
INTRODUCTION
Apoptosis is a critical cellular process in maintaining normal tissue homeostasis [1]. Proliferating cells in the absence of apoptosis is considered as an initial hallmark of cancer [2, 3]. As a mediator of the mitochondrial apoptotic pathway [4], p53 upregulated modulator of apoptosis (PUMA) is known to result in the displacement and activation of Bax/Bak through its binding to anti-apoptotic Bcl-2 proteins, leading to mitochondrial dysfunction and caspase activation, hence initiating apoptosis [5]. Absence of PUMA could efficiently protect normal cells (including thymocyte and lymphocyte) from the apoptosis induced by γ-irradiation and DNA-damaging drugs [6]. In many cancer cells, PUMA-dependent apoptosis is typically blocked by the elevated expression of myeloid cell leukemia-1 (Mcl-1, one of the most frequently overexpressed anti-apoptotic proteins) and the resulting deregulation of the Mcl-1-PUMA interaction [7]. While activation of Mcl-1 leads to survival of cancer cells, inhibition of Mcl-1 has been found to eliminate a series of cancer cells [8–11]. Therefore, small-molecule inhibitors of Mcl-1 can be useful as potential therapeutic agents for cancer treatments [10, 12, 13]. However, many Mcl-1 inhibitors obtained from prior efforts are potent to activate the mitochondrial apoptotic pathway by upregulating PUMA [10, 14], leading to apoptosis in both cancer and normal cells. The PUMA-dependent apoptosis of non-cancerous cells is a common side-effect of major agents used in current cancer therapy [15]. Therefore, it is challenging but rewarding to develop anti-cancer agents of high effectiveness and safety. As mentioned above, the antagonism between Mcl-1 and PUMA is vital to the survival of cells [16, 17]. When PUMA is upregulated, cells tend to apoptosis, on the contrary, when Mcl-1 is upregulated, cells tend to survival. The high-affinity interaction between Mcl-1 and PUMA has been verified by series of studies including yeast two-hybrid assay, co-immunoprecipitation studies and structural studies [16]. PUMA is reported to bind as an amphipathic helix in a deep hydrophobic groove on the surface of Mcl-1 through its BH3 domain [18, 19]. Targeting the druggable interface of Mcl-1-PUMA is promising to discover dual-acting compounds that could disrupt the interaction between Mcl-1 and PUMA, providing a way to settle the conflicts between effectiveness and side-effects of anti-cancer drugs [13, 20, 21].
In this work, we for the first time used a combination of computer modelling and biological assays, to investigate the fundamental Mcl-1-PUMA interactions and to seek potential small molecules with high anti-cancer activity and safety. While some inhibitors of anti-apoptotic proteins (like ABT-263 and ABT-199) show promise in clinical trials for multiple cancers including acute myeloid leukemia and small-cell lung cancer [22, 23], our compound 8 (Comp8, Scheme 1) distinguishes itself in its dual activity to both suppress cancer cells and inhibit PUMA-dependent apoptosis of non-cancerous cells. This work not only offers validation for targeting Mcl-1-PUMA interface as a reliable strategy to develop novel anti-cancer drugs with both pronounced efficacy against cancers and low side effect in normal tissues, but also serves as a probe to achieve safer cancer therapies by modulating Mcl-1-PUMA interaction.
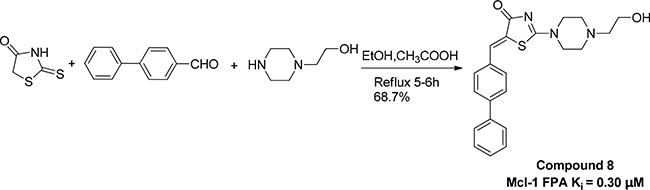
Scheme 1: Preparation of compound 8.
RESULTS AND DISCUSSION
The starting point for our study is the determination of compounds that potentially act on the Mcl-1-PUMA interface, using pharmacophore-based virtual screening. Molecular dynamics (MD) simulations of a 50 ns timescale were performed with the Mcl-1-PUMA complex models to provide the energetics and plasticity details of the interface. Using structural data (revealed by pairwise per residue free energy decomposition method in Supplementary Table 1) of the MD representative structure of Mcl-1-PUMA complex (Figure 1A) and the common features (revealed by multi-conformational alignment method) of the eight PUMA modulators (Supplementary Figure 2), we constructed a structure-based (Figure 1C left) and ligand-based (Figure 1D) pharmacophore model, respectively. A prototype of the combined pharmacophore model was further developed (Figure 1C right) by merging the two models. To improve the combined prototype for high sensitivity and specificity, the combined model was finally optimized to one hydrophobic feature (I144PUMA-H205Mcl-1) in the structure-based model, two hydrogen bond features (hydrogen bond acceptor and hydrogen bond donor) in the ligand-based model, and three shared pharmacophore features (R142PUMA-D237Mcl-1, D146PUMA-R244Mcl-1 and A145PUMA-G243Mcl-1) of the two models. Compounds constitute the SPECS database, were queried by the final pharmacophore model (Figure 1E). The eight highest-scoring compounds (namely Comp 1–8, see Supplementary Table 3) were directly purchased from SPECS and prepared for further biological tests.
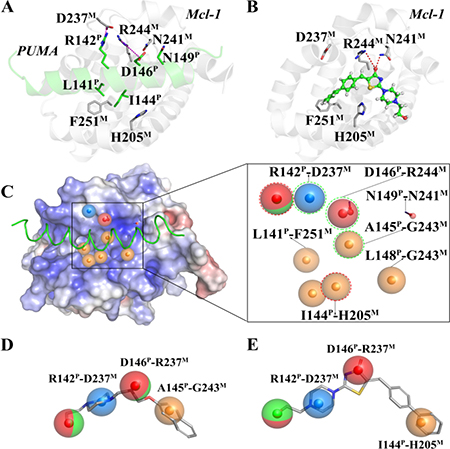
Figure 1: Key interactions at the interfaces of (A) Mcl-1-PUMA and (B) Mcl-1-Comp8 complexes. (C) The structure-based pharmacophore model (Left); The pharmacophore prototype merging (C) and (D) (Right). (D) The best ligand-based pharmacophore model. (E) The final pharmacophore model for prospective virtual screening. Details are present in the Supplementary Information.
Given the crucial role of R244Mcl-1 in developing Mcl-1 inhibitors [12, 19, 24], we firstly tested the inhibitory effects of these eight compounds on Mcl-1 by FPA competition assay. As shown in Figure 2A, Mcl-1 is significantly deactivated by Comp8 (Ki = 0.30 ± 0.04 μM). The Ki value is comparable with or lower than the counterparts of many other known Mcl-1 inhibitors (like UMI-77, Ki = 0.49 μM) [12, 19, 25, 26]. As for other seven compounds, they shed little effect on inhibiting Mcl-1 (Supplementary Figure 3). In the stable complex (Figure 1B) obtained by docking Comp8 into the binding center of Mcl-1 (Supplementary Figure 4) [27], Comp8 binds within the BH3-binding groove of Mcl-1 and is anchored by two polar contacts formed between the carboxylate group (Comp8) and N241/R244 (Mcl-1). Moreover, van der Waals interactions provided by the residue H205Mcl-1 are pivotal to the stability of Mcl-1-Comp8 complex. The stable occupation of Comp8 in the PUMA-binding groove of Mcl-1 possibly suppresses the Mcl-1-dependent anti-apoptosis of multiple cancer cells. To verify this, Comp8 was tested alongside a known BH3 mimetic ABT-263 for its activity to induce apoptosis in three cancer cell lines (A2780, MCF-7, SMMC-7721). These are well-known cancer cells that depend on Mcl-1 for continuous survival. As shown, on A2780, MCF-7 and SMMC-7721, the inhibitory effect of Comp8 is comparable with the counterpart of ABT-263 (IC50 in the range between 20 to 50 μM, see Figure 3) which is currently under clinical trials in numerous malignancies [28, 29]. Thereafter, flow cytometry detection (FCM) and western blot (WB) analysis of three apoptotic marker proteins (Caspase-3, Cyto-c and Bcl-2) were performed. According to the results of FCM, a substantial increase of apoptotic cells was observed in cancer cells treated with Comp8 (Supplementary Figure 5). WB analysis also revealed similar results (Supplementary Figure 6), the changes of three apotosis indicators fitted well with the characteristics of apoptosis. All the results suggested that Comp8 inhibited cancer cells by promoting apoptosis. The consistency of FPA competition assays and cell tests suggests that Comp8 can be developed as a potential lead compound against cancer through inhibition of Mcl-1.
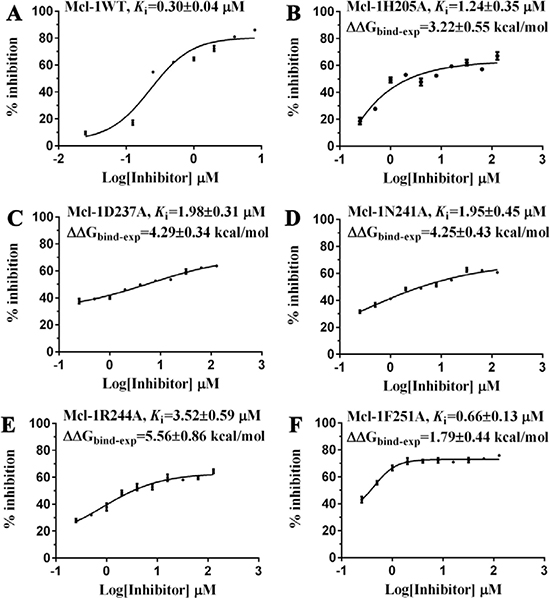
Figure 2: The inhibitory effects of Comp8 on (A) wild and (B–F) mutant types of Mcl-1. Details are present in the Supplementary Information.
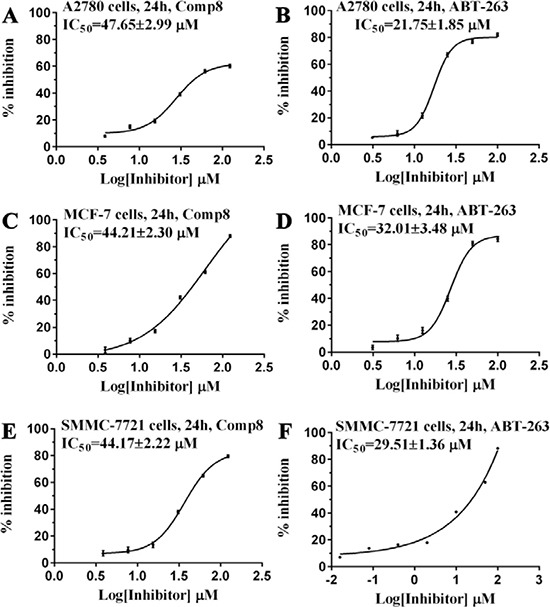
Figure 3: Comparison of (A, C, E) Comp8 and (B, D, F) ABT-263 on the activity to induce the apoptosis of three cancer cell lines.
By overlaying the two representative structures of Mcl-1-PUMA and Mcl-1-Comp8 complexes from MD simulations (Supplementary Figure7), we find that Mcl-1 residues involved in the interactions with PUMA and Comp8 are greatly overlapped [18]. Like the hydrogen bond formed between R244 and the carboxylate group of Comp8, the same R244 forms a hydrogen bond with a conserved Asp of BH3 domain derived from BH3-only Bcl-2 proteins (PUMA as a representative) as well [12, 18].
Before testing the activity of Comp8 to disrupt the Mcl-1-PUMA interaction, it is necessary to determine whether these overlapped residues are key sites. Five Mcl-1 residues (H205, N241, R244, F251 and D237) which contribute significantly to Comp8/PUMA binding to Mcl-1 are subjected to site-directed mutagenesis (Supplementary Table 1). As revealed by FPA assays, the affinity between Mcl-1 and Comp8 is decreased at varying degree due to the mutation of each residue (Figure 2), while such mutations shed little effects on the binding of the fluorescence probe we used in FPA (Supplementary Table 4). Thereinto, D237, N241 and R244 are regarded as key residues in the Mcl-1-Comp8 interaction with the experimental binding free energy changes (ΔΔGbind-exp, Figure 2) beyond the threshold of hot-spot (> 4.0 kcal/mol) [30]. Likewise, these residues also contribute to the stabilization of the Mcl-1-PUMA complex [18]. This great overlap of key residues in Mcl-1-Comp8 and Mcl-1-PUMA complexes supports the postulation that Comp8 can disrupt Mcl-1-PUMA interaction. The overexpression of PUMA can cause severe death of DLD-1 cells (Figure 4A) [31]. However, the PUMA-dependent apoptosis is significantly inhibited by Comp8 (IC50= 38.93 ± 0.91 μM) (Figure 4B), which suggests its high selectivity to target the BH3 domain of PUMA (Figure 4D). In our tests, normal cells were fairly sensitive to the upregulation of PUMA, HUVECs infected with Ad-PUMA (Adeovirus overexpressing PUMA) exhibted rapid apoptosis. Even so, the addition of low-dose Comp8 (less than 12.5 μM) still shed inhibitory effect on the PUMA-induced apoptosis of HUVECs (Supplementary Figure 8).
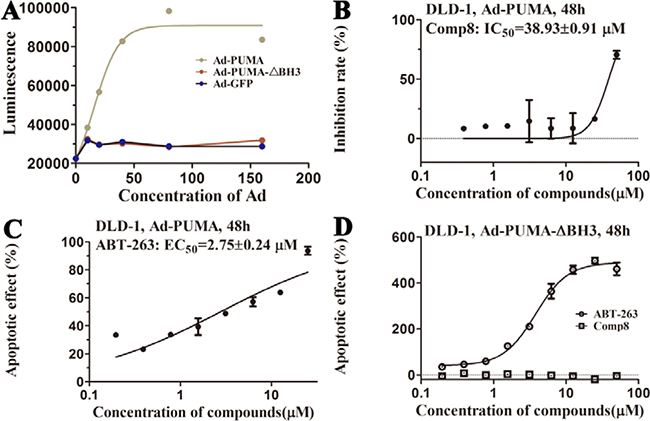
Figure 4: (A) Apoptosis is induced by PUMA with BH3 domain. The action curves of (B) Comp8 and (C) ABT-263 on apoptosis. (D) The effects of ABT-263 and Comp8 on DLD-1 cells infected with Ad-PUMA-ΔBH3. PUMA-dependent apoptosis can be effectively blocked by Comp8 which selectively targeting PUMA BH3.
In summary, we have identified a dual-acting compound, Comp8, at the same time to reduce multiple cancer cells and to inhibit PUMA-mediated apoptosis. Despite the comparable activity with most previously reported compounds in Mcl-1 inhibition [12, 19, 24], Comp8 displays crucial, unique activity to also inhibit PUMA-dependent apoptosis. This work represents the first study that fully considers such dual activity in the discovery of potential anti-cancer compounds. Moreover, apart from R244, the binding site of Comp8 is quite different from other Mcl-1 inhibitors [10, 19]. The discovery of dual-acting Comp8 sheds light on the discovery of new anti-cancer drugs as well as the improvement of current cancer treatments, especially when considering the PUMA-dependent apoptosis caused by γ-radiation and/or chemotherapeutic agents [32–35]. The convenient synthesis and purification of Comp8 (Scheme 1) together with its small molecular size further enhance its potential value in the the development of safer cancer therapeutics targeting Mcl-1-PUMA interface.
MATERIALS AND METHODS
The Mcl-1-PUMA complex was retrieved from the crystal structure of the mouse Mcl-1 complexed with PUMA (PDB ID, 2ROC) [18]; The Mcl-1 complexed with compound 8 (Comp8) was constructed by molecular docking simulations using the program GOLD5.3 [27]; The molecular dynamic (MD) simulations for Mcl-1-PUMA and Mcl-1-Comp8 complexes were carried out by Amber12 package [36]; The final pharmacophore model for prospective virtual screening was designed in combination with the structure- and ligand-based pharmacophore modeling by using Ligandscout4.09 [37]; The pharmacophore-based virtual screening was performed using the Iscreen module provided by LigandScout4.09 [37], Only the compounds that matched all pharmacophore features were considered as a hit; The candidate compounds were subjected to FPA assay and cell tests to check their corresponding activities on disrupting Mcl-1-PUMA interface. All the details of experimental procedures were present in the Supplementary Information.
Author contributions
S.Y.Z., H.C., J.N.L and J.Y.L. conceived the project. J.Y.L. and Z.T. designed the experiment. J.Y.L., Z.T., N.Z., X.Y.L. and C.Y.L. performed the experiments and prepared the manuscript. H.C. and S.Y.Z. supervised the study and contributed reagents/materials. All authors contributed to data analysis.
ACKNOWLEDGMENTS
The research was supported by the National Natural Science Foundation of China (grant number 81573343), the National Natural Science Foundation of China (grant number 21503272), and the General Financial Grant from the China Postdoctoral Science Foundation (grant number 2015M572753).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
REFERENCES
1. Schwartzman RA, Cidlowski JA. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocr Rev. 1993; 14:133–51.
2. Rudin CM, Thompson CB. Apoptosis and disease: regulation and clinical relevance of programmed cell death. Annu Rev Med. 1997; 48:267–81.
3. Favaloro B, Allocati N, Graziano V, Di Ilio C, De Laurenzi V. Role of apoptosis in disease. Aging (Albany NY). 2012; 4:330–49.
4. Bouchet BP, Caron de Fromentel C, Puisieux A, Galmarini CM. p53 as a target for anti-cancer drug development. Crit Rev Oncol Hematol. 2006; 58:190–207.
5. Yu J, Zhang L. PUMA, a potent killer with or without p53. Oncogene. 2008 (Suppl 1); 27:S71–83.
6. Yu J, Zhang L. No PUMA, no death: implications for p53-dependent apoptosis. Cancer Cell. 2003; 4:248–49.
7. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9:47–59.
8. Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009; 66:1326–36.
9. Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, Alexander WS, Lowe SW, Robb L, Strasser A. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012; 26:120–25.
10. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, Bruno A, Csekei M, Paczal A, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016; 538:477–82.
11. Wei SH, Dong K, Lin F, Wang X, Li B, Shen JJ, Zhang Q, Wang R, Zhang HZ. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008; 62:1055–64.
12. Bruncko M, Wang L, Sheppard GS, Phillips DC, Tahir SK, Xue J, Erickson S, Fidanze S, Fry E, Hasvold L, Jenkins GJ, Jin S, Judge RA, et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J Med Chem. 2015; 58:2180–94.
13. Akçay G, Belmonte MA, Aquila B, Chuaqui C, Hird AW, Lamb ML, Rawlins PB, Su N, Tentarelli S, Grimster NP, Su Q. Inhibition of Mcl-1 through covalent modification of a noncatalytic lysine side chain. Nat Chem Biol. 2016; 12:931–36.
14. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson ÅB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev. 2013; 27:1365–77.
15. Hanusova V, Skalova L, Kralova V, Matouskova P. Potential anti-cancer drugs commonly used for other indications. Curr Cancer Drug Targets. 2015; 15:35–52.
16. Mei Y, Du W, Yang Y, Wu M. Puma(*)Mcl-1 interaction is not sufficient to prevent rapid degradation of Mcl-1. Oncogene. 2005; 24:7224–37.
17. Delbridge AR, Opferman JT, Grabow S, Strasser A. Antagonism between MCL-1 and PUMA governs stem/progenitor cell survival during hematopoietic recovery from stress. Blood. 2015; 125:3273–80.
18. Day CL, Smits C, Fan FC, Lee EF, Fairlie WD, Hinds MG. Structure of the BH3 domains from the p53-inducible BH3-only proteins Noxa and Puma in complex with Mcl-1. J Mol Biol. 2008; 380:958–71.
19. Friberg A, Vigil D, Zhao B, Daniels RN, Burke JP, Garcia-Barrantes PM, Camper D, Chauder BA, Lee T, Olejniczak ET, Fesik SW. Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J Med Chem. 2013; 56:15–30.
20. Thiel P, Kaiser M, Ottmann C. Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angew Chem Int Ed Engl. 2012; 51:2012–18.
21. Arkin M, Tang Y, Wells J. Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol. 2004; 3:301–17.
22. Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, Dive C, Enschede SH, Nolan C, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol. 2011; 29:909–16.
23. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, Cortes J, DeAngelo DJ, Debose L, Mu H, Döhner H, Gaidzik VI, Galinsky I, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014; 4:362–75.
24. Burke JP, Bian Z, Shaw S, Zhao B, Goodwin CM, Belmar J, Browning CF, Vigil D, Friberg A, Camper DV, Rossanese OW, Lee T, Olejniczak ET, Fesik SW. Discovery of tricyclic indoles that potently inhibit Mcl-1 using fragment-based methods and structure-based design. J Med Chem. 2015; 58:3794–805.
25. Leverson JD, Zhang H, Chen J, Tahir SK, Phillips DC, Xue J, Nimmer P, Jin S, Smith M, Xiao Y, Kovar P, Tanaka A, Bruncko M, et al. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis. 2015; 6:e1590–1590.
26. Abulwerdi F, Liao C, Liu M, Azmi AS, Aboukameel A, Mady AS, Gulappa T, Cierpicki T, Owens S, Zhang T, Sun D, Stuckey JA, Mohammad RM, Nikolovska-Coleska Z. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol Cancer Ther. 2014; 13:565–75.
27. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997; 267:727–48.
28. Wang F, Fesik SW. 16. Discovery of inhibitors of protein-protein interactions using fragment-based methods: Wiley-VCH Verlag GmbH & Co. KGaA).
29. Kipps TJ, Eradat H, Grosicki S, Catalano J, Cosolo W, Dyagil IS, Yalamanchili S, Chai A, Sahasranaman S, Punnoose E, Hurst D, Pylypenko H. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma. 2015; 56:2826–33.
30. Moreira IS, Fernandes PA, Ramos MJ. Computational alanine scanning mutagenesis—an improved methodological approach. J Comput Chem. 2007; 28:644–54.
31. Peddaboina C, Jupiter D, Fletcher S, Yap JL, Rai A, Tobin RP, Jiang W, Rascoe P, Rogers MK, Smythe WR, Cao X. The downregulation of Mcl-1 via USP9X inhibition sensitizes solid tumors to Bcl-xl inhibition. BMC Cancer. 2012; 12:541.
32. Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, Look AT. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005; 123:641–53.
33. Yu H, Shen H, Yuan Y, XuFeng R, Hu X, Garrison SP, Zhang L, Yu J, Zambetti GP, Cheng T. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose gamma-irradiation. Blood. 2010; 115:3472–80.
34. Qiu W, Leibowitz B, Zhang L, Yu J. Growth factors protect intestinal stem cells from radiation-induced apoptosis by suppressing PUMA through the PI3K/AKT/p53 axis. Oncogene. 2010; 29:1622–32.
35. Mustata G, Li M, Zevola N, Bakan A, Zhang L, Epperly M, Greenberger JS, Yu J, Bahar I. Development of small-molecule PUMA inhibitors for mitigating radiation-induced cell death. Curr Top Med Chem. 2011; 11:281–90.
36. Case DA, Betz RM, Botello-Smith W, Cerutti DS, Cheatham TE 3rd, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, et al. AMBER 12. University of California, San Francisco. 2012.
37. Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. 2005; 45:160–69.