
Preclinical Background
PPARγ
Inhibition of PPARγ through a dominant-negative transgene or by pharmacologic intervention enables a transition from an ER- to an ER+ lineage enrichment in breast cancer
The evidence: The role of PPARγ in lineage specification is often thought of in the context of its ability to regulate several tumor suppressor genes. Evidence to support a role for PPARγ in the development of the ER+ lineage was provided by transgenic mice expressing the fusion protein Pax8PPARγ, a dominant-negative form of PPARγ [4, 5], that is expressed in follicular thyroid cancer as a result of a t(2;3)(q13;p25) translocation between the paired-box transcription factor Pax8 and PPARγ [4]. Induction of mammary carcinogenesis in this transgenic model led to the appearance of ER+ tumors that were exquisitely sensitive to the ER antagonist fulvestrant [6] (Figure 1). These findings led us to determine whether the irreversible PPARγ antagonist GW9662 could act as a pharmacologic mimic of Pax8PPARγ and similarly induce the appearance of ER+ tumors in an otherwise ER- animal model. GW9662 did in fact replicate many of the phenotypic features of Pax8PPARγ transgenic mice and similarly rendered tumors sensitive to fulvestrant [7] (Figure 1). Thus, it was now possible to pharmacologically manipulate tumor lineage by inhibiting PPARγ, and in essence achieve a synthetic lethal effect [8] to endocrine therapy.
Since Pax8PPARγ induced a progenitor cell phenotype by PPARγ suppression, we examined if the converse would be true, viz. whether deficiency of the progenitor cell factor Stem Cell Antigen-1 (Sca-1/Ly6a) would upregulate the expression of PPARγ. Induction of mammary carcinogenesis in Sca-1 knockout mice led to a marked increase in PPARγ expression and to a synthetic lethal effect by the PPARγ agonist GW7845 [9].
Further insight into how PPAR could modulate the ER+ tumor lineage was suggested by the coactivator/corepressor dynamics of the ER [10]. PPARγ interferes with ER transactivation by binding to canonical ER response elements [11, 12] in a fashion similar to ER inhibition of PPAR response element (PPRE)-dependent transcription [13]. PPARγ and PPARδ have opposing actions either by direct competition [14], coactivator competition [15] and/or ligand-dependent activation and repression [16]. Additional studies using MMTV-AIB1 transgenic mice support this notion, where AIB1coactivator expression led to the development of ER+ tumors [17, 18]. This phenotype is similar to what we have recently reported for MMTV-PPARδ mice [19], and supports the concept that ligand-dependent recruitment of coactivators to PPARδ promotes ER+ progenitor cell expansion and oncogenesis by blocking the negative regulatory effects of PPARγ on this lineage (Figure 1). Interestingly, tumorigenesis in both AIB1 and PPARδ mice was dependent on mTOR activation downstream of phospholipid catabolism and an inflammatory phenotype [17], which may suggest a possible link between lipid biosynthesis, ER+ breast cancer and obesity, particularly in postmenopausal women [20].
PPARδ
Overexpression of the PPARδ transgene in the presence of an agonist enables a transition from ER- to an ER+ lineage enrichment in breast cancer
The evidence: PPARδ was shown to stimulate mitotic clonal expansion of progenitor cells more than a decade ago [21, 22]. It was conceivable, therefore, that the selective PPARδ agonist GW501516 [23] would act as a tumor promoter in mammary carcinogenesis, which proved to be the case [24] (reviewed in [2, 25]). Conversely, disruption of PPARδ expression reduced tumorigenesis in experimental breast cancer models [26]. It is interesting to note that 50% of invasive breast cancers expressed moderate to high levels of PPARδ protein [2] and that 65% of this type of breast cancer express increased PPARδ mRNA, whereas, the reverse is true for normal breast (www.oncomine.org;TCGA database). Thus, the majority of aggressive breast cancers would be expected to be responsive to a PPARδ agonist or PPARγ antagonist that could reverse the negative regulation by PPARγ on the ER+ lineage.
Etiological factors relevant to breast cancer that are directly relevant to pharmacological modulation of PPARs are obesity and inflammation [27]. These processes ultimately provide a milieu for the biosynthesis of endogenous PPARδ ligands [28], including arachidonic metabolites PGI2 [29] under the control of the PPRE-regulated gene Cox2/Pges2 [30], 15-HETE [31] and polyunsaturated fatty acids [32-34], under the control of PPAR-responsive genes. In this context, PPARγ activation by GW501516 increased arachidonic and linoleic acids in mammary tumors [35], and elicited an inflammatory gene signature in other animal models [36, 37], which are also consistent with PPARδ-mediated repression of PPARγ [14]. Additionally, metabolomic analysis of these mice revealed increased levels of lysophosphatidic acid and phosphatidic acid, both positive effectors of mTOR activity, and rendered PPARδ mice sensitive to the antitumor activity of the rapamycin analog everolimus [17].
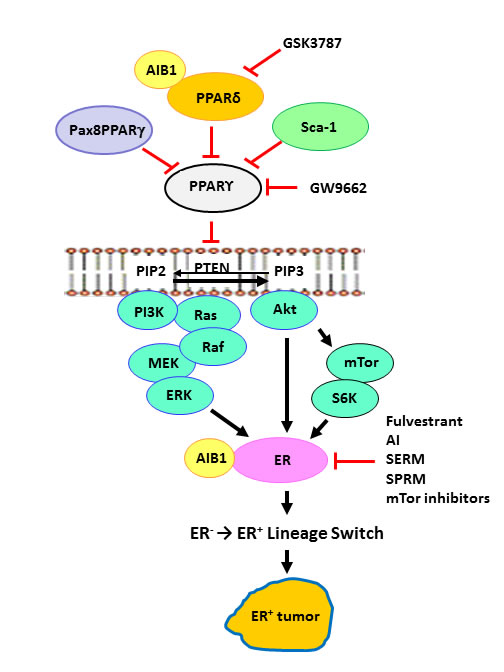
Figure 1: PPARs and the ER+ lineage. Dominant-negative Pax8-PPARg, PPARd, Sca-1/Ly6a and PPARg inhibitor GW9662 each result in attenuation of the tumor suppressor effects of PPARg, eg. PTEN expression [5-7, 9, 19, 24], which was previously shown to occur transcriptionally [39]. Higher ratios of PPARd/PPARg promote the expansion of the ER+ progenitor lineage, leading to development of ER+ tumors. This paradigm suggests that negative regulation of PPARg or positive regulation of PPARd will enhance sensitivity to endocrine and targeted therapy by a mechanism analogous to synthetic lethality. GSK3787, PPARd inhibitor; GW9662, PPARg inhibitor; AI, aromatase inhibitors; SERM, selective ER modulators; SPRM, selective PR modulators.
Clinical Relevance
The ability of PPAR agonists and antagonists to modulate oncogenic signaling pathways provides a therapeutic paradigm through which presentation of the ER+ lineage is favored, leading to several important clinical strategies for the treatment of ER- breast cancer.
First, PPARγ inhibition should enhance the appearance of the ER+ tumor lineage, enabling sensitivity to anti-ER therapy.
Second, PPARδ activation should similarly promote the development of ER+ oncogenesis through its negative regulatory effect on PPARγ, while simultaneously activating of mTor signaling.
Third, the opposing actions of PPARγ and PPARδ provide a framework to test this proposition, which would lead to a synthetic lethal approach to therapy, whereby phenotypically ER- tumors are rendered ER+ by treatment with a PPARγ antagonist and/or a PPARδ agonist to enable targeting the ER with endocrine therapy.
Fourth, Potential candidate populations might include patients with clinically overt triple-negative breast cancer or luminal B breast cancer with low ER expression.
Treatment Scheduling
We envision a therapeutic paradigm where patients would receive treatment with a PPARγ antagonist and/or a PPARδ agonist to activate the ER+ lineage simultaneously with chemotherapy plus an ER inhibitor such as fulvestrant or an aromatase inhibitor. Since we have also observed the dependence of PPAR-mediated tumorigenesis on mTOR signaling [19], combining endocrine therapy with a rapamycin analog should further block tumor progression as found in experimental models [38].
Acknowledgments
The authors acknowledge support from the National Institutes of Health and the Avon Foundation for Women.
Conflicts of Interest
The authors declare no conflicts of interest.
References
1. Kopelovich L, Fay JR, Glazer RI, Crowell JA. Peroxisome proliferator-activated receptor modulators as potential chemopreventive agents. Molecular Cancer Therapeutics. 2002; 1:357-363.
2. Glazer RI, Yuan H, Xie Z, Yin Y. PPARgamma and PPARdelta as modulators of neoplasia and cell fate. PPAR Res. 2008; 2008:247379.
3. Glazer RI. PPARdelta as a Metabolic Initiator of Mammary Neoplasia and Immune Tolerance. PPAR Res. 2016; 2016:3082340.
4. Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma. Science. 2000; 289:1357-1360.
5. Yin Y, Yuan H, Wang C, Pattabiraman N, Rao M, Pestell RG, Glazer RI. 3-Phosphoinositide-Dependent Protein Kinase-1 Activates the Peroxisome Proliferator-Activated Receptor-{gamma} and Promotes Adipocyte Differentiation. Mol Endocrinol. 2006; 20:268-278.
6. Yin Y, Yuan H, Zeng X, Kopelovich L, Glazer RI. Inhibition of peroxisome proliferator-activated receptor gamma increases estrogen receptor-dependent tumor specification. Cancer Res. 2009; 69:687-694.
7. Yuan H, Kopelovich L, Yin Y, Lu J, Glazer RI. Drug-targeted inhibition of peroxisome proliferator-activated receptor-gamma enhances the chemopreventive effect of anti-estrogen therapy. Oncotarget. 2012; 3:345-356. doi: 10.18632/oncotarget.457.
8. Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nature reviews Cancer. 2005; 5:689-698.
9. Yuan H, Upadhyay G, Yin Y, Kopelovich L, Glazer RI. Stem cell antigen-1 deficiency enhances the chemopreventive effect of peroxisome proliferator-activated receptor{gamma} activation. Cancer Prev Res (Phila). 2012; 5:51-60.
10. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000; 103:843-852.
11. Houston KD, Copland JA, Broaddus RR, Gottardis MM, Fischer SM, Walker CL. Inhibition of proliferation and estrogen receptor signaling by peroxisome proliferator-activated receptor gamma ligands in uterine leiomyoma. Cancer Res. 2003; 63:1221-1227.
12. Keller H, Givel F, Perroud M, Wahli W. Signaling cross-talk between peroxisome proliferator-activated receptor/retinoid X receptor and estrogen receptor through estrogen response elements. Mol Endocrinol. 1995; 9:794-804.
13. Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, Giordano F, Ando S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005; 11:6139-6147.
14. Shi Y, Hon M, Evans RM. The peroxisome proliferator-activated receptor delta, an integrator of transcriptional repression and nuclear receptor signaling. Proc Natl Acad Sci U S A. 2002; 99:2613-2618.
15. Gustafsson MC, Knight D, Palmer CN. Ligand modulated antagonism of PPARgamma by genomic and non-genomic actions of PPARdelta. PloS one. 2009; 4:e7046.
16. Adhikary T, Kaddatz K, Finkernagel F, Schonbauer A, Meissner W, Scharfe M, Jarek M, Blocker H, Muller-Brusselbach S, Muller R. Genomewide analyses define different modes of transcriptional regulation by peroxisome proliferator-activated receptor-beta/delta (PPARbeta/delta). PloS One. 2011; 6:e16344.
17. Torres-Arzayus MI, Font de Mora J, Yuan J, Vazquez F, Bronson R, Rue M, Sellers WR, Brown M. High tumor incidence and activation of the PI3K/AKT pathway in transgenic mice define AIB1 as an oncogene. Cancer Cell. 2004; 6:263-274.
18. Torres-Arzayus MI, Yuan J, DellaGatta JL, Lane H, Kung AL, Brown M. Targeting the AIB1 oncogene through mammalian target of rapamycin inhibition in the mammary gland. Cancer Res. 2006; 66:11381-11388.
19. Yuan H, Lu J, Xiao J, Upadhyay G, Umans R, Kallakury B, Yin Y, Fant ME, Kopelovich L, Glazer RI. PPARdelta induces estrogen receptor-positive mammary neoplasia through an inflammatory and metabolic phenotype linked to mTOR activation. Cancer Res. 2013; 73:4349-4361.
20. Khan S, Shukla S, Sinha S, Meeran SM. Role of adipokines and cytokines in obesity-associated breast cancer: therapeutic targets. Cytokine Growth Factor Rev. 2013; 24:503-513.
21. Hansen JB, Zhang H, Rasmussen TH, Petersen RK, Flindt EN, Kristiansen K. Peroxisome proliferator-activated receptor delta (PPARdelta )-mediated regulation of preadipocyte proliferation and gene expression is dependent on cAMP signaling. J Biol Chem. 2001; 276:3175-3182.
22. Jehl-Pietri C, Bastie C, Gillot I, Luquet S, Grimaldi PA. Peroxisome-proliferator-activated receptor delta mediates the effects of long-chain fatty acids on post-confluent cell proliferation. Biochem J. 2000; 350:93–98.
23. Pelton P. GW-501516 GlaxoSmithKline/Ligand. Curr Opin Investig Drugs. 2006; 7:360-370.
24. Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, Kopelovich L, Glazer RI. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005; 65:3950-3957.
25. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nature Reviews Cancer. 2012; 12:181-195.
26. Ghosh M, Ai Y, Narko K, Wang Z, Peters JM, Hla T. PPARdelta is pro-tumorigenic in a mouse model of COX-2-induced mammary cancer. Prostaglandins Other Lipid Mediat. 2009; 88:97-100.
27. Hursting SD, Hursting MJ. Growth signals, inflammation, and vascular perturbations: mechanistic links between obesity, metabolic syndrome, and cancer. Arterioscler Thromb Vasc Biol. 2012; 32:1766-1770.
28. Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006; 116:590-597.
29. Gupta RA, Tan J, Krause WF, Geraci MW, Willson TM, Dey SK, DuBois RN. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc Natl Acad Sci U S A. 2000; 97:13275-13280.
30. Yoshinaga M, Kitamura Y, Chaen T, Yamashita S, Tsuruta S, Hisano T, Ikeda Y, Sakai H, Nakamura K, Takayanagi R, Muto Y. The simultaneous expression of peroxisome proliferator-activated receptor delta and cyclooxygenase-2 may enhance angiogenesis and tumor venous invasion in tissues of colorectal cancers. Dig Dis Sci. 2009; 54:1108-1114.
31. Naruhn S, Meissner W, Adhikary T, Kaddatz K, Klein T, Watzer B, Muller-Brusselbach S, Muller R. 15-hydroxyeicosatetraenoic acid is a preferential peroxisome proliferator-activated receptor beta/delta agonist. Mol Pharmacol. 2010; 77:171-184.
32. Forman BM, Chen J, Evans RM. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc Natl Acad Sci U S A. 1997; 94:4312-4317.
33. Xu HE, Lambert MH, Montana VG, Parks DJ, Blanchard SG, Brown PJ, Sternbach DD, Lehmann JM, Wisely GB, Willson TM, Kliewer SA, Milburn MV. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol Cell. 1999; 3:397-403.
34. Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995; 270:23975-23983.
35. Pollock CB, Yin Y, Yuan H, Zeng X, King S, Li X, Kopelovich L, Albanese C, Glazer RI. PPARdelta activation acts cooperatively with 3-phosphoinositide-dependent protein kinase-1 to enhance mammary tumorigenesis. PloS One. 2011; 6:e16215.
36. Pollock CB, Rodriguez O, Martin PL, Albanese C, Li X, Kopelovich L, Glazer RI. Induction of metastatic gastric cancer by peroxisome proliferator-activated receptordelta activation. PPAR Res. 2010; 2010:571783.
37. Romanowska M, Reilly L, Palmer CN, Gustafsson MC, Foerster J. Activation of PPARbeta/delta causes a psoriasis-like skin disease in vivo. PloS one. 2011; 5:e9701.
38. Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, Chen S, Lane HA. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005; 11:5319-5328.
39. Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001; 11:764-768.