
INTRODUCTION
Stomach cancer is one of common malignancies of digestive tract carcinoma. More than 950,000 new stomach cancer cases and 720,000 deaths occur in 2012 worldwide [1]. It is divided into stomach adenocarcinoma (STAD) and stomach squamous cell carcinoma (SSCC) based on the histopathologic feature. STAD is the predominant subtype in stomach cancer.
The regions of highest rates of stomach cancer are in Eastern Asia countries especially in China, Japan and Korea; stomach cancer is almost twice more common in males than females [1]. Numerous articles indicate that the onset and progression of stomach cancer is likely attributed to the interconnection of H. pylori infection, genetic factors and environment exposure. H. pylori is responsible for 90% new noncardia stomach cancer cases worldwide [2]. Higher expression of β2-AR, VEGF and PODXL is significantly associated with poorer survival and higher risk of recurrence in patients with stomach cancer, [3–5]. rs2494752 AG/GG variant genotype in AKT1, rs1034528 CG/CC and rs3806317 GA/GG variants in mTOR, and copy-number loss of CYCLOPS and STOP genes are related to stomach cancer susceptibility and carcinogenesis [6–8]. However, pathological mechanism of STAD is largely unknown.
microRNAs (miRNAs) with 20–25 nucleotides belong to non-coding RNAs, which function as promoting or repressing roles in cellular process tumorigenesis [9, 10]. miRNA-222 promotes cell migration by suppressing p27kip1 in STAD [11]. Over-expression of miR-296-5p promotes cell proliferation in stomach cancer by decreased expression of Caudal-related [12]. Piwi-interaction RNAs (piRNA), as a class of non-coding RNAs, is involved in tumorigenesis of several cancers. piR-823 expression is markedly reduced in gastric cancer tissues compared to non-cancerous tissues and over-expression of piR-823 suppresses cell growth in gastric cancer [13]. piR-651 is significantly up-regulated in gastric cancer and higher expression of piR-651 predicts advanced TNM stage. Knock down of piR-651 inhibits cell growth and arrests G2/M phase in gastric cancer [14]. The transcripts with longer than 200 nucleotides, not translated into proteins, are defined as long non-coding RNAs (lncRNAs) [15]. lncRNAs are known to implicate in splicing, recruiting transcription factors and mRNA stability [16]. Mounting reports demonstrate that aberrant expression of lncRNAs emerge as vital regulator of diverse biological processes in cancers, such as tumorigenesis, development and metastasis [15].
In this study, we used high-throughput RNA-sequencing to obtain lncRNA and mRNA expression profile in STAD, identified differentially expressed lncRNAs/mRNAs in STAD compared to adjacent non-tumor tissues and constructed lncRNA/mRNA co-expression network. The diagnostic value of candidate DEmRNAs was validated in TCGA database. Our study might provide valuable information for exploring tumorigenesis mechanism in STAD and identification of potential biomarkers for STAD diagnosis.
RESULTS
RNA-sequencing of STAD tissues
STAD and adjacent non-tumor tissues obtained from three patients were applied for RNA-sequencing. All of three patients were diagnosed as diffuse Lauren type and T4N1M0 stage of STAD (Supplementary Table 1). After raw reads trim, respective 1.05 × 108, 0.95 × 108, 1.15 × 108 clean reads were generated from three STAD samples, and 1.04 × 108, 0.89 × 108, 0.968 × 108 clean reads were generated from three corresponding adjacent non-tumor tissues, as Supplementary Table 2 shown. All of the clean reads were aligned with the human reference genome (hg.19). The mapped ratio of all samples was above 70% (Supplementary Table 3).
lncRNAs and mRNAs expression profile
Respective 15929 lncRNAs and 20205 mRNAs were mapped with the human reference genome hg. 19. Total of 74 significantly differential expression lncRNAs (DElncRNAs) with at least 2-fold change were identified in STAD compared with adjacent non-tumor tissues (Supplementary Table 4). 43 DElncRNAs were up-regulated and 31 DElncRNAs were down-regulated in STAD, which were described as volcano plot (Figure 1A). As Figure 1B shown, 449 DEmRNAs including 238 up- and 211 down-regulated DEmRNAs were screened in STAD (Supplementary Table 5).
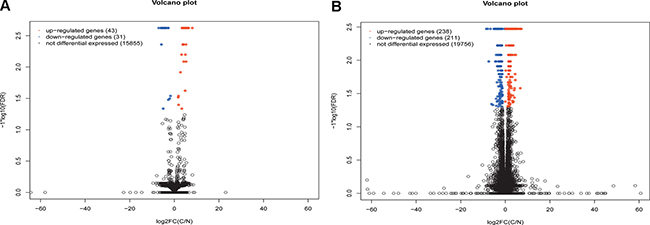
Figure 1: Volcano spot of lncRNA and mRNA expression in stomach adenocarcinoma. (A) volcano spot of lncRNA expression in cancer group and control group; (B) volcano spot of mRNA expression in cancer group and control group. C indicated stomach adenocarcinoma and N indicated adjacent non-tumor samples. Red spot and blue spot respectively indicated up- and down-regulated genes in stomach adenocarcinoma.
Hierarchical clustering of the lncRNAs and mRNAs expression profile was performed by using hcluster in R language. Spearman Pearson correlation between 6 samples were calculated. Hierarchical clustering of the expression of the 74 lncRNAs and 449 mRNAs displayed that there were obvious discrimination between STAD and adjacent non-tumor tissues (Figure 2 and Supplementary Figure 1).The most significantly up-regulated DElncRNA was LOC105377924 and the most significantly down-regulated DElncRNA was SEMA3B-AS1 in STAD (Table 1). S100A2 and NTRK3 were the most significantly up- and down-regulated DEmRNAs in stomach STAD, as Table 2 shown.
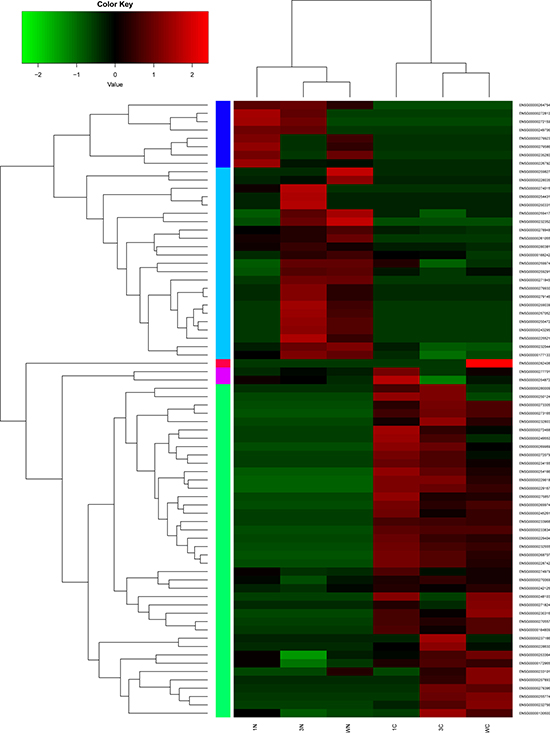
Figure 2: Hierarchical clustering analysis of the expression levels of DElncRNAs in stomach adenocarcinoma and adjacent non-tumor tissues. Row and column represented DElncRNAs and tissue samples. The color scale indicated log10FPKM of the expression levels of DElncRNAs. Red and green indicated up- and down-regulation. C represented stomach adenocarcinoma tissues and N represented adjacent non-tumor tissues.
Table 1: The top 15 up- and down-regulated DElncRNAs in STAD
Ensembl Gene ID |
Gene Symbol |
FDR |
log2FC |
|---|---|---|---|
Up-regulation |
|||
ENSG00000282408 |
LOC105377924 |
0.002386 |
8.040191 |
ENSG00000229618 |
AC011288.2 |
0.002386 |
6.394161 |
ENSG00000254166 |
CASC19 |
0.002386 |
6.274223 |
ENSG00000229167 |
RP11-73M7.1 |
0.002386 |
6.002959 |
ENSG00000250124 |
CTC-261N6.2 |
0.002386 |
5.666884 |
ENSG00000269989 |
RP11-635N19.3 |
0.002386 |
5.654653 |
ENSG00000272468 |
RP1-86C11.7 |
0.002386 |
5.428779 |
ENSG00000248103 |
CTC-338M12.9 |
0.002386 |
5.305212 |
ENSG00000273165 |
RP11-1057B6.1 |
0.008203 |
5.254069 |
ENSG00000230316 |
FEZF1-AS1 |
0.002386 |
5.249582 |
ENSG00000268707 |
RP11-247A12.7 |
0.004375 |
5.209937 |
ENSG00000232803 |
SLCO4A1-AS1 |
0.002386 |
5.176534 |
ENSG00000275857 |
AC009133.21 |
0.002386 |
5.173935 |
ENSG00000249550 |
LINC01234 |
0.002386 |
5.157594 |
ENSG00000269974 |
RP11-932O9.10 |
0.002386 |
5.095659 |
Down-regulation |
|||
ENSG00000232352 |
SEMA3B-AS1 |
0.002386 |
–6.99292701 |
ENSG00000259417 |
LINC01314 |
0.002386 |
–6.006864475 |
ENSG00000272159 |
RP11-350N15.6 |
0.002386 |
–5.912151637 |
ENSG00000272810 |
U91328.22 |
0.004375 |
–5.801065509 |
ENSG00000259827 |
RP11-343H19.2 |
0.002386 |
–5.593494209 |
ENSG00000225521 |
AC005237.4 |
0.002386 |
–5.526766822 |
ENSG00000274015 |
CTD-2302E22.6 |
0.002386 |
–5.401633879 |
ENSG00000259039 |
RP11-409I10.2 |
0.002386 |
–5.179666299 |
ENSG00000250331 |
LINC01340 |
0.002386 |
–5.097315823 |
ENSG00000250472 |
TRIM36-IT1 |
0.002386 |
–5.061832349 |
ENSG00000264754 |
CTD-2653B5.1 |
0.002386 |
–5.041799308 |
ENSG00000232044 |
LINC01105 |
0.046115 |
–5.001686931 |
ENSG00000177133 |
LINC00982 |
0.002386 |
–4.981227991 |
ENSG00000257052 |
RP11-881M11.2 |
0.002386 |
–4.971847005 |
ENSG00000254431 |
RP11-550A5.2 |
0.002386 |
–4.806200304 |
DElncRNA: differentiated expressed long non-coding RNA; STAD: stomach adenocarcinoma; FDR: false discovery rate; FC: fold change.
Table 2: The top 15 up- and down-regulated DEmRNAs in STAD
Ensembl Gene ID |
Gene Symbol |
FDR |
log2FC |
|---|---|---|---|
Up-regulation |
|||
ENSG00000196754 |
S100A2 |
0.00337364 |
7.183158953 |
ENSG00000186832 |
KRT16 |
0.00337364 |
6.789708057 |
ENSG00000128422 |
KRT17 |
0.0262905 |
6.716559208 |
ENSG00000241794 |
SPRR2A |
0.00337364 |
6.546179879 |
ENSG00000170373 |
CST1 |
0.00337364 |
6.349039672 |
ENSG00000143546 |
S100A8 |
0.00337364 |
5.623496687 |
ENSG00000119547 |
ONECUT2 |
0.00337364 |
5.255570479 |
ENSG00000188100 |
FAM25A |
0.00337364 |
5.221544589 |
ENSG00000159516 |
SPRR2G |
0.00337364 |
4.984393393 |
ENSG00000171711 |
DEFB4A |
0.00337364 |
4.874560923 |
ENSG00000143536 |
CRNN |
0.00337364 |
4.823932742 |
ENSG00000125780 |
TGM3 |
0.00830821 |
4.667539055 |
ENSG00000149968 |
MMP3 |
0.00337364 |
4.616511381 |
ENSG00000163347 |
CLDN1 |
0.0198214 |
4.494385576 |
ENSG00000165474 |
GJB2 |
0.00337364 |
4.389336874 |
Down-regulation |
|||
ENSG00000140538 |
NTRK3 |
0.003374 |
–8.49189937 |
ENSG00000182333 |
LIPF |
0.003374 |
–7.91251678 |
ENSG00000157017 |
GHRL |
0.010417 |
–7.41649025 |
ENSG00000196482 |
ESRRG |
0.003374 |
–6.64138873 |
ENSG00000164128 |
NPY1R |
0.045544 |
–6.1867236 |
ENSG00000168079 |
SCARA5 |
0.047017 |
–5.56246897 |
ENSG00000185615 |
PDIA2 |
0.003374 |
–5.084125 |
ENSG00000157445 |
CACNA2D3 |
0.003374 |
–4.95423687 |
ENSG00000187045 |
TMPRSS6 |
0.003374 |
–4.93371202 |
ENSG00000167779 |
IGFBP6 |
0.003374 |
–4.81612684 |
ENSG00000166165 |
CKB |
0.003374 |
–4.69931608 |
ENSG00000196616 |
ADH1B |
0.003374 |
–4.66470595 |
ENSG00000159197 |
KCNE2 |
0.003374 |
–4.53444785 |
ENSG00000159212 |
CLIC6 |
0.003374 |
–4.51924504 |
ENSG00000100604 |
CHGA |
0.003374 |
–4.4627063 |
DEmRNA: differentially expressed mRNA; STAD: stomach adenocarcinoma; FDR: false discovery rate; FC: fold change.
In our study, 74 identified DElncRNAs were widely distributed in all autosomes and chromosomes X (Figure 3A). 449 DEmRNAs were distributed in all autosomes, chromosomes X and chromosomes Y (Figure 3A). Based on the annotation results, the biotype of 74 DElncRNAs was divided into 5 categories. 22 DElncRNAs (29.7%) were natural antisense, 35 DElncRNAs (47.3%) were lincRNAs, 4 DElncRNAs (5.4%) were processed transcripts, 5 DElncRNAs (6.8%) was sense intronic and 8 DElncRNAs (10.8%) were others (Figure 3B).
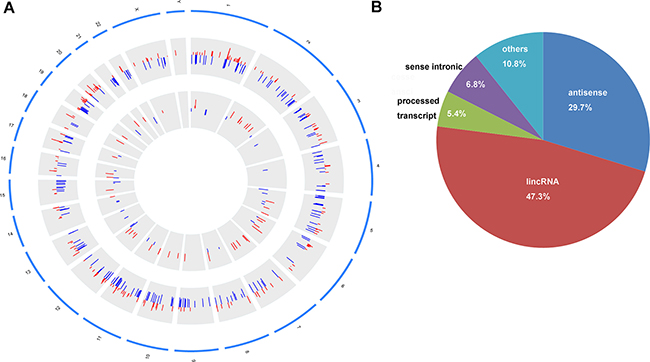
Figure 3: Circos plot of the distribution of DElncRNAs/DEmRNAs on chromosomes and the biotype of DElncRNAs. (A) circos plot. The outer layer of blue cycle was the chromosome map of the human genome. The larger inner layer and smaller inner layer represented the distribution of DEmRNAs and DElncRNAs on different chromosome, respectively. Red color indicated up-regulation and blue color indicated down-regulation. The height of the bar in the larger inner layer and in the smaller inner layer represented log2FC of expression levels of DElncRNAs and DEmRNAs. (B) The biotype of 74 DElncRNAs was classified into five categories. lincRNA indicated long intergenic non-protein coding RNA.
DElncRNA/DEmRNA co-expression network
In order to investigate the potential functions of DElncRNAs in STAD, the Pearson correlation coefficient (PCC) indicating the co-expression relationship between 74 DElncRNAs and 449 DEmRNAs were calculated based on the expression levels of DElncRNAs and DEmRNAs. DElncRNA/DEmRNA co-expression pairs with |PCC| ≥ 0.9 were our concerns. In Supplementary Figure 2, total of 74 DElncRNAs and 445 DEmRNAs were involved in the co-expressed network, which consisted of 519 nodes and 2993 edges. LINC01105 (ENSG00000232044, Figure 4B), RP11-440D17.4 (ENSG00000273305), RP11-617F23.1 (ENSG00000259291) and LINC00261 (ENSG0000 0259974, Figure 4A) were the hub DElncRNAs in the network, which had high connectivity with DEmRNAs.
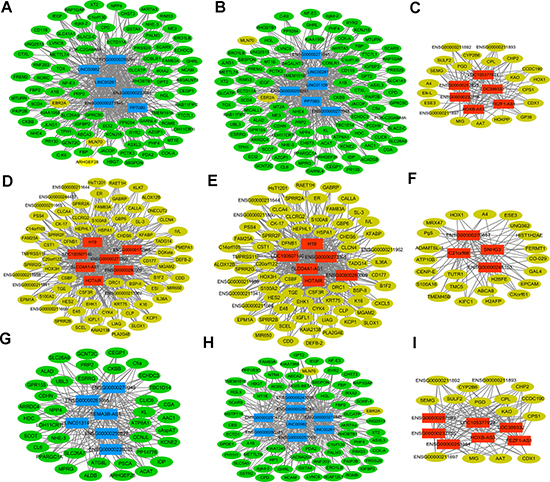
Figure 4: Sub-networks of DElncRNAs/DEmRNAs co-expression. (A) the sub-network of LINC00261; (B) the sub-network of LINC01105; (C) the sub-network of FEZF1-AS1; (D) the sub-network of H19; (E) the sub-network of HOTAIR; (F) the sub-network of SNHG3; (G) the sub-network of SEMA3B-AS1; (H) the sub-network of LINC00982; (I) the sub-network of LINC105377924. Circular node represented DEmRNA and rectangle node represented DElncRNA. Yellow was up-regulated DEmRNA and green was down-regulated DEmRNAs; Red indicated up-regulated DElncRNA and blue indicated down-regulated DElncRNA.
GO and KEGG pathway enrichment analyses
In order to predict the potential functions of DEmRNAs co-expressed with DElncRNAs, GO and KEGG pathway were adopted to demonstrate it. 445 DEmRNAs co-expressed with DElncRNAs were significantly enriched in extracellular matrix and extracellular region of cell component, cell adhesion and biological adhesion in biological process, calcium ion binding and receptor binding in molecular function (Table 3). Those DEmRNAs were significantly enriched in several KEGG signaling pathways. Gastric acid secretion, complement and coagulation cascades, pancreatic secretion, cytokine-cytokine receptor interaction and Jak-STAT signaling pathway were the top pathways associated with STAD (Table 4).
Table 3: GO term enrichment of dysregulated mRNAs
GO ID |
GO terms |
P-value |
FDR |
|---|---|---|---|
Cell component (top 10) |
|||
GO:0031012 |
extracellular matrix |
8.79E-11 |
4.84E-07 |
GO:0005576 |
extracellular region |
1.19E-10 |
6.57E-07 |
GO:0031988 |
membrane-bounded vesicle |
1.60E-10 |
8.79E-07 |
GO:0005615 |
extracellular space |
1.64E-10 |
9.05E-07 |
GO:0031982 |
vesicle |
2.14E-10 |
1.18E-06 |
GO:0044421 |
extracellular region part |
2.22E-10 |
1.22E-06 |
GO:0070062 |
extracellular vesicular exosome |
2.78E-10 |
1.53E-06 |
GO:0043230 |
extracellular organelle |
2.80E-10 |
1.54E-06 |
GO:0065010 |
extracellular membrane-bounded organelle |
2.80E-10 |
1.54E-06 |
GO:0005578 |
proteinaceous extracellular matrix |
6.97E-09 |
3.84E-05 |
Biological process (top 10) |
|||
GO:0007155 |
cell adhesion |
1.83E-10 |
1.01E-06 |
GO:0022610 |
biological adhesion |
2.16E-10 |
1.19E-06 |
GO:0044763 |
single-organism cellular process |
2.75E-10 |
1.52E-06 |
GO:0044699 |
single-organism process |
3.15E-10 |
1.73E-06 |
GO:0050896 |
response to stimulus |
2.85E-09 |
1.57E-05 |
GO:0006950 |
response to stress |
6.20E-09 |
3.42E-05 |
GO:0032101 |
regulation of response to external stimulus |
7.94E-09 |
4.37E-05 |
GO:0030198 |
extracellular matrix organization |
8.31E-09 |
4.58E-05 |
GO:0043062 |
extracellular structure organization |
8.84E-09 |
4.87E-05 |
GO:0009611 |
response to wounding |
1.42E-08 |
7.85E-05 |
Molecular function (top 10) |
|||
GO:0005509 |
calcium ion binding |
2.63E-08 |
0.000145 |
GO:0005102 |
receptor binding |
1.16E-07 |
0.000636 |
GO:0004866 |
endopeptidase inhibitor activity |
5.07E-06 |
0.0279 |
GO:0061135 |
endopeptidase regulator activity |
7.81E-06 |
0.043 |
GO:0005254 |
chloride channel activity |
8.67E-06 |
0.0478 |
GO:0030414 |
peptidase inhibitor activity |
8.97E-06 |
0.0494 |
FDR: false discovery rate.
Table 4: KEGG pathway enrichment of dysregulated mRNAs in STAD
Items |
Items_Details |
FDR |
Genes |
|---|---|---|---|
hsa04971 |
Gastric acid secretion |
1.98E-05 |
SLC9A4, CALML3, CFTR, ADCY2, KCNE2, KCNJ15, SLC26A7, KCNQ1, SSTR2 |
hsa04610 |
Complement and coagulation cascades |
5.21E-05 |
F10, F2R, SERPINA1, C5, C8A, F5, CR2, C7 |
hsa04972 |
Pancreatic secretion |
9.20E-05 |
CFTR, CLCA4, ADCY2, CLCA2, KCNQ1, RYR2, TRPC1, CCKAR, PLA2G4E |
hsa04640 |
Hematopoietic cell lineage |
0.001457 |
KIT, ITGA2, CSF3R, CSF3, CR2, MME, IL11RA |
hsa05322 |
Systemic lupus erythematosus |
0.001691 |
HIST1H2BM, C5, C8A, HIST1H2AE, HIST1H2BE, FCGR3A, C7 |
hsa05020 |
Prion diseases |
0.001992 |
C5 ,C8A, C7 |
hsa00480 |
Glutathione metabolism |
0.002214 |
IDH2, GSTP1, RRM2, PGD, GSTA4 |
hsa00980 |
Metabolism of xenobiotics by cytochrome P450 |
0.002473 |
AKR1C4, CYP2B6, DHDH, GSTP1, GSTA4, ADH1B |
hsa04060 |
Cytokine-cytokine receptor interaction |
0.002843 |
KIT, CXCL9, CXCL5, CXCL1, CCR8, LIFR, EDA, TNFSF15, CSF3R, CSF3, IL11RA |
hsa00590 |
Arachidonic acid metabolism |
0.002877 |
CYP2B6, ALOX12B, PTGIS, PLA2G4E, PTGS2 |
hsa04640 |
Hematopoietic cell lineage |
0.002996 |
KIT, CSF3R, CSF3, IL11RA |
hsa00010 |
Glycolysis / Gluconeogenesis |
0.00308 |
FBP2, ALDOB, HKDC1 |
hsa00051 |
Fructose and mannose metabolism |
0.00308 |
FBP2, ALDOB, HKDC1 |
hsa04970 |
Salivary secretion |
0.003109 |
CALML3, ADCY2, MUC5B, DMBT1, AQP5, CST1 |
hsa04978 |
Mineral absorption |
0.003124 |
MT2A, SLC26A9, HEPH, MT1E, SLC9A3 |
hsa04020 |
Calcium signaling pathway |
0.003165 |
ADCY2, RYR2, TRPC1, CCKAR |
hsa05200 |
Pathways in cancer |
0.003449 |
HIF1A, KIT, LAMA2, ITGA2, CSF3R, FGF13, GSTP1, WNT5A, BRCA2, LAMC2, PTGS2, MMP1 |
hsa05412 |
Arrhythmogenic right ventricular cardiomyopathy |
0.007626 |
RYR2, LAMA2, ITGA2, CACNA2D3, CDH2 |
hsa04630 |
Jak-STAT signaling pathway |
0.007986 |
CSF3R, CSF3, IL11RA |
hsa00532 |
Glycosaminoglycan biosynthesis - chondroitin sulfate |
0.00833 |
XYLT2, B3GAT1, CHST3 |
STAD: stomach adenocarcinoma; FDR: false discovery rate.
Moreover, those DEmRNAs (underlined genes in Table 4) co-expressed with DElncRNAs shown in Figure 3 were significantly enriched in abovementioned signaling pathways. KCNE2 (co-expressed with LINC01105 and SEM3B-AS1), KCNJ15 (co-expressed with LINC01105 and LINC00261), SLC26A7 (co-expressed with SEM3B-AS1) and SSTR2 (co-expressed with LINC00982) were significantly enriched in gastric acid secretion; CLCA4 co-expressed with H19 and HOTAIR was significantly enriched in pancreatic secretion; CSF3R co-expressed with H19 and HOTAIR was significantly enriched in pathways in cancer, Jak-STAT signaling pathway and cytokine-cytokine receptor interaction; CXCL5 co-expressed with H19 and HOTAIR was significantly enriched in cytokine-cytokine receptor interaction; RYR2 co-expressed with LINC00261 and LINC01105 was significantly enriched in pancreatic secretion and calcium signaling pathway.
qRT-PCR validation of DElncRNA and DEmRNAs
DElncRNAs and DEmRNAs were identified in our study through high throughout sequencing and bioinformatics analysis. qRT-PCR was subjected to validate the expression levels of DElncRNAs and DEmRNAs in ten STAD tissues and ten matched adjacent tumor tissues. Three DElncRNAs and one DEmRNAs were chose for verification. As Figure 5A and 5C shown, LINC00982 and LINC00261 (P < 0.01) were down-regulated in STAD compared with controls. SHNG3 was significantly up-regulated in STAD tissues in Figure 5B (P < 0.01). In our DEmRNA expression profile analysis, FOXA2 was not significantly down-regulated (FDR > 0.05), but it had the down-regulated tendency in STAD. In the qRT-PCR validation, FOXA2 (P < 0.01) was significantly down-regulated in STAD (Figure 5D). In generally, the verification results of qRT-PCR were consistent with our bioinformatics analysis.
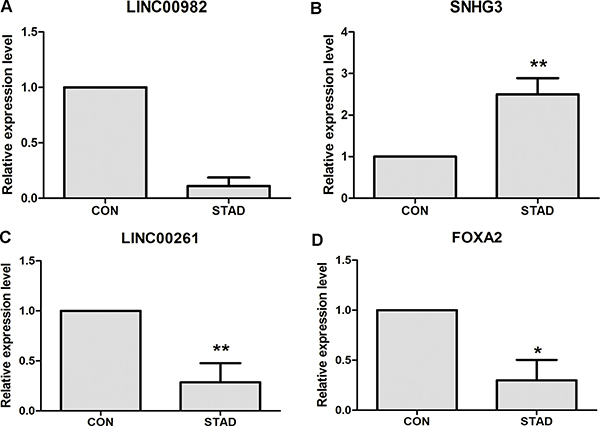
Figure 5: qRT-PCR validation of dysregulated DElncRNAs and DEmRNAs in STAD compared with matched non-tumor tissues. (A) The expression level of lncRNA LINC00982; (B) The expression level of lncRNA SNHG3; (C) The expression level of lncRNA LINC00261; (D) The expression level of FOXA2. STAD indicated stomach adenocarcinoma; CON indicated paired adjacent non-tumor tissues. * represented P < 0.05 and ** represented P < 0.01.
The expression levels of candidate DElncRNAs were analyzed in the TCGA datasets
The lncRNA expression profile of 285 STAD tissues (case group) and 33 adjacent non-tumor tissues (control group) were retrieved from TCGA database. The difference of expression levels of the nine DElncRNAs between STAD and non-tumor tissues were analyzed and were depicted using box-plots, which was visually illustrated by median and inter-quartile range. As Figure 6B, 6G and 6H shown, the expression levels of LINC01105 (P = 0.002724), SEMA3B-AS1 (P = 0.003923) and LINC00982 (P = 0.001156) were significantly down-regulated in case group compared with control group. In addition, the expression levels of FEZF1-AS1 (P = 2.2e-16), H19 (P = 2.37e-07), HOTAIR (P = 2.2e-16) and LINC01234 (P = 6.78e-16) in case group were significantly higher than that in control group, as Figure 6C–6E and 6I revealed. In addition, the difference of expression levels of LINC00261 and SNHG3 between case group and control group was not significant, but LINC00261 expression had the down-regulated tendency and SNHG3 expression had the up-tendency in case group compared with control group (Figure 6A and 6F). In general, the results of cross validation indicated that the expression patterns of candidate DElncRNAs in TCGA database was compatible with our RNA-sequencing and bioinformatics analyses.
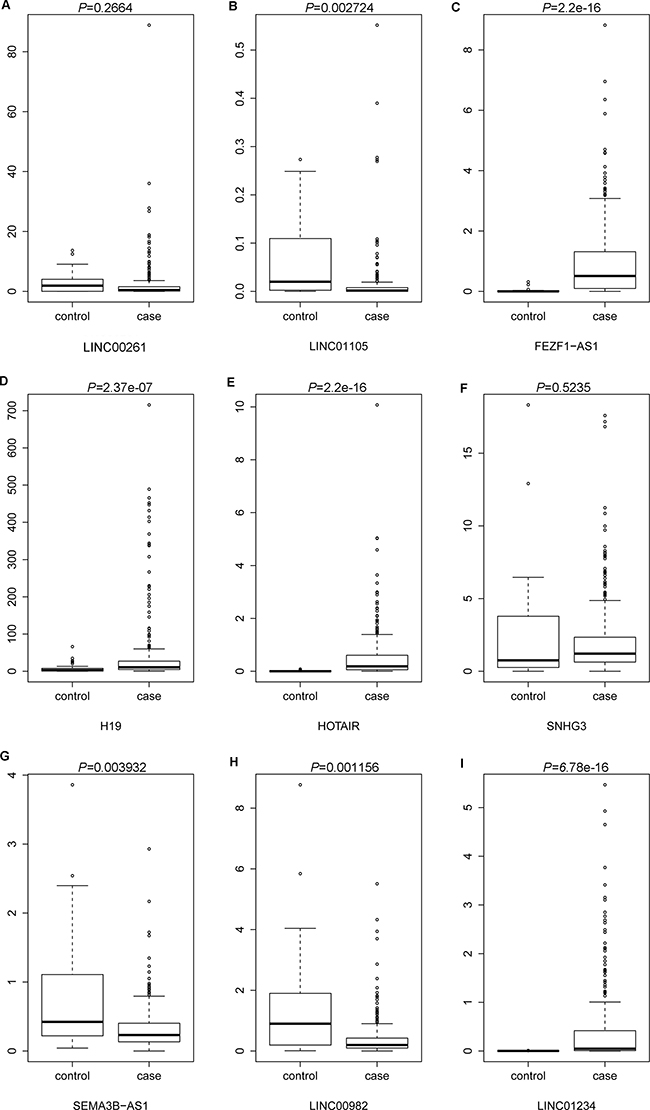
Figure 6: The cross validation of the expression levels of candidate DElncRNAs in STAD tissues based on TCGA database. (A) LINC00261; (B) LINC01105; (C) FEZF1-AS1; (D) H19; (E) HOTAIR; (F) SNHG3; (G) SEMA3B-AS1; (H) LINC00982; (I) LINC01234. Case group and control group indicated STAD tissues and adjacent non-tumor tissues.
ROC curve analysis
In order to access the discriminatory ability of the nine candidate DElncRNAs among 285 STAD tissues and 33 adjacent non-tumor tissues generated from TCGA database, ROC curve analyses were conducted and AUC were calculated. As Figure 7B–7E, 7H and 7I shown, the AUC of LINC01105 (0.731), FEZF1-AS1 (0.914), H19 (0.709), HOTAIR (0.923), LINC00982 (0.740) and LINC01234 (0.894) was more than 0.7; the AUC of LINC00261 (0.591, Figure 7A), SNHG3 (0.568, Figure 7F) and SEMA3B-AS1 (0.694, Figure 7G) was less than 0.7.
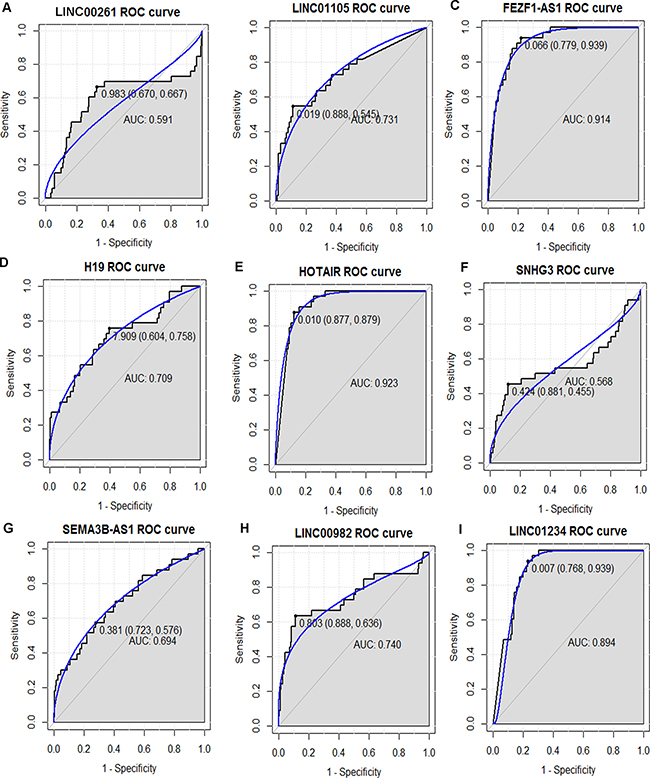
Figure 7: The discriminatory ability of the evaluated DElncRNAs between STAD tissues and adjacent non-tumor tissues was accessed with ROC curve. (A) LINC00261; (B) LINC01105; (C) FEZF1-AS1; (D) H19; (E) HOTAIR; (F) SNHG3; (G) SEMA3B-AS1; (H) LINC00982; (I) LINC01234.
FEZF1-AS1, HOTAIR and LINC01234 had the largest AUC in those nine DElncRNAs. For STAD diagnosis, the sensitivity and specificity of FEZF1-AS1 was 93.9% and 77.9% (Figure 7C); the sensitivity and specificity of HOTAIR was 87.9% and 87.7% (Figure 7E); the sensitivity and specificity of LINC01234 was 93.9% and 76.8% (Figure 7I), respectively.
DISCUSSION
LncRNAs are long non-coding transcripts with longer than 200 nucleotides. They are expressed at lower level compared to protein-coding mRNA [17]. Mounting evidence demonstrate that DElncRNAs are linked to multiple human diseases, such as colorectal cancer, breast cancer and lung cancer [18].
In our study, the landscape of lncRNAs in STAD tissues was obtained by RNA-sequencing . Total of 74 DElncRNAs and 449 DEmRNAs were identified in STAD. The expression levels of LINC00982, LINC00261 and SNHG3 of DElncRNAs and FOXA2 of DEmRNA were verified through qRT-PCR, the results of which were generally consistent with high-throughput sequencing. DElncRNAs distributed on all chromosomes except Y chromosomes and DEmRNAs distributed on all chromosomes including X and Y chromosomes. The DElncRNA/DEmRNA co-expression network was constructed for predicting the potential functions of DElncRNAs in STAD.
In accordance with the previous studies, lncRNA H19 and lncRNA HOTAIR were up-regulated and LINC00261 was down-regulated in stomach cancer. In our data, H19 and HOTAIR was up-regulated with 23.5 and 17.5 fold in STAD, respectively. H19 co-expressed with 72 DEmRNAs and 6 DElncRNAs (Figure 4D). Increased expression of H19 contributes to tumor suppressor p53 inactivation in stomach cancer, which promotes cell proliferation, migration, invasion and metastasis in stomach cancer [19, 20]. Increased H19 is significantly correlated to advanced TNM stage and lymph node metastasis, while leads to poorer overall survival in patients with STAD [21]. Circulating H19 is up-regulated in plasma of patients with STAD compared to healthy controls and it might be promising biomarker of STAD patients [22]. In the DElncRNA/DEmRNA co-expression network, HOTAIR positively correlated with 67 DEmRNAs and 5 DElncRNAs, such as H19 (Figure 4E). Over-expression of HOTAIR promotes the epithelia-to-mesenchymal transition, cell invasion, cell metastasis and it is associated with unfavorable survival of patients with STAD [23–26].
LINC00982 and LINC00261 were down-regulated and SNHG3 significantly up-regulated in SATD (Figure 3), which were consistent with our high-throughput sequencing. The recent study demonstrates that decreased LINC000982 contributes to cell proliferation, cell cycle progression, poor overall survival and disease-free survival; and its over-expression suppresses cell proliferation and renders cell cycle arrest in stomach cancer [27]. LINC00261 is down-regulated in stomach cancer and co-expressed with 85 DEmRNAs in our study (Figure 4A). Lower expression of LINC00261 correlates with deeper tumor invasion, poorer differentiation and lymph node metastasis in stomach cancer [28]. Currently, the potential functions and mechanism of SNHG3 in STAD are largely unclear. Our study was the first time to report that SNHG3 was over-expressed in STAD. It is reported that SNHG3 is significantly up-regulated in hepatocellular carcinoma (HCC) compared with paired non-tumor tissues; up-regulation of SNHG3 is correlated with overall survival, recurrence-free survival and relapse in HCC [29]. The aforementioned information suggests that increased SNHG3 might contribute to the carcinogenesis of STAD.
FEZF1-AS1 is up-regulated in colorectal carcinoma (CRC), which promotes aggressive behaviors of CRC cells, including cell proliferation, cell migration and cell metastasis [30]. In our results, FEZF1-AS1 was one of top 10 significantly up-regulated DElncRNAs with 38 fold up-regulation in STAD. FEZF1-AS1 was identified as a novel lncRNA associated with STAD tumorigenesis in our study. LINC01234 is up-regulated in esophageal squamous cell carcinoma (ESCC) and the three-lncRNA signature including LINC01234 could accurately predict survival of patients with ESCC [31, 32]. LINC01234 had almost 36 fold up-regulation in STAD. The potential roles of LINC01234 in STAD tumorigenesis need to be explored in the future work.
445 out of 449 DEmRNAs were co-expressed with 74 DElncRNAs. In order to predict the potential functions of DElncRNAs, GO terms and KEGG signaling pathways of 445 DEmRNAs were enriched, respectively. The results of significant GO annotation, such as cell adhesion, biological adhesion, calcium ion binding and receptor binding, indicated that DElncRNAs and DEmRNAs associated with these GO terms might contribute to STAD tumorigenesis. Several KEGG pathways were enriched from DEmRNAs, such as gastric acid secretion, complement and coagulation cascades, pancreatic secretion, cytokine-cytokine receptor interaction and Jak-STAT signaling pathway, suggesting that DElncRNAs and DEmRNAs might play essential roles through involving in those pathways in STAD.
Except of H19, HOTAIR, LINC000982 and LINC00261, most of DElncRNAs were identified as novel lncRNAs associated with STAD, such as the most significantly up-regulated LOC105377924 (Figure 4I), the most significantly down-regulated SEMA3B-AS1 (Figure 4G), and LINC01105 (ENSG00000232044, Figure 4B), which had the highest degree with DEmRNAs in the co-expression network. Whether those novel DElncRNAs refer to the initiation and development of STAD needs to be clarified through in vivo and in vitro experiments in the future work. The ROC curve analyses indicated that lncRNA FEZF1-AS1, HOTAIR and LINC01234 might be potential biomarkers for STAD diagnosis. In the further work, large cohort of STAD patients needs to be enrolled and the discriminatory ability of those DElncRNAs in STAD diagnosis needs to be further validated in clinical practice.
Our work reported the dysregulated lncRNAs involved in STAD by using high-throughput RNA sequencing. Our study might be the foundation for future investigation for diagnostic biomarkers for STAD and illumination of tumorigenesis mechanisms of STAD.
MATERIALS AND METHODS
Patients and samples
STAD tissues and adjacent non-tumor tissues were obtained from patients received the tumor radical resection in the Fourth Affiliated Hospital of Hebei Medical University. Patients were received none of chemo- or radiotherapy before gastrectomy. The pathological type of patients with stomach cancer was explicitly diagnosed by histopathologic examination. The details of clinical features including gender, age, Lauren type, histologic type and TNM stage were collected. Lastly, 3 subjects with STAD were enrolled into our study.
The study was approved by the Ethics Committee of the Fourth Affiliated Hospital of Hebei Medical University and informed written consent was obtained from all patients. The research complied with the principles of the Declaration of Helsinki.
Library preparation and high-throughput sequencing
Total RNA of collected specimens was extracted by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manual instruction. Firstly, total RNA was subjected to ribosomal RNA removal by Ribo-Zero Magnetic kit (EpiCentre, Mandison, WI,USA); secondly, cDNA library was constructed by Truseq RNA sample Prep Kit (Illumina, Inc., San Diego, CA, USA). RNA was purified and fragmented into 200 base pairs; RNA fragment primed with random hexamer primers were applied for the first cDNA strand synthesis; then the second cDNA strand was synthesized and dUTP was used instead of dTTP; the double cDNA strands were underwent end pair, 3′end adenylation and adapter ligation; the second cDNA strand was digested by UNG enzyme (Illumina, Inc., San Diego, CA, USA). Thirdly, libraries were amplified through polymerase chain reaction for 15 cycles, purified through Certified Low Range Ultra Agarose (Bio-Rad) and quantified through Picogreen (Molecular probes) on TBS380 (Turner Biosystems). Then bridge PCR was performed on cBot. Lastly, each library was loaded into one lane of the Illumina HiSeq 4000 for sequencing.
Quality control of raw sequence
The raw image data obtained from high-throughput RNA-sequencing was translated into raw FASTQ sequence data by Base Calling. Raw RNA-Seq data were filtered using FASTxtool SeqPrep (https://github.com/jstjohn/SeqPrep) and Sickle (https://github.com/najoshi/sickle) according to the following criteria: (1) reads containing sequencing adaptors; (2) nucleotides with a quality score < 20 were trimmed from the end of the sequence; once the quality value of reads after being trimmed less than 10, the whole sequence was abandoned; (3) N base rate of raw reads with more than 10% were discarded; (4) raw reads with base pairs less than 20 after adapter removal and trim was abnegated.
Clean reads mapping
Clean reads were aligned with the human reference genome, Ensemble GRCh38 v 84 (hg19), by using TopHat and Ensemble gene annotation. TopHat allows a maximum of two mismatches when mapping the reads to the reference genome. Fragment assembly and the relative expression of the reads with the normalized RNA-Seq fragment counts were processed by Cufflinks [33]. The assembled transcripts were merged to an integrated transcriptome of by using Cuffmerge. Fragments per kilobase of exon per million mapped reads (FPKM) were used to determine the transcription abundance of lncRNA and mRNA.
Differentially expressed lncRNA and differentially expressed mRNA analysis
Cuffdiff was used to calculate the FPKM of each reads. Paired t-tests were carried out for expression testing between STAD and adjacent non-tumor tissues [33]. FDR < 0.05 and |log2FC > 1| was selected as the criteria for DElncRNAs and FDR < 0.05 was set as the threshold of DEmRNAs after applying Benjamini-Hochberg correction for multiple test.
Co-expression analysis and network construction between DElncRNAs and DEmRNAs
DElncRNA-DEmRNA co-expression network was performed to explore the critical roles of DElncRNA and DEmRNA in progression of STAD. Pearson correlation coefficient (PCC) reflecting co-expression relationship between DElncRNAs and DEmRNAs were calculated according to their expression levels. DElncRNA-DEmRNA pairs with |PCC| ≥ 0.90 were retained for network construction [34]. The DElncRNA-DEmRNA network was visualized by Cytoscape 3.1 (http://cytoscape.org/) [35].
Gene Ontology and KEGG enrichment analysis
Gene Ontology (GO) function and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were used to predict the biological function of DEmRNAs [36]. Online GeneCoDis3 (http://genecodis.cnb.csic.es/analysis) was used to enrich GO terms and KEGG pathways [37, 38]. FDR < 0.05 was set as the cut-off for significant GO terms and KEGG pathways.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA of STAD and adjacent non-tumor tissues were extracted using Trizol (Invitrogen, CA, USA) according to the manual instructions. The M-MLV Reverse Transcriptase kit (Promega, WI, USA) was used to synthesize the cDNA. qRT-PCR reactions were performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on the Rotor gene 3000 (Corbett, Brisbane, Australia). β-actin was used as internal control for mRNA detection. The relative expression of candidate genes was calculated using the 2–ΔΔCT equation [39]. The PCR primers were shown as Supplementary Table 6. At least triple experiments were subjected to qRT-PCR verification.
The expression levels of DElncRNAs were validated through lncRNA expression profile generated from The Cancer Genome Atlas database
The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/tcga/) is a public funded project and has produced multidimensional data at the DNA, RNA and protein levels for a broad range of human tumor types. The Atlas of Noncoding RNAs in Cancer (TANRIC, http://ibl.mdanderson.org/tanric/_design/basic/index.html) is an open resource for interactive exploration of lncRNAs in the context of TCGA clinical and genomic data. In our study, TANRIC database was employed to retrieve the lncRNA expression profile of STAD based on RNA-sequencing generated from TCGA data. Lastly, the lncRNA expression data of 285 STAD tissues and 33 adjacent non-tumor tissues were retrieved. The difference of expression levels of DElncRNAs between STAD tissues and adjacent non-tumor tissues were compared and were delineated by box-plot.
Receiver operating characteristic analyses
In order to assess the diagnostic value of candidate DElncRNAs, receiver operating characteristic (ROC) analyses were performed using pROC package in R language. The area under the curve (AUC) under binomial exact confidence interval was calculated to generate the ROC curve.
Statistical analysis
Mean ± standard deviation and independent-samples t-test were used in the statistical analysis. P < 0.05 was considered as significant difference. To group samples based on expression values of mRNA and lncRNAs, we performed hierarchical cluster analysis [40]. These analyses were performed by R platform (http://www.r-project.org/) [41]. Circos plot was described as the distribution of DElncRNAs and DEmRNAs in chromosomes.
CONFLICTS OF INTEREST
All of authors declare that they have no conflicts of interest.
REFERENCES
1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10–29.
2. Plummer M, Franceschi S, Vignat J, Forman D, de Martel C. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer. 2015; 136:487–90. doi: 10.1002/ijc.28999.
3. Takahashi K, Kaira K, Shimizu A, Sato T, Takahashi N, Ogawa H, Yoshinari D, Yokobori T, Asao T, Takeyoshi I, Oyama T. Clinical significance of beta2-adrenergic receptor expression in patients with surgically resected gastric adenocarcinoma. Tumour Biol. 2016; 37:13885–92. doi: 10.1007/s13277-016-5139-2.
4. Lin C, Zhang Z, Xu Y, Wang R, Chen S, Gao J, Wang D, Huang Q, Tu X, Wang L. High Tumor Vascular Endothelial Growth Factor Expression Is Associated With Poorer Clinical Outcomes in Resected T3 Gastric Adenocarcinoma. Am J Clin Pathol. 2016; 146:278–88. doi: 10.1093/ajcp/aqw110.
5. Borg D, Hedner C, Nodin B, Larsson A, Johnsson A, Eberhard J, Jirstrom K. Expression of podocalyxin-like protein is an independent prognostic biomarker in resected esophageal and gastric adenocarcinoma. BMC Clin Pathol. 2016; 16:13. doi: 10.1186/s12907-016-0034-8.
6. Wang MY, He J, Zhu ML, Teng XY, Li QX, Sun MH, Wang XF, Yang YJ, Wang JC, Jin L, Wang YN, Wei QY. A Functional Polymorphism (rs2494752) in the AKT1 Promoter Region and Gastric Adenocarcinoma Risk in an Eastern Chinese Population. Sci Rep. 2016; 6: 20008. doi: 10.1038/srep20008.
7. Wang MY, Li QX, He J, Qiu LX, Wang YN, Li J, Sun MH, Wang XF, Yang YJ, Wang JC, Jin L, Wei QY. Genetic variations in the mTOR gene contribute toward gastric adenocarcinoma susceptibility in an Eastern Chinese population. Pharmacogenet Genomics. 2015; 25:521–30. doi: 10.1097/fpc.0000000000000163.
8. Cutcutache I, Wu AY, Suzuki Y, McPherson JR, Lei Z, Deng N, Zhang S, Wong WK, Soo KC, Chan WH, Ooi LL, Welsch R, Tan P, Rozen SG. Abundant copy-number loss of CYCLOPS and STOP genes in gastric adenocarcinoma. Gastric Cancer. 2016; 19:453–65. doi: 10.1007/s10120-015-0514-z.
9. Moazed D. Small RNAs in transcriptional gene silencing and genome defence. Nature. 2009; 457:413–20. doi: 10.1038/nature07756.
10. Rupaimoole R, Calin GA, Lopez-Berestein G, Sood AK. miRNA Deregulation in Cancer Cells and the Tumor Microenvironment. Cancer Discov. 2016; 6:235–46. doi: 10.1158/2159-8290.cd-15-0893.
11. Lloyd KA, Moore AR, Parsons BN, O’Hara A, Boyce M, Dockray GJ, Varro A, Pritchard DM. Gastrin-induced miR-222 promotes gastric tumor development by suppressing p27kip1. Oncotarget. 2016; 7:45462–78. doi: 10.18632/oncotarget.9990.
12. Li T, Lu YY, Zhao XD, Guo HQ, Liu CH, Li H, Zhou L, Han YN, Wu KC, Nie YZ, Shi YQ, Fan DM. MicroRNA-296-5p increases proliferation in gastric cancer through repression of Caudal-related homeobox 1. Oncogene. 2014; 33:783–93. doi: 10.1038/onc.2012.637.
13. Cheng J, Deng H, Xiao B, Zhou H, Zhou F, Shen Z, Guo J. piR-823, a novel non-coding small RNA, demonstrates in vitro and in vivo tumor suppressive activity in human gastric cancer cells. Cancer Lett. 2012; 315:12–7. doi: 10.1016/j.canlet.2011.10.004.
14. Cheng J, Guo JM, Xiao BX, Miao Y, Jiang Z, Zhou H, Li QN. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin Chim Acta. 2011; 412:1621–5. doi: 10.1016/j.cca.2011.05.015.
15. Dey BK, Mueller AC, Dutta A. Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription. 2014; 5:e944014. doi: 10.4161/ 21541272.2014.944014.
16. Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013; 14:699–712. doi: 10.1038/nrm3679.
17. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012; 22:1775–89. doi: 10.1101/gr.132159.111.
18. Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014; 5:8027–38. doi: 10.18632/oncotarget.2469.
19. Yang F, Bi J, Xue X, Zheng L, Zhi K, Hua J, Fang G. Up-regulated long non-coding RNA H19 contributes to proliferation of gastric cancer cells. FEBS J. 2012; 279:3159–65. doi: 10.1111/j.1742-4658.2012.08694.x.
20. Li H, Yu B, Li J, Su L, Yan M, Zhu Z, Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014; 5:2318–29. doi: 10.18632/oncotarget.1913.
21. Chen JS, Wang YF, Zhang XQ, Lv JM, Li Y, Liu XX, Xu TP. H19 serves as a diagnostic biomarker and up-regulation of H19 expression contributes to poor prognosis in patients with gastric cancer. Neoplasma. 2016; 63:223–30. doi: 10.4149/207_150821n454.
22. Hashad D, Elbanna A, Ibrahim A, Khedr G. Evaluation of the Role of Circulating Long Non-Coding RNA H19 as a Promising Novel Biomarker in Plasma of Patients with Gastric Cancer. J Clin Lab Anal. 2016; 30:1100–05. doi: 10.1002/jcla.21987.
23. Zhao J, Liu Y, Zhang W, Zhou Z, Wu J, Cui P, Zhang Y, Huang G. Long non-coding RNA Linc00152 is involved in cell cycle arrest, apoptosis, epithelial to mesenchymal transition, cell migration and invasion in gastric cancer. Cell Cycle. 2015; 14:3112–23. doi: 10.1080/15384101. 2015.1078034.
24. Liu YW, Sun M, Xia R, Zhang EB, Liu XH, Zhang ZH, Xu TP, De W, Liu BR, Wang ZX. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015; 6:e1802. doi: 10.1038/cddis.2015.150.
25. Xu ZY, Yu QM, Du YA, Yang LT, Dong RZ, Huang L, Yu PF, Cheng XD. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int J Biol Sci. 2013; 9:587–97. doi: 10.7150/ijbs.6339.
26. Zhang S, Chen S, Yang G, Gu F, Li M, Zhong B, Hu J, Hoffman A, Chen M. Long noncoding RNA HOTAIR as an independent prognostic marker in cancer: a meta-analysis. PLoS One. 2014; 9:e105538. doi: 10.1371/journal.pone.0105538.
27. Fei ZH, Yu XJ, Zhou M, Su HF, Zheng Z, Xie CY. Upregulated expression of long non-coding RNA LINC00982 regulates cell proliferation and its clinical relevance in patients with gastric cancer. Tumour Biol. 2016; 37:1983–93. doi: 10.1007/s13277-015-3979-9.
28. Fan Y, Wang YF, Su HF, Fang N, Zou C, Li WF, Fei ZH. Decreased expression of the long noncoding RNA LINC00261 indicate poor prognosis in gastric cancer and suppress gastric cancer metastasis by affecting the epithelial-mesenchymal transition. J Hematol Oncol. 2016; 9:57. doi: 10.1186/s13045-016-0288-8.
29. Zhang T, Cao C, Wu D, Liu L. SNHG3 correlates with malignant status and poor prognosis in hepatocellular carcinoma. Tumour Biol. 2016; 37:2379–85. doi: 10.1007/s13277-015-4052-4.
30. Chen N, Guo D, Xu Q, Yang M, Wang D, Peng M, Ding Y, Wang S, Zhou J. Long non-coding RNA FEZF1-AS1 facilitates cell proliferation and migration in colorectal carcinoma. Oncotarget. 2016; 7:11271–83. doi: 10.18632/oncotarget.7168.
31. Tong YS, Wang XW, Zhou XL, Liu ZH, Yang TX, Shi WH, Xie HW, Lv J, Wu QQ, Cao XF. Identification of the long non-coding RNA POU3F3 in plasma as a novel biomarker for diagnosis of esophageal squamous cell carcinoma. Mol Cancer. 2015; 14:3. doi: 10.1186/1476-4598-14-3.
32. Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F, Shi S, Feng X, Sun N, Liu Z, Skogerboe G, et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014; 63:1700–10. doi: 10.1136/gutjnl-2013-305806.
33. Ghosh S, Chan CK. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol Biol. 2016; 1374:339–61. doi: 10.1007/978-1-4939-3167-5_18.
34. Benesty J, Chen J, Huang Y, Cohen I. (2009). Pearson correlation coefficient. Noise reduction in speech processing: Springer. pp1–4.
35. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–504. doi: 10.1101/gr.1239303.
36. Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–62.
37. Zheng Q, Wang XJ. GOEAST: a web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008; 36:W358–W63.
38. Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011; 39:W316–22. doi: 10.1093/nar/gkr483.
39. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3:1101–8.
40. Zhao S, Guo Y. Advanced heat map and clustering analysis using heatmap3. 2014; 2014:986048. doi: 10.1155/2014/986048.
41. Gentleman R, Carey V, Huber W, Irizarry R, Dudoit S. Bioinformatics and computational biology solutions using R and Bioconductor. Springer Science & Business Media. 2006.