, Lee G, Hall GF. Interneuronal transfer of human tau between Lamprey central neurons in situ. J Alzheimers Dis. 2010; 19:647-664.
19. Borroni B, Gardoni F, Parnetti L, Magno L, Malinverno M, Saggese E, Calabresi P, Spillantini MG, Padovani A, Di Luca M. Pattern of Tau forms in CSF is altered in progressive supranuclear palsy. Neurobiol Aging. 2009; 30:34-40.
20. Dai CL, Chen X, Kazim SF, Liu F, Gong CX, Grundke-Iqbal I, Iqbal K. Passive immunization targeting the N-terminal projection domain of tau decreases tau pathology and improves cognition in a transgenic mouse model of Alzheimer disease and tauopathies. J Neural Transm. 2015; 122:607-617.
21. Barthélemy NR, Gabelle A, Hirtz C, Fenaille F, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Junot C, Becher F, Lehmann S. Differential Mass Spectrometry Profiles of Tau Protein in the Cerebrospinal Fluid of Patients with Alzheimer’s Disease, Progressive Supranuclear Palsy, and Dementia with Lewy Bodies. J Alzheimers Dis. 2016; 51:1033-1043.
22. Gómez-Ramos A, Díaz-Hernández M, Rubio A, Díaz-Hernández JI, Miras-Portugal MT, Avila J. Characteristics and consequences of muscarinic receptor activation by tau protein. Eur Neuropsychopharmacol. 2009; 19:708–717.
23. Fá M, Puzzo D, Piacentini R, Staniszewski A, Zhang H, Baltrons MA, Li Puma DD, Chatterjee I, Li J, Saeed F, Berman HL, Ripoli C, Gulisano W, et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci Rep. 2016; 6:19393.
24. Amadoro G, Corsetti V, Stringaro A, Colone M, D’Aguanno S, Meli G, Ciotti M, Sancesario G, Cattaneo A, Bussani R, Mercanti D, Calissano P. A NH2 tau fragment targets neuronal mitochondria at AD synapses: possible implications for neurodegeneration. J Alzheimers Dis. 2010; 21:445-470.
25. Amadoro G, Corsetti V, Atlante A, Florenzano F, Capsoni S, Bussani R, Mercanti D, Calissano P. Interaction between NH(2)-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol Aging. 2012; 33:833.e1-25.
26. Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, Della Valle F, Borreca A, Manca A, Meli G, Ferraina C, Feligioni M, D’Aguanno S, et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet. 2015; 24:3058-3081.
27. Amadoro G, Corsetti V, Sancesario GM, Lubrano A, Melchiorri G, Bernardini S, Calissano P, Sancesario G. Cerebrospinal fluid levels of a 20-22 kDa NH2 fragment of human tau provide a novel neuronal injury biomarker in Alzheimer’s disease and other dementias. J Alzheimers Dis. 2014; 42:211-226.
28. Sokolow S, Henkins KM, Bilousova T, Gonzalez B, Vinters HV, Miller CA, Cornwell L, Poon WW, Gylys KH. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J Neurochem. 2015; 133:368-379.
29. Avila J, de Barreda EG, Fuster-Matanzo A, Simón D, Llorens-Martín M, Engel T, Lucas JJ, Díaz-Hernández M, Hernández F. Looking for novel functions of tau. Biochem Soc Trans. 2012; 40:653-655.
30. Pooler AM, Noble W, Hanger DP. A role for tau at the synapse in Alzheimer’s disease pathogenesis. Neuropharmacology. 2014; 76:1-8.
31. Avila J, Simón D, Díaz-Hernández M, Pintor J, Hernández F. Sources of extracellular tau and its signaling. J Alzheimers Dis. 2014; 40:S7-S15.
32. Regan P, Whitcomb DJ, Cho K. Physiological and Pathophysiological Implications of Synaptic Tau. Neuroscientist. 2016 Feb 23. [Epub ahead of print].
33. Amadoro G, Serafino AL, Barbato C, Ciotti MT, Sacco A, Calissano P, Canu N. Role of N-terminal tau domain integrity on the survival of cerebellar granule neurons. Cell Death Differ. 2004; 11:217-230.
34. Amadoro G, Ciotti MT, Costanzi M, Cestari V, Calissano P, Canu N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc Natl Acad Sci U S A. 2006; 103:2892-2897.
35. Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009; 7:e34.
36. von Bergen M, Barghorn S, Jeganathan S, Mandelkow EM, Mandelkow E. Spectroscopic approaches to the conformation of tau protein in solution and in paired helical filaments. Neurodegener Dis. 2006; 3:197-206.
37. Georgieva ER, Xiao S, Borbat PP, Freed JH, Eliezer D. Tau binds to lipid membrane surfaces via short amphipathic helices located in its microtubule-binding repeats. Biophys J. 2014; 107:1441-1452.
38. Anunciado D, Rai DK, Qian S, Urban V, O’Neill H. Small-angle neutron scattering reveals the assembly of alpha-synuclein in lipid membranes. Biochim Biophys Acta. 2015; 1854:1881-1889.
39. Maas T, Eidenmüller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem. 2000; 275:15733-15740.
40. Eidenmüller J, Fath T, Maas T, Pool M, Sontag E, Brandt R. Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochem J. 2001; 357:759-767.
41. Pooler AM, Hanger DP. Functional implications of the association of tau with the plasma membrane. Biochem Soc Trans. 2010; 38:1012-1015.
42. Elbaum-Garfinkle S, Ramlall T, Rhoades E. The role of the lipid bilayer in tau aggregation. Biophys J. 2010; 98:2722-2730.
43. Jones EM, Dubey M, Camp PJ, Vernon BC, Biernat J, Mandelkow E, Majewski J, Chi EY. Interaction of tau protein with model lipid membranes induces tau structural compaction and membrane disruption. Biochemistry. 2012; 51:2539-2550.
44. Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993; 10:1089-1099.
45. Busciglio J, Lorenzo A, Yeh J, Yankner B. Abeta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995; 14:879-888.
46. Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, Hanger D. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging. 2012; 33:431.e27-38.
47. Sciacca MF, Chillemi R, Sciuto S, Pappalardo M, La Rosa C, Grasso D, Milardi D. Interactions of two O-phosphorylresveratrol derivatives with model membranes. Arch Biochem Biophys. 2012; 521:111-116.
48. Pappalardo G, Milardi D, Magrì A, Attanasio F, Impellizzeri G, La Rosa C, Grasso D, Rizzarelli E. Environmental factors differently affect human and rat IAPP: conformational preferences and membrane interactions of IAPP17-29 peptide derivatives. Chemistry. 2007; 13:10204-10215.
49. Di Natale G, Pappalardo G, Milardi D, Sciacca MF, Attanasio F, La Mendola D, Rizzarelli E. Membrane interactions and conformational preferences of human and avian prion N-terminal tandem repeats: the role of copper(II) ions, pH, and membrane mimicking environments. J Phys Chem B. 2010; 114:13830-13838.
50. Grasso D, Grasso G, Guantieri V, Impellizzeri G, La Rosa C, Milardi D, Micera G, Osz K, Pappalardo G, Rizzarelli, Sanna D, Sóvágó I. Environmental effects on a prion’s helix II domain: copper(II) and membrane interactions with PrP180-193 and its analogues. Chemistry. 2005; 12:537-547.
51. Cañadas O, Casals C. Differential scanning calorimetry of protein-lipid interactions. Methods Mol Biol. 2013; 974:55-71.
52. Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004; 24:6715-6723.
53. Pantusa M, Vad B, Lillelund O, Kjær L, Otzen D, Bartucci R. Alpha-synuclein and familial variants affect the chain order and the thermotropic phase behavior of anionic lipid vesicles. Biochim Biophys Acta. 2016; 1864:1206-1214.
54. Broemstrup T, Reuter N. Molecular dynamics simulations of mixed acidic/zwitterionic phospholipid bilayers. Biophys J. 2010; 99:825-833.
55. Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008; 31:454-463.
56. Marambaud P, Dreses-Werringloer U, Vingtdeux V. Calcium signaling in neurodegeneration. Mol Neurodegener. 2009; 4:20.
57. Vázquez E, Sánchez-Prieto J. Presynaptic modulation of glutamate release targets different calcium channels in rat cerebrocortical nerve terminals. Eur J Neurosci. 1997; 9:2009-2018.
58. Cousin MA, Robinson PJ. Ca(2+) influx inhibits dynamin and arrests synaptic vesicle endocytosis at the active zone. J Neurosci. 2000; 20:949-957.
59. Nimmervoll B, Flucher BE, Obermair GJ. Dominance of P/Q-type calcium channels in depolarization-induced presynaptic FM dye release in cultured hippocampal neurons. Neuroscience. 2013; 253:330-340.
60. Ghijsen WE, Leenders AG, Lopes da Silva FH. Regulation of vesicle traffic and neurotransmitter release in isolated nerve terminals. Neurochem Res. 2003; 28:1443-1452.
61. Gamblin TC, Berry RW, Binder LI. Modeling tau polymerization in vitro. A review and synthesis. Biochemistry. 2003; 42: 15009–15017.
62. Gómez-Ramos A, Díaz-Hernández M, Cuadros R, Hernández F, Avila J. Extracellular tau is toxic to neuronal cells. FEBS Lett. 2006; 580: 4842-4850.
63. Reynolds MR, Berry RW, Binder LI. Site -specific nitration differentially influences tau assembly in vitro. Biochemistry. 2005; 44: 13997–14009.
64. Shammas SL, Garcia GA, Kumar S, Kjaergaard M, Horrocks MH, Shivji N, Mandelkow E, Knowles TP, Mandelkow E, Klenerman D. A mechanistic model of tau amyloid aggregation based on direct observation of oligomers. Nat Commun. 2015; 6:7025.
65. Nayak SV, Rondé P, Spier AD, Lummis SC, Nichols RA. Calcium changes induced by presynaptic 5-hydroxytryptamine-3 serotonin receptors on isolated terminals from various regions of the rat brain. Neuroscience. 1999; 91:107-117.
66. Flach K, Hilbrich I, Schiffmann A, Gärtner U, Krüger M, Leonhardt M, Waschipky H, Wick L, Arendt T, Holzer M. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem. 2012; 287:43223-43233.
67. Ion Channel Formation by Tau Protein: Implications for Alzheimer’s Disease and Tauopathies. Biochemistry. 2015; 54:7320-7325.
68. Berrocal M, Corbacho I, Vázquez-Hernández M, Ávila J, Sepúlveda MR, Mata AM. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim Biophys Acta. 2015; 1852:1465-1476.
69. Schneggenburger R, Neher E. Presynaptic calcium and control of vesicle fusion. Curr Opin Neurobiol. 2005; 15:266-274.
70. Paula-Lima AC, Brito-Moreira J, Ferreira ST. Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer’s disease. J Neurochem. 2013; 126:191-202.
71. Daniel JA, Malladi CS, Kettle E, McCluskey A, Robinson PJ. Analysis of synaptic vesicle endocytosis in synaptosomes by high-content screening. Nat Protoc. 2012; 7:1439-1455.
72. Ivannikov MV, Sugimori M, Llinás RR. Synaptic vesicle exocytosis in hippocampal synaptosomes correlates directly with total mitochondrial volume. J Mol Neurosci. 2013; 49:223-230.
73. Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005; 6:57-69.
74. Raiteri M, Angelini F, Levi GA. Simple apparatus for studying the release of neurotransmitters from synaptosomes. Eur J Pharmacol. 1974; 25:411-414.
75. Musante V, Neri E, Feligioni M, Puliti A, Pedrazzi M, Conti V, Usai C, Diaspro A, Ravazzolo R, Henley JM, Battaglia G, Pittaluga A. Presynaptic mGlu1 and mGlu5 autoreceptors facilitate glutamate exocytosis from mouse cortical nerve endings. Neuropharmacology. 2008; 55:474-482.
76. Summa M, Di Prisco S, Grilli M, Marchi M, Pittaluga A. Hippocampal AMPA autoreceptors positively coupled to NMDA autoreceptors traffic in a constitutive manner and undergo adaptative changes following enriched environment training. Neuropharmacology. 2011; 61:1282-1290.
77. Di Prisco S, Merega E, Milanese M, Summa M, Casazza S, Raffaghello L, Pistoia V, Uccelli A, Pittaluga A. CCL5-glutamate interaction in central nervous system: Early and acute presynaptic defects in EAE mice. Neuropharmacology. 2013; 75:337-346 .
78. Nisticò R, Florenzano F, Mango D, Ferraina C, Grilli M, Di Prisco S, Nobili A, Saccucci S, D’Amelio M, Morbin M, Marchi M, Mercuri NB, Davis RJ, et al. Presynaptic c-Jun N-terminal Kinase 2 regulates NMDA receptor-dependent glutamate release. Sci Rep. 2015; 5:9035.
79. Overk CR, Masliah E. Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem Pharmacol. 2014; 88:508-516.
80. Sze CI, Bi H, Kleinschmidt-DeMasters BK, Filley CM, Martin LJ. Selective regional loss of exocytotic presynaptic vesicle proteins in Alzheimer’s disease brains. J Neurol Sci. 2000; 175:81-90.
81. Honer WG. Pathology of presynaptic proteins in Alzheimer’s disease: more than simple loss of terminals. Neurobiol Aging. 2003; 24:1047-1062.
82. Reddy PH, Mani G, Park BS, Jacques J, Murdoch G, Whetsell W Jr, Kaye J, Manczak M. Differential loss of synaptic proteins in Alzheimer’s disease: implications for synaptic dysfunction. J Alzheimers Dis. 2005; 7:103-117.
83. Savioz A, Leuba G, Vallet PG. A framework to understand the variations of PSD-95 expression in brain aging and in Alzheimer’s disease. Ageing Res Rev. 2014; 18:86-94.
84. Leuba G, Savioz A, Vernay A, Carnal B, Kraftsik R, Tardif E, Riederer I, Riederer BM. Differential changes in synaptic proteins in the Alzheimer frontal cortex with marked increase in PSD-95 postsynaptic protein. J Alzheimers Dis. 2008; 15:139-151.
85. Muntané G, Dalfó E, Martinez A, Ferrer I. Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience. 2008; 152:913-923.
86. Roy S, Zhang B, Lee VM, Trojanowski JQ. Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol. 2005; 109:5-13.
87. Mandelkow EM, Stamer K, Vogel R, Thies E, Mandelkow E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging. 2003; 24:1079-1085.
88. Griffin JW, Watson DF. Axonal transport in neurological disease. Ann Neurol. 1988; 23:3-13.
89. Lopes S, Teplytska L, Vaz-Silva J, Dioli C, Trindade R, Morais M, Webhofer C, Maccarrone G, Almeida OF, Turck CW, Sousa N, Sotiropoulos I, Filiou MD. Tau Deletion Prevents Stress-Induced Dendritic Atrophy in Prefrontal Cortex: Role of Synaptic Mitochondria. Cereb Cortex. 2016; 27:2580-2591.
90. Matamoros AJ, Baas PW. Microtubules in health and degenerative disease of the nervous system. Brain Res Bull. 2016; 126: 217-225.
91. Tsai HH, Lee JB, Shih YC, Wan L, Shieh FK, Chen CY. Location and conformation of amyloid β(25-35) peptide and its sequence-shuffled peptides within membranes: implications for aggregation and toxicity in PC12 cells. Chem Med Chem. 2014; 9:1002-1011.
92. Bernado P, Svergun DI. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol Biosyst. 2012; 8:151-167.
93. Bernado P, Svergun DI. Analysis of intrinsically disordered proteins by small-angle X-ray scattering. Methods Mol Biol. 2012; 896:107-122.
94. Kikhney AG, Svergun DI. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015; 589: 2570-2577.
95. Mylonas E, Hascher A, Bernadó P, Blackledge M, Mandelkow E, Svergun DI. Domain conformation of tau protein studied by solution small-angle X-ray scattering. Biochemistry. 2008; 47: 10345-10353.
96. Chung PJ, Choi MC, Miller HP, Feinstein HE, Raviv U, Li Y, Wilson L, Feinstein SC, Safinya CR. Direct force measurements reveal that protein Tau confers short-range attractions and isoform-dependent steric stabilization to microtubules. Proc Natl Acad Sci U S A. 2015; 112: E6416-6425.
97. Bianconi A, Ciasca G, Tenenbaum A, Battisti A, Campi G. Temperature and solvent dependence of the dynamical landscape of tau protein conformations. J Biol Phys. 2012; 38:169-179 .
98. Ciasca G, Campi G, Battisti A, Rea G, Rodio M, Papi M, Pernot P, Tenenbaum A, Bianconi A. Continuous thermal collapse of the intrinsically disordered protein tau is driven by its entropic flexible domain. Langmuir. 2012; 28:13405-13410.
99. Shkumatov AV, Chinnathambi S, Mandelkow E, Svergun DI. Structural memory of natively unfolded tau protein detected by small-angle X-ray scattering. Proteins. 2011; 79: 2122-2131.
100. Battisti A, Ciasca G, Grottesi A, Bianconi A, Tenenbaum A. Temporary secondary structures in tau, an intrinsically disordered protein. Molecular Simulation. 2012; 38:525-533.
101. Battisti A, Ciasca G, Tenenbaum A. Transient tertiary structures in tau, an intrinsically disordered protein. Molecular Simulation. 2013; 39:1084-1092.
102. Ryan TM, Kirby N, Mertens HD, Roberts B, Barnham KJ, Cappai R, Pham Cle L, Masters CL, Curtain CC. Small angle X-ray scattering analysis of Cu(2+)-induced oligomers of the Alzheimer’s amyloid beta peptide. Metallomics. 2015; 7:536-543.
103. Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci U S A. 2004; 101:12491-12496.
104. von Bergen M, Barghorn S, Biernat J, Mandelkow EM, Mandelkow E. Tau aggregation is driven by a transition from random coil to beta sheet structure. Biochim Biophys Acta. 2005; 1739: 158-166.
105. Pristerà A, Saraulli D, Farioli-Vecchioli S, Strimpakos G, Costanzi M, di Certo MG, Cannas S, Ciotti MT, Tirone F, Mattei E, Cestari V, Canu N. Impact of N-tau on adult hippocampal neurogenesis, anxiety, and memory. Neurobiol Aging. 2013; 34:2551-2563.
106. Pilz I , Glatter O, Kratky O. Small-angle X-ray scattering. Methods Enzymol. 1979; 61:148-249.
107. Hofmann H, Soranno A, Borgia A, Gast K, Nettels D, Schuler B. Polymer scaling laws of unfolded and intrinsically disordered proteins quantified with single-molecule spectroscopy. Proc Natl Acad Sci U S A. 2012; 109:16155-16160.
108. Jowitt TA, Murdoch AD, Baldock C, Berry R, Day JM, Hardingham TE. Order within disorder: Aggrecan chondroitin sulphate-attachment region provides new structural insights into protein sequences classified as disordered. Proteins. 2010; 78:3317–3327.
109. Kovacs GG. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015; 41:3-23.
110. Wang Y, Garg S, Mandelkow EM, Mandelkow E. Proteolytic processing of tau. Biochem Soc Trans. 2010; 38:955-961.
111. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010; 6:131-144.
112. Brier MR, Gordon B, Friedrichsen K, McCarthy J, Stern A, Christensen J, Owen C, Aldea P, Su Y, Hassenstab J, Cairns NJ, Holtzman DM, Fagan AM, et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 2016; 8:338ra66.
113. Valera E, Spencer B, Masliah E. Immunotherapeutic Approaches Targeting Amyloid-β, α-Synuclein, and Tau for the Treatment of Neurodegenerative Disorders. Neurotherapeutics. 2016; 13:179-189.
114. Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007; 68:1501-1508.
115. Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009; 29:10741-10749.
116. Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu GQ, Masliah E, Mucke L. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One. 2012; 7:e45881.
117. Erez H, Shemesh OA, Spira ME. Rescue of tau-induced synaptic transmission pathology by paclitaxel. Front Cell Neurosci. 2014; 8:34.
118. Decker JM, Krüger L, Sydow A, Zhao S, Frotscher M, Mandelkow E, Mandelkow EM. Pro-aggregant Tau impairs mossy fiber plasticity due to structural changes and Ca(++) dysregulation. Acta Neuropathol Commun. 2015; 3:23.
119. Tai HC, Wang BY, Serrano-Pozo A, Frosch MP, Spires-Jones TL, Hyman BT. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol Commun. 2014; 2:146.
120. Moreno H, Choi S, Yu E, Brusco J, Avila J, Moreira JE, Sugimori M, Llinás RR. Blocking Effects of Human Tau on Squid Giant Synapse Transmission and Its Prevention by T-817 MA. Front Synaptic Neurosci. 2011; 3:3.
121. Moreno H, Morfini G, Buitrago L, Ujlaki G, Choi S, Yu E, Moreira JE, Avila J, Brady ST, Pant H, Sugimori M, Llinás RR. Tau pathology-mediated presynaptic dysfunction. Neuroscience. 2016; 325:30-38.
122. Taverna E, Saba E, Rowe J, Francolini M, Clementi F, Rosa P. Role of lipid microdomains in P/Q-type calcium channel (Cav2.1) clustering and function in presynaptic membranes. J Biol Chem. 2004; 279:5127-5134.
123. Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2004; 24: 3801-3809.
124. Ronzitti G, Bucci G, Emanuele M, Leo D, Sotnikova TD, Mus LV, Soubrane CH, Dallas ML, Thalhammer A, Cingolani LA, Mochida S, Gainetdinov RR, Stephens GJ, et al. Exogenous α-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J Neurosci. 2014; 34:10603-10615.
125. Emanuele M, Esposito A, Camerini S, Antonucci F, Ferrara S, Seghezza S, Catelani T, Crescenzi M, Marotta R, Canale C, Matteoli M, Menna E, Chieregatti E. Exogenous Alpha-Synuclein Alters Pre- and Post-Synaptic Activity by Fragmenting Lipid Rafts. EBioMedicine. 2016; 7:191-204.
126. Mochida S, Westenbroek RE, Yokoyama CT, Zhong H, Myers SJ, Scheuer T, Itoh K, Catterall WA. Requirement for the synaptic protein interaction site for reconstitution of synaptic transmission by P/Q-type calcium channels. Proc Natl Acad Sci U S A. 2003; 100:2819-2824.
127. Müller WE, Kirsch C, Eckert GP. Membrane-disordering effects of β-amyloid peptides. Biochemical Society Transactions. 2001; 29:617-623.
128. Bollmann JH, Sakmann B. Control of synaptic strength and timing by the release-site Ca2+ signal. Nat Neurosci. 2005; 8:426-434.
129. Fayed N, Modrego PJ, Rojas-Salinas G, Aguilar K. Brain glutamate levels are decreased in Alzheimer’s disease: a magnetic resonance spectroscopy study. Am J Alzheimers Dis Other Demen. 2011; 26:450-456.
130. Menkes-Caspi N, Yamin HG, Kellner V, Spires-Jones TL, Cohen D, Stern EA. Pathological tau disrupts ongoing network activity. Neuron. 2015; 85:959-966.
131. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007; 53:337-351.
132. Leroy K, Bretteville A, Schindowski K, Gilissen E, Authelet M, De Decker R, Yilmaz Z, Buée L, Brion JP. Early axonopathy preceding neurofibrillary tangles in mutant tau transgenic mice. Am J Pathol. 2007; 171:976-992.
133. Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta. 2012; 1822:639-649.
134. Jadhav S, Katina S, Kovac A, Kazmerova Z, Novak M, Zilka N. Truncated tau deregulates synaptic markers in rat model for human tauopathy. Front Cell Neurosci. 2015; 9:24.
135. Pir GJ, Choudhary B, Mandelkow E, Mandelkow EM. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol Neurodegener. 2016; 11:33.
136. Michel CH, Kumar S, Pinotsi D, Tunnacliffe A, St George-Hyslop P, Mandelkow E, Mandelkow EM, Kaminski CF, Kaminski Schierle GS. Extracellular monomeric tau protein is sufficient to initiate the spread of tau protein pathology. J Biol Chem. 2014; 289:956-967.
137. Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, Schachter J, Renger JJ, Parmentier-Batteur S. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J Neurosci. 2015; 35:14234-14250.
138. Atlante A, Amadoro G, Bobba A, de Bari L, Corsetti V, Pappalardo G, Marra E, Calissano P, Passarella S. A peptide containing residues 26-44 of tau protein impairs mitochondrial oxidative phosphorylation acting at the level of the adenine nucleotide translocator. Biochim Biophys Acta. 2008; 1777:1289-1300.
139. Liu C, Song X, Nisbet R, Götz J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J Biol Chem. 2016; 291:8173-8188.
140. Dujardin S, Lécolle K, Caillierez R, Bégard S, Zommer N, Lachaud C, Carrier S, Dufour N, Aurégan G, Winderickx J, Hantraye P, Déglon N, Colin M, et al. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol Commun. 2014; 2:14.
141. Díaz-Hernández M, Gómez-Ramos A, Rubio A, Gómez-Villafuertes R, Naranjo JR, Miras-Portugal MT, Avila J. Tissue-nonspecific alkaline phosphatase promotes the neurotoxicity effect of extracellular tau. J Biol Chem. 2010; 285:32539-32548.
142. Yamada K, Holth JK, Liao F, Stewart FR, Mahan TE, Jiang H, Cirrito JR, Patel TK, Hochgrafe K, Mandelkow EM, Holtzman DM. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014; 211: 387–393.
143. Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013; 80:402-414.
144. Subramanian S, Savanur G, Madhavadas S. Passive immunization targeting the N-terminal region of phosphorylated tau (residues 68-71) improves spatial memory in okadaic acid induced tauopathy model rats. Biochem Biophys Res Commun. 2016; 483:585-589.
145. Gilson V, Mbebi-Liegeois C, Sellal F, de Barry J. Effects of Low Amyloid-β (Aβ) Concentration on Aβ1-42 Oligomers Binding and GluN2B Membrane Expression. J Alzheimers Dis. 2015; 47:453-466.
146. Nicholson AM, Methner DN, Ferreira A. Membrane cholesterol modulates {beta}-amyloid-dependent tau cleavage by inducing changes in the membrane content and localization of N-methyl-D-aspartic acid receptors. J Biol Chem. 2011; 286:976-986.