
INTRODUCTION
According with the recently published statistics on cancer in USA, B cell-chronic lymphocytic leukemia (CLL) remains the most common form of adult leukemia in the Western world, with 18,960 new cases expected in 2016 and 4,660 estimated death [1]. CLL is defined a malignant lymphoproliferative disorder of mature clonal B lymphocytes (B-CLL) that accumulate in the blood and other lymphoid tissues. The diagnosis of CLL occurs when B-CLL count is > 5,000/μL. Other features of the disease are established and periodically reviewed by the international working group of CLL (iwCLL) [2, 3]. Although the origin of B-CLL is still controversial, recent evidence suggest that genetic and epigenetic alterations occurring in pluripotent haematopoietic stem cells (HSCs) in the earliest phase of B lymphocytes may possibly lead to CLL [2, 4]. According to this model, antigenic stimulation of CLL HSCs may lead to selection and expansion of mature B cells, with the generation of oligoclonal populations. Additional genetic and epigenetic changes, as well as micro-environmental factors and BCR (B cell receptor) stimulation generate the population of cells which are currently considered the precursor of CLL, i.e., monoclonal B cell lymphocytosis (MBL) [5, 2]. CLL is classified into two major subgroups, in relation to the presence or absence of mutations in the Ig heavy chain variable region (IGHV) genes, to whom correspond different pathogenesis and prognosis. The IGHV-mutated (IGHV-M) forms of CLL are less aggressive with a median survival of 20 years, respect to the 8 years for the IGHV-unmutated (IGHV-UM) CLL [6, 7]. However, despite the importance of the IGHV mutational status as prognostic index of the disease, its determination is expensive and not always available in routine laboratories. These practical difficulties have been partially bypassed by the introduction of alternative biomarkers, such as the expression of Zap70 and CD38, both easily detectable by flow cytometry. The tyrosine kinase Zap70 is involved in T-cell receptor signaling and regulation, while CD38 is a surface antigen. Both markers are highly expressed in IGHV-UM CLL and their over-expression is associated with poor diagnosis and shorter time to treatment and survival, although the correlation with the aggressiveness of the disease and resistance to therapy is more pronounced for CLL expressing Zap-70, than for those with high level of CD38 whose expression vary over time [8, 9].
In the recent years, the efficacy of therapeutic treatments against CLL significantly improved. Currently, in patients with advanced stage disease (Rai III and IV or Binet C) [10, 11], the combination immunochemotherapy consisting of fludarabine, cyclophosphamide and the anti-CD20 monoclonal antibody rituximab (FC-R protocol) resulted in an ORR (overall response rate) of 95%, CR (complete response) rates of 72%, a higher eradication of minimal residual disease and longer duration of response [12, 13]. Unfortunately, despite these significant therapeutic improvements, complete remission remains rare and treatment failure rate is still high, especially in fludarabine-refractory patients and in those carrying TP53 inactivation [8, 14].
Searching for novel treatments, the interest of several groups in the last 10–15 years focused on the possibility to re-establish apoptosis sensitivity in CLL [15–17]. In general, the strategy consists in inhibiting the activity, or lowering the expression of anti-apoptotic factors, mainly belonging to the Bcl-2 and IAP families, which are often over-expressed in CLL as a consequence of the constitutive activation of pro-survival pathways [15]. Among the several natural and synthetic compounds tested, those belonging to so-called group of “BH3-mimetic” deserved particular attention [18, 19]. This term indicates small compounds able to “mimic” the high binding affinity of BH3-only proteins, such as Bim, Bid, PUMA, NOXA, which are natural ligands and inhibitors of pro-survival Bcl-2 family members, which sequester pro-apoptotic Bax and Bak factors. As a consequence of the binding between BH3-only agents and anti-apoptotic Bcl-2 factors, Bax and Bak are released, oligomerize and form pores into the mitochondrial outer membrane. Free cytochrome c activates caspases and induces apoptosis [20, 21]. ABT-737 can be considered the founder of this class of pharmacological agents [22]. It binds with high affinity to Bcl-2, Bcl-XL and Bcl-w, but does not antagonize other anti-apoptotic members, such as Mcl-1 or Bfl-1/A1, which determine resistance to ABT-737 in CLL [23]. The [Mcl-1 + Bfl-1]/Bcl-2 ratio has been validated in a panel of leukemic cell lines as an index to predict the response of CLL to ABT-737 [24]. The orally available analog of ABT-737, i.e. ABT-263 (Navitoclax), showed promising activity in a phase I study with a partial response rate of 35% in patients with relapsed/refractory CLL. However, the frequent dose-dependent thrombocytopenia induced by ABT-263, due to its capacity to inhibit Bcl-XL, a key survival protein in platelets, represented a limit to its pharmacological application [25]. A new derivative, ABT-199 (Venetoclax), specifically binds to Bcl-2, instead of multiple Bcl-2 family members, avoiding the Navitoclax’s adverse effects [26]. In a phase I clinical study in patients with relapsed or refractory CLL or small lymphocytic lymphoma, response rates ranged from 71 to 79% in subjects with resistance to fludarabine, chromosome 17p deletions, and IGHV-UM. Complete remissions occurred in 20% of the patients [27, 28].
The mechanisms responsible for the over-expression of antiapoptotic Bcl-2 factors in CLL are still unclear. At least in the case of patients carrying del13q14, the explanation may reside in the deletion of microRNAs miR-15a and miR-16, which down-regulate Bcl-2 expression [29]. The redundancy of the Bcl-2 family members and the high affinity of BH3-mimetics only for specific anti-apoptotic factors generate resistance in CLL. As an example, we and others demonstrated that Mcl-1 over-expression confers resistance to ABT-737 and its down-regulation increases ABT-737 lethality in human leukemia cells and CLL [23, 30–32]. Similarly, overexpressed Mcl-1 can also be responsible for ABT-263 and ABT-199 resistance in CLL cells. In fact, high levels of Mcl-1 inversely correlated with treatment response in the phase I study of ABT-263 [25] and protected hematological malignancies from ABT-199 [33, 34].
Based on these evidence, we demonstrated that bypassing Mcl-1 mediated resistance, represents a useful strategy to sensitize leukemic cell lines and CLL to apoptosis. In fact, the combined treatment with quercetin and several apoptotic inducers, including fludarabine and death ligands (e.g., recombinant TRAIL and anti-CD95) resulted in increased levels of cell death [35–38]. Quercetin (3,3′,4′,5,7-pentahydroxyflavone) is a natural flavonoid, subclass flavonol, widely present in fruits and beverages. It is a functionally pleiotropic molecule with multiple potential anticancer properties [39–41]. In the case of Mcl-1, we demonstrated that quercetin can down-regulate Mcl-1 acting on its mRNA stability and protein degradation, but can also inhibit the PI3K-Akt pathway, which leads to Mcl-1 activation [31, 32, 42]. When associated with ABT-737, quercetin synergistically induced apoptosis in B-cells isolated from CLL patients and in five leukemic cell lines, as demonstrated by the calculation of the Combination Index [31].
Quercetin belongs to the wide group of natural, anticancer compounds proposed as apoptotic inducers in chemotherapy or in adjuvant chemotherapy when associated with other drugs [15, 43, 44]. Here, we identified in protein kinase CK2 the primary and direct target of quercetin in HG3 cells, derived from a CLL clone immortalized by EBV infection [45]. This cell line was obtained from a patient with IGVH-UM phenotype and biallelic 13q14 deletions with genomic loss of DLEU7, miR15a/miR16–1, resembling the poor prognostic CLL patients. Moreover, HG3 is the first cell line that resembles human B1 cells with regard to surface marker profile (CD5+CD20+CD27+CD43+) and spontaneous Ab-secretion [45].
CK2 is a constitutively active dual specificity kinase that phosphorylates serine/threonine and tyrosine residues with multiple functions in normal and malignant cells (reviewed in [46, 47]). CK2 is composed of two catalytic (α and/or α′) and two regulatory (β) subunits, present in the cells and differently associated to form tetrameric complexes (α2β2, α′2β2, or α′αβ2), or as single monomers. CK2 phosphorylates multiple physiological and non-physiological substrates, characterized by the presence of a highly specific consensus site, i.e., a region containing acidic residues surrounding the phosphor-acceptor amino acid [46]. CK2 has been identified as a player in the pathogenesis of hematopoietic tumors including CLL [48, 49]. Firstly, elevated levels of CK2 β-subunit phosphorylated on Ser209 have been detected in B-cells isolated from 44 CLL patients [50]. It is worthwhile to note that Ser209 phosphorylation can positively regulate the activity of the holoenzyme contributing to its stability [47]. Secondly, inhibition of CK2 activity resulted in reduced phosphorylation of PTEN (phosphatase and tensin homolog) at Ser380 and of Akt at Ser473 and was associated to apoptosis. Similarly, when both CK2 and PI3K inhibitors where combined, cytotoxicity of B-cells increased. The authors concluded that CK2, by phosphorylating, bona fide, PTEN on Ser380 blocks it capacity to convert PIP3 into PIP2 maintaining fully active the pro-survival and anti-apoptotic PI3K/Akt pathway. Inhibition of CK2 can rescue PTEN activity increasing apoptosis in CLL [50]. This scenario was confirmed by a parallel work showing that CK2 was overexpressed and hyperactive in CLL and its inhibition correlated with increased PTEN activity and inactivation of PKC, a PI3K target. Importantly, the same authors reported that the cytotoxicity of CK2 inhibitors (TBBz, DRB and CX-4945) was minimal on normal T and B lymphocytes, while B-cells isolated from advanced stage (Binet B or C) of CLL patients showed a much higher sensitivity to these drugs [51, 52]. It is worthwhile to note that CX-4945, a potent and selective orally bioavailable inhibitor of CK2, alone [53] or in association with fludarabine [54], induced apoptosis in CLL. CX-4945 is capable to attenuate the PI3K/Akt signaling by dephosphorylating Akt on the CK2-specific Ser129 site and on the canonical Ser473 and Thr308 regulatory sites [55].
In the present work, we demonstrate that quercetin directly inhibits CK2 and PI3K enzymatic activities in a CLL-derived cell line. This mechanism contributes to restore ABT-737 sensitivity and increase lethality in human leukemia cells.
RESULTS
Synergistic effect of quercetin associated with ABT-737 in HG3 cell line
We previously demonstrated that quercetin is able to restore sensitivity to apoptosis in leukemic cell lines and B-cells from CLL patients when associated with ABT-737 [31]. We verified if this effect was confirmed in HG3 cells. Figure 1A shows that this cell line was resistant to increasing concentration of ABT-737 (0.25–1 μM) and slightly sensitive to 10–30 μM quercetin with a significant cytotoxicity ranging between 20–30%. When quercetin and ABT-737 were associated, the cytotoxic effect increased to about 30, 40 and 60% at the indicated combinations (Figure 1A). The concentrations of quercetin applied have been selected based on previous studies [31] and with the purpose to limit its toxicity in HG3 cells. The combined treatment was synergistic as demonstrated by the calculation of the Combination Index (C.I.) which resulted < 1 for all the fraction affected (Fa). The isobologram in Figure 1B has been extrapolated using a constant ratio between the concentration of quercetin and ABT-737 (i.e., 40:1). The cytotoxic effect of the combined treatment was associated with the activation of an apoptotic process, as demonstrated by caspase-3 cleavage (Figure 1C) and Annexin-V exposure (Figure 1D). Values of caspase-3 activation and Annexin-V exposure increased time-dependently and picked at 6 and 16–18 h, respectively (data not shown).
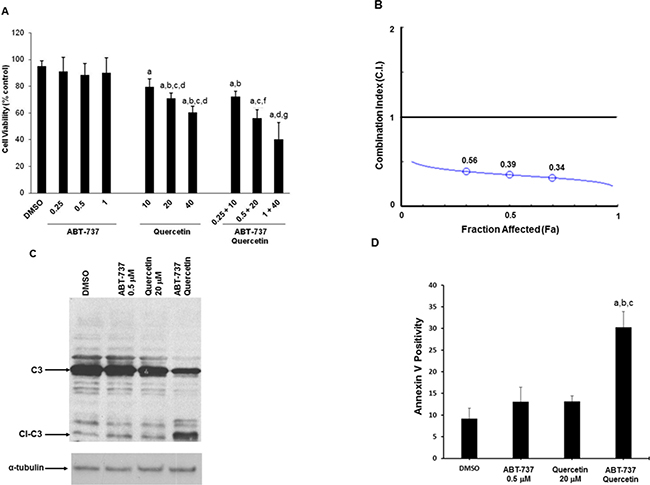
Figure 1: Quercetin in association with ABT-737 induces apoptosis in HG3 cells. (A) Cells (0.5 × 106/ml) were treated with different doses of ABT-737, quercetin or their combination as indicated for 24 h. Cell death, measured by neutral red assay, is reported as percentage of DMSO (0.1% v/v) treated cells, as described in Materials and Methods. Symbols (a, b, c, d, f, g) indicate significance with respect to DMSO (a) and treated cells (b = 0.25 μM ABT-737; c = 0.5 μM ABT-737; d = 1 μM ABT-737; e = 10 μM quercetin; f = 20 μM quercetin, g = 40 μM quercetin); p < 0.001 for all determinations except for a versus e, where p < 0.05 (one-way ANOVA test). (B) Combination Index (C.I.) isobologram. C.I. values, obtained from neutral red experiment (panel A) using a 1:40 concentration ratio of ABT-737 and quercetin, were plotted against the fraction affected (Fa). (C) Proteolytic activation of caspase-3 was measured after 6 h of incubation with the indicated concentrations of ABT-737 and quercetin and their combination. Immunoblot was performed using a specific antibody against caspase-3 (C3 = caspase-3; Cl-C3 = cleaved caspase-3). (D) Annexin V measurement in HG3 cells after 18 h incubation with quercetin (20 μM), ABT-737 (0.5 μM) and their combination, as described in Materials and Methods. Symbols (a, b, c) indicate significance; p < 0.001 with respect to DMSO (a) and treated cells (b = 0.5 μM ABT-737; c = 20 μM quercetin; d = ABT-737 + quercetin) (one-way ANOVA test).
Quercetin inhibits the PI3K-Akt-Mcl-1 pathway
We previously reported the capacity of quercetin to sensitize leukemic cells to apoptosis inducing Mcl-1 degradation [31, 32, 38]. In addition, it is well known that Mcl-1 is activated by multiple pathways in CLL, including PI3K/Akt signaling [56]. In HG3 cells, the expression of Mcl-1 following quercetin treatment (25 μM) was reduced of about 5-fold after 2 h of treatment and correlated with inhibition of the activating phosphorylation of Akt on Ser473 (Figure 2). It is worthwhile to note the extremely rapid effect of quercetin on Akt de-phosphorylation (3-fold decrease after 5 min), suggesting a fast uptake of the molecule and/or the presence of a substrate able to bind quercetin with high affinity.
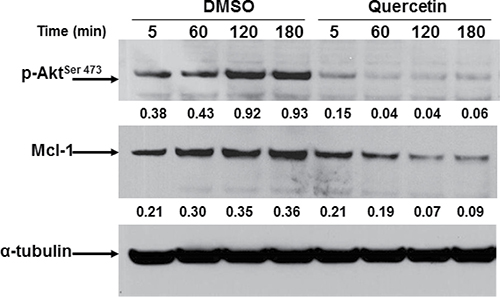
Figure 2: Quercetin down-regulates Mcl-1 and inhibits Akt phosphorylation in HG3. Cells (0.5 × 106/ml) were treated for the indicated time (min) with quercetin (25 μM) or DMSO (0.1% v/v). Immunoblots were incubated for 16 h at 4°C with anti-phospho-Akt (pAkt) antibody (upper panel), stripped and re-probed with anti-Mcl-1 antibody (lower panel). Densitometric analyses were obtained measuring optical density of bands normalized respect to the expression of α-tubulin (numbers below top and middle panels). Immunoblots are representative of at least four independent experiments.
Quercetin uptake in HG3 cells
To verify if quercetin was bioavailable in HG3, we treated cells with increasing concentrations of the molecule and measured its time-dependent incorporation. As reported in Table 1, quercetin was clearly measurable even at 5 min from treatment at all concentrations tested. Treatment with 25 μM quercetin resulted in an incorporation of 38.47 ± 16.46 ng/2 × 106 cells, very shortly after its addition to the cell culture medium (5 min). The uptake depended upon concentrations applied and quercetin stability decreased over time. In fact, as reported in Figure 3A, quercetin decreased of about 4-fold after 15 h from treatment at 25 μM. The presence of quercetin in HG3 cells was also easily and clearly evidenced loading cells with DPBA, a dye which specifically binds flavonols (Figure 3B).
Table 1: Quercetin uptake in HG3 cell line
Control |
Treatment (min) |
Quercetin concentrations applied |
||
|---|---|---|---|---|
5 μM |
25 μM |
50 μM |
||
0.35 ± 0.03* |
5 |
8.89 ± 1.04* |
38.47 ± 16.46* |
103.16 ± 15.72* |
ND |
60 |
7.54 ± 0.44* |
29.52 ± 15.11* |
79.67 ± 14.45* |
*numbers indicate ng quercetin/2 × 106 cells + s.d.
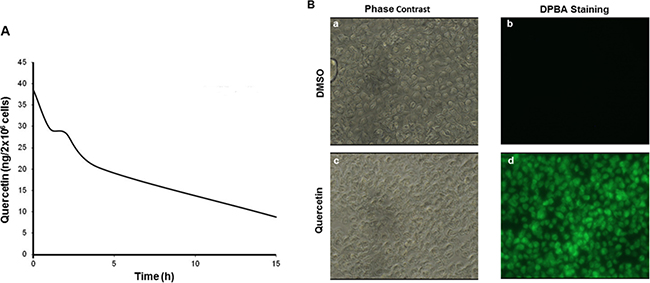
Figure 3: Quercetin stability in HG3 cells. (A) Cells were incubated in the presence of 25 μM quercetin or vehicle control (DMSO) at different times (5, 60, 120, 240 min until 18 h); subsequently, quercetin was quantified in cells as described in Materials and Methods. (B) Cells (1 × 106/ml) were treated with 25 μM quercetin or DMSO (0.1% v/v) before DPBA staining (5 min) performed as described in Materials and Methods. Cells were visualized using a fluorescent microscopy and photographed in phase contrast (a and c) and in FITC filter with 400× magnification (b and d).
PI3K enzymatic activity is directly inhibited by quercetin
Based on the previous results, we hypothesized that PI3K could represent an early target of quercetin in HG3. This conclusion was based on several experimental evidence: i. the fast uptake of quercetin (Table 1 and Figure 3); ii. the rapid deactivation of Akt (Figure 2); iii. the capacity of quercetin to target the ATP binding of PI3K and inhibit its enzymatic activity [57, 58]. To confirm this hypothesis, we firstly demonstrated that quercetin was able to inhibit the recombinant catalytic subunit of PI3K, with an IC50 of about 14 μM (Figure 4A). This assay was performed in vitro employing the recombinant enzyme present in the commercially available PI3K assay kit (see Methods section). Subsequently, we immunoprecipitated PI3K from quercetin treated HG3 cells using an antibody able to recognize the p85-α and -β regulatory subunits of class I PI3Ks. As reported in Figure 4B, treatment with 25 μM quercetin reduced of about 50% the immunoprecipitated enzymatic activity of the bona fide PI3K isoforms expressed in HG3 cells, after 1 h of treatment. The decreased enzymatic activity was not due to a reduced expression or amount of the immunoprecipitated kinase, since the immunoblot in Figure 4C does not show any significant changes in p85 levels, confirming the capacity of quercetin to inhibit PI3K enzymatic activity.
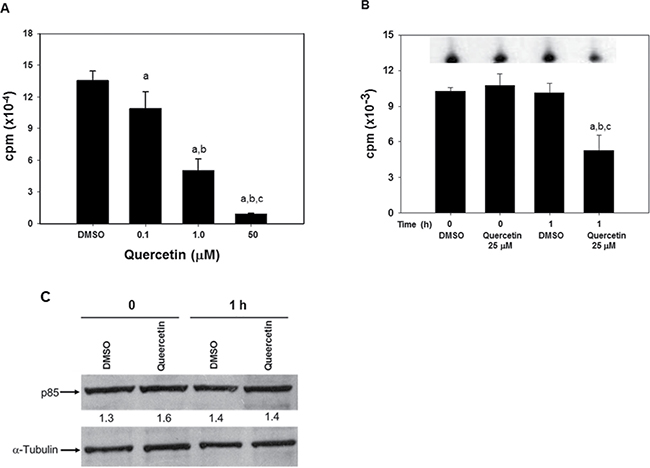
Figure 4: Quercetin inhibits PI3K activity in vitro and in HG3 cells. (A) In vitro kinase assay was performed using a commercially available kit (Abcam) by measuring the amount of radioactively labeled [γ-32P] ATP incorporated into a lipid substrate, as described in the manufacturer’s protocol in the presence of the range of quercetin concentrations indicated. The enzymatic activity was determined using as catalytic subunit the human recombinant PI3K-γ (His tagged) produced in sf9 insect cells and included in the kit. Symbols (a, b, c, d) indicate significance with respect to DMSO (a) and treated cells (b = 0.1 μM quercetin; c = 1 μM quercetin; d = 50 μM quercetin); p < 0.01 for all determinations, except for a versus b where p < 0.05 (one-way ANOVA test). (B) Cells (5 × 106/ml) were incubated in the presence of 25 μM quercetin, or DMSO (0.1% v/v) as control, for the indicated time (0 and 1 h). Immunoprecipitation was performed as reported in Materials and Methods section on 500 μg of total proteins using an antibody reacting against the p85-α and -β regulatory subunits of class I PI3Ks. The immunoprecipitates were used as enzymatic source to detect PI3K activity using the commercial kit described in panel A. The insert on top of the graph shows a representative autoradiogram of 32P-phosphorylated lipid substrate, following separation using TLC, as reported in Materials and Methods. Symbols (a, b, c) indicate significance; p < 0.05 with respect to DMSO t = 0 (a); DMSO t = 1 h (c) and treated cells (b = 25 μM quercetin at t = 0; d = 25 μM quercetin at t = 1 h) (one-way ANOVA test). (C). After the kinase assay, the same immunoprecipitates used in panel B were washed to eliminate the reaction buffer remaining after the kinase reactions, added with loading buffer, boiled for 5 minutes and loaded on a 4–12% pre-cast gel before immunoblotting. The membrane was incubated with primary antibody against p85-α and -β regulatory subunits of class I PI3Ks, or α-tubulin. Band intensities were quantified measuring optical density on Gel Doc 2000 and analysed by Multi-Analyst Software. Numbers between the panels indicate p85 expression normalized respect to α-tubulin. The immunoblot is representative of at least two independent experiments.
CK2 enzymatic activity is directly inhibited by quercetin upstream of PI3K
The observation that the uptake of quercetin and the dephosphorylation of Akt “preceded” the inhibition of PI3K, suggested the possibility that an upstream modulator of PI3K/Akt pathway could be also targeted by quercetin. It is known that PTEN is a negative regulator of PI3K [59] and CK2 phosphorylates PTEN on several residues inducing its functional inhibition [60]. In addition, we and others previously reported that pure CK2 enzyme can be competitively inhibited by quercetin [61]. As reported in Figure 5A, CK2 activity was inhibited in HG3 cells treated with 25 μM quercetin, soon after addition of the flavonol (in the range of 0–2 min, indicated as 0 min in Figure 5A). The inhibition persisted for several hours and the inhibitory effect of quercetin treatment was superimposable to the effect of a CK2 specific inhibitor, TBBz [62]. The immunoblot on the bottom of Figure 5A shows that the expression of CK2α does not change significantly upon treatment with quercetin, confirming that the decreased kinase activity was due to quercetin inhibition, not to changes in the expression of the catalytic subunit. The consequence of CK2 inhibition by quercetin on the PI3K-Akt pathway was confirmed by the immunoblot in Figure 5B which shows PTEN dephosphorylation on Ser380, one of the phosphorylation site triggered by CK2, at 2–5 min following both quercetin and TBBz treatments. As expected, PTEN dephosphorylation coincided with Akt inactivation at the same experimental times. Also in this case, the control immunoblottings on the right side of Figure 5B show that changes in phospho-PTEN and phospho-Akt levels were not due to reduced expression of the total intracellular PTEN and Akt proteins. It is worthwhile to note that comparing the different times of quercetin and TBBz treatments (5 versus 60 min), the capacity of TBBz to reduce the activating Akt phosphorylation on Ser473 significantly decreased at 60 min (Figure 5C) compared to 5 min (Figure 5B). On the contrary, inactivation of Akt by quercetin increased at 60 min and was superimposable to CAL-101 (Figure 5C), a well-known and specific inhibitor of PI3K-Akt pathway. Finally, we demonstrated that TBBz was unable to complement quercetin in the association with ABT-737, resulting in increased sensitivity to apoptosis. In fact, Figure 5D shows that the effect of TBBz plus ABT-737 on cell viability was not synergic, but barely additive. This was also confirmed by the calculation of the C.I. which remained close to 1 (data not shown).
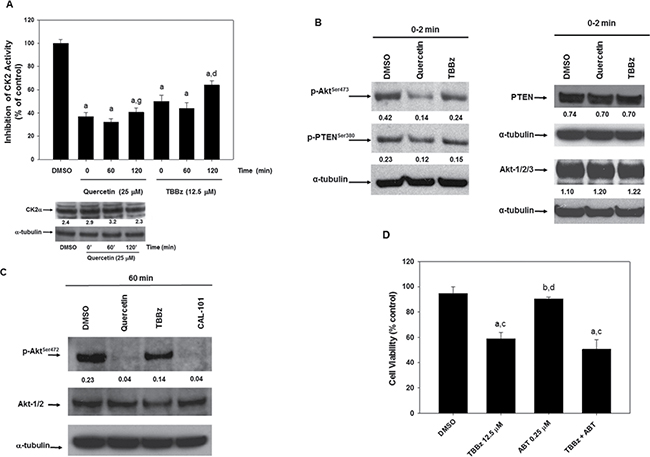
Figure 5: Quercetin inhibits CK2 activity restoring the control of PTEN on PI3K-Akt pathway. (A) HG3 cells, after treatment with 0.1% DMSO, 25 μM quercetin, 12.5 μM TBBz for the indicated times, were lysed and used to assay CK2 activity as described in Materials and Methods. Symbols (a, d) indicate significance with respect to DMSO (a) and treated cells (d = 25 μM quercetin at t = 120 min; g = 12.5 μM TBBz at t = 120 min); p < 0.001 for all determinations, except for d versus g where p < 0.05 (one-way ANOVA test). The immunoblot on the bottom of the panel shows the expression of CK2α subunit detected using a not commercial, anti-CK2α antibody. The membrane was re-probed with the anti α-tubulin antibody. Band intensities were quantified measuring optical density on Gel Doc 2000 and analysed by Multi-Analyst Software. Numbers between the panels indicate CK2α expression normalized respect to α-tubulin. The image is representative of one out of two experiments performed. (B) and (C) HG3 cells were treated as in panel A for 0–2 min (B) or 60 min (C). After immunoblotting, the membranes were incubated for 16 h at 4°C with anti-phospho-Akt (p-AktSer473), anti-phospho-PTEN (p-PTENSer380), anti-Akt1/2/3, anti-PTEN, or α-tubulin antibodies. In panel C, the treatment with CAL-101 (5 μM) was also included. Band intensities were quantified measuring optical density on Gel Doc 2000 and analysed by Multi-Analyst Software. In panel B, numbers between panels indicate the expression of p-AktSer473 and p-PTENSer380, Akt1/2/3 and PTEN normalized respect to α-tubulin. In panel C, numbers between top and middle panels indicate the expression of p-AktSer473 normalized respect to Akt1/2/3. Immunoblots are representative of at least three independent experiments performed. (D) HG3 cells were treated at the concentrations indicated for 24 h and the effect of TBBz, ABT-737 and their combination on cell viability (neutral red assay) was measured. Symbols (a, b, c, d) indicate significance with respect to DMSO (a) and treated cells (b = 12.5 μM TBBz; c = 0.25 μM ABT-737; d = TBBz+ABT-737); p < 0.001 (one-way ANOVA test).
Quercetin is selective for BH3-mimetic compound triggering Bcl-2/Bcl-XL
We reasoned that quercetin efficacy in restoring sensitivity to the apoptotic effect of ABT-737 was dependent upon destabilization of Mcl-1, which is not a specific target of ABT-737. However, ABT-737 is essential to target and inactivate Bcl-2 and Bcl-XL. Therefore, using BH3-mimetics with limited affinity for Mcl-1, but able to selectively inhibit the pro-survival functions of Bcl-2 and Bcl-XL, we expected that these compounds would behave similarly to ABT-737 plus quercetin. In fact, when ABT-263 (Navitoclax) and Wehi-539, which bind with higher efficiency than ABT-737 to Bcl-2 and Bcl-XL, respectively [63, 64], were associated with quercetin in HG3 cells, they showed a synergistic effect on apoptotic induction, comparable to the co-treatment ABT-737 plus quercetin. Table 2 reports the calculation of the C.I. for the two combinations ABT-263 plus quercetin and Wehi-539 plus quercetin. In both cases, the C.I. resulted < 1 and in the same range of those reported in Figure 1B for ABT-737 and quercetin. Increased apoptosis, determined by measuring the activation of caspase-3, was detected in all co-treatments investigated (Figure 6).
Table 2: C.I. values for different BH3-mimetics when associated with quercetin in HG3 cell line
Total dose* |
Fa |
C.I. |
|
|---|---|---|---|
ABT-236/Quercetin [1/80] |
10.12 |
0.58 |
0.368 |
20.25 |
0.72 |
0.364 |
|
WHEI-539/Quercetin [1/80] |
10.12 |
0.23 |
0.465 |
20.25 |
0.35 |
0.310 |
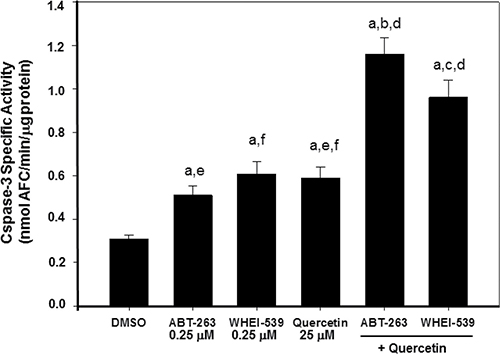
Figure 6: Quercetin and BH3-mimetics enhance caspase-3 activity in HG3 cells. Activation of caspase-3 was measured after 6 h of incubation with the indicated reagents (0.25 μM ABT-263, 0.25 μM Whei, 25 μM quercetin) using the enzymatic assay described in Materials and Methods. Specific caspase-3 activity was expressed as nmol AFC/min/μg protein. Symbols (a, b, c, d, f, g) indicate significance with respect to DMSO (a) and treated cells (b = 0.25 μM ABT-263; c = 0.25 μM WHEI-539; d = 25 μM quercetin; e = 0.25 μM ABT-263 + 25 μM quercetin; f = 0.25 μM WHEI-539 + 25 μM quercetin); p < 0.001 for all determinations except for a versus b where p < 0.05 (one-way ANOVA test).
As a corollary of the results obtained with ABT-263 and Wehi-539, we observed that quercetin loses its synergistic capacity when associated with TW-37, a BH3- mimetic compound with higher affinity for Mcl-1 [65]. In fact, the calculated C.I. value indicated only an additive effect (even antagonist at high Fa) and the apoptotic capacity of quercetin plus TW-37 was also strongly reduced (data not shown).
DISCUSSION
In the Introduction, we reported that one of the possible approaches against CLL is the design of new therapeutic strategies to bypass the acquired resistance to apoptosis occurring in B-cells. This goal can be achieved with the recurrence to a new generation of BH3-mimetics compounds, Venetoclax is an example [27]. Alternatively, novel protocols can be applied, based on the combination of chemotherapy and immunotherapy in relation to the Binet or Rai stages of the disease, the physical fit of the patients, the presence of a del(17p) or TP53 mutation (reviewed in [66, 67]). As an example, in the latter situation, significant improvements in terms of progression-free survival, response rate and overall survival have been obtained with the combination of Idelalisib (formerly known as CAL-101 or GS-1101), a first-in-class inhibitor of the PI3K delta isoform, in combination with rituximab [68]. The case of Idelalisib further highlights the importance of inhibiting the PI3K-Akt pathway for an efficient therapy against CLL.
The present article reinforces the notion that plant-derived compounds can enhance apoptosis in CLL, as extensively reviewed elsewhere [15, 69]. However, we added new contributions: 1. quercetin represents one of the few compounds (if not the unique) where its direct molecular target(s) in CLL-derived cells have been identified; 2. quercetin requires the presence of specific BH3-mimetics compounds to “synergistically” enhance apoptosis, at least in in vitro and ex vivo models. In the next paragraphs, we will deeply discuss these two issues.
Quercetin ability to act as “not specific” inhibitor of serine-threonine kinases, with a Ki in the micromolar range, is known since the late nineties [70]. We and others demonstrated that quercetin directly binds and inhibits recombinant or purified forms of catalytically active CK2 and PI3K; this inhibition occurs also on the cellular enzymes (this paper; [58, 61, 71]). Here, we focused on the “timing” of quercetin effects, being CK2 enzymatic activity inhibited “immediately” after the cellular uptake of the flavonoid, while PI3K was inhibited later, but, in any case, within 1 h from quercetin treatment. This represents an important novelty in considering the multifunctional effects of naturally occurring compounds, whose capacity to inhibit cell growth has been often erroneously defined without carefully considering: i. their active intracellular concentration; ii. their specific molecular target(s); iii. their “affinity” for one pathway over others that can also be triggered, but with less specificity or at later times. The demonstration that CK2, and, consequently, the CK2/PTEN pathway (Figure 7), represents the primary target of quercetin in HG3 cells, being inhibited at time 0–2 min from treatment, does not mean that the same mechanism occurs in other cellular systems, where quercetin may show higher affinity for other substrates/pathways besides CK2/PTEN. More in general, our finding opens the road to re-evaluate the specificity of plant-derived compounds in cancer cells.
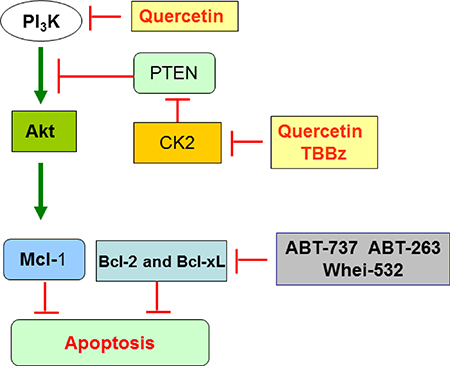
Figure 7: Scheme summarizing the key targets of quercetin in HG3 cells. The double inhibitory activity of quercetin on CK2 and PI3K converges on Akt pathway, which is inactivated and unable to phosphorylate and stabilize anti-apoptotic Mcl-1. In parallel, ABT-737 (or other BH3-mimetics) can trigger and inhibit different anti-apoptotic members of Bcl-2 family. The combined effect of quercetin plus ABT-737 restores sensitivity to apoptosis in HG3 and CLL (see text for details).
Regarding quercetin binding and inhibition of CK2 activity, strong structural evidence suggest that quercetin competes with the ATP binding site. Elegant experiments addressed this issue. The planarity of the molecule, pre flavone scaffold with at least two hydroxyl groups at positions 7 and 4′ ensure the competitive inhibition with respect to ATP [72]. Using a different approach, a biotinylated version of quercetin has been employed as intrinsic photo-affinity proteomics probe to capture CK2 as substrate from both an in vitro pull-down experiments on the pure enzyme and from CK2 present in cellular extracts. In this case, it has been proposed that quercetin may bind to the allosteric cleft between α/β surface of CK2, an interacting region between α, β subunits crucial to the function of CK2 holoenzyme [73]. Based on this evidence, it is plausible that similar inhibition mechanisms are active in vivo to explain the rapid inactivation of CK2 observed in HG3 cells following quercetin treatment.
Regarding the inhibition of PI3K by quercetin, it is worthwhile to mention that X-ray crystallographic structure of PI3K-γ isoform bound to quercetin indicates that the molecule fits into the ATP binding site of the kinase with a Kd value of 0.28 μM. It is also interesting that the orientation of quercetin into the ATP binding site is different compared to myricetin, a quercetin analog which only differs for the addition of a hydroxyl at the 5′-OH of the phenyl moiety, and LY294002, a specific PI3K inhibitor whose structure has been designed using quercetin as lead compound [58]. This observation indicates a possible specificity of quercetin compared to other PI3K inhibitors. Probably, the same competitive enzymatic inhibition of PI3K isoforms occurs in vivo to explain the results shown in Figure 4. We do not know exactly which PI3K isoform(s) were present in the immunoprecipitation shown in Figure 4B, since we employed an antibody which recognizes the p85 regulatory subunit of class IA PI3K enzymes, able to bind the catalytic subunits p110-α, -β and –δ. However, considering that class I PI3Ks are present in all cell types, with p110–δ and –γ enriched in leukocytes and B-CLL [59, 74], we can bona fide conclude that PI3K-δ is present in HG3 cells, as confirmed by the observation that the treatment with CAL-101(Idelasib) resulted in a strong reduction of Akt phosphorylated on the activating residue Ser473 (Figure 5C).
As schematically summarized in the cartoon shown in Figure 7, the double inhibitory activity of quercetin on CK2 and PI3K converges on Akt, which is inactivated and unable to phosphorylate and stabilize anti-apoptotic Mcl-1 [75]. In parallel, ABT-737 (or other BH3-mimetics) can trigger and inhibit different anti-apoptotic members of Bcl-2 family. The combined effect of quercetin plus ABT-737 restores sensitivity to apoptosis in HG3 and possibly CLL. The present model raises several spontaneous questions: i. what is the advantage to use quercetin over more specific kinase inhibitors for CK2 and/or PI3K, such as TBBz and CAL-101, respectively? ii. why does the combined treatment of quercetin plus ABT-737 generate a synergistic effect instead of an additive one? About the first issue, theoretically, quercetin allows “to hit two birds with one stone”, since the molecule inhibits two signaling pathways, both converging on Akt inactivation and Mcl-1 destabilization. The use of a single compound (quercetin), instead of the combination of two (TBBz + CAL-101), is useful in reducing toxicity and, generally, unwanted side effects. As shown in Figure 5C, quercetin efficacy in reducing phospho-AktSer473 was comparable to CAL-101. In addition, the combination ABT-737 plus TBBz reduced cell viability additively compared to the synergistic effect of ABT-737 plus quercetin (consider Figure 1 versus Figure 5D). This observation opens the question on why and how the latter treatment is synergistic. Several studies report that understanding synergism may explain the mechanism of action of even a single agent [76]. In the case of quercetin and ABT-737, to be synergic, we can image that the two pathways, respectively triggered by these compounds, must converge on a common, downstream target which, in turns, amplifies the apoptotic signal. Currently, this hypothesis is not supported by strong experimental evidence. As an alternative explanation, we can image that quercetin triggers other substrates involved in the resistance to apoptosis, lowering the threshold necessary to favor a pro-apoptotic response. Three circumstantial evidence support this view: i. we previously demonstrated that in U-937 cells, quercetin downregulates Mcl-1 acting directly or indirectly on its mRNA stability and protein degradation.[32]; ii. similarly to other phytochemicals [77], quercetin may up-regulate NOXA which is important in CLL since the ratio NOXA/Mcl-1 dictates CLL sensitivity to ABT-737 [78] (Russo M. et al., unpublished); iii. recently, it has been described the capacity of quercetin to directly bind the BH3 domain of Bcl-2 and Bcl-XL proteins, inhibiting their activity and promoting cancer cell apoptosis. Structural similarities seem to exist in the interactions between the Bcl-XL/quercetin compared to Bcl-XL/ABT-737 complexes, showing that quercetin can bind to Bcl-XL as ABT-737 [79]. This latter approach, although largely based on in vitro data, opens new perspectives to the interpretation of the mechanism of action of quercetin in B-CLL and deserves further investigations.
Is it possible to consider a clinical outcome of quercetin, in combination with BH3- mimetic antagonists? In the case of a therapeutic use of quercetin against CLL, its intravenously administration in patients undergoing chemotherapeutic treatment will ensure high circulating concentrations of the free aglycone and avoid the formation of conjugates. In this respect, a unique phase I clinical trial of quercetin has been so far completed, suggesting a recommended a dose of 1400 mg m−2, which corresponds to about 2.5 g for a 70 kg individual, administered via intravenous infusion at three-weeks or weekly intervals [80]. In addition, if quercetin becomes a molecule of pharmacological interest in CLL and/or other forms of cancer, its chemical bioavailability can be improved using different biotechnological approaches. To this aim, the recent literature is extremely fertile on proposing engineered formulations (e.g., silica, chitosan, PLGA and PLA nanoparticles, liposomes, micelles, etc.) of quercetin [81, 82]. Of course, these approaches require not only an enhanced target specific delivery of the nanoformulations, but also a high level of specificity towards cancer cells to prevent unwanted side effects on normal cells.
In conclusion, this study represents one of the few examples in the literature where the direct targets of quercetin in a specific form of cancer have been identified. It also highlights the importance to evaluate the intracellular uptake and stability of the molecule and to identify the earlier events related to its biological activity. This preclinical study may open new perspectives for a clinical trial on CLL patients where quercetin can be included in the chemotherapeutic protocol as adjuvant agent.
MATERIALS AND METHODS
Reagents
Roswell Park Medium Institute (RPMI) medium, L-glutamine 200 mM, penicillin 5000 IU/ml/streptomycin 5000 μg/ml and PBS (phosphate buffer saline) tablets were purchased from Invitrogen (S. Giuliano Milanese, MI, Italy); fetal bovine serum from Cambrex (Milano, Italy). Neutral red solution (0.33% v/v), trypan blue solution (0.4% v/v, propidium iodide, quercetin, TBBz (4, 5, 6, 7-tetrabromobenzimidazole) and dimetylsulfoxide (DMSO) were from Sigma-Aldrich (Milano, Italy). Cal-101 (GS1101) was purchased from Sellechem (Aurogene, Rome, Italy). ABT-737, ABT-263, Whei-532 and TW-32 7 were kindly provided by Apex-Bio (USA) and dissolved in DMSO, aliquoted and stored at −20°C. Methanol and formic acid HPLC grade were obtained from Merck (Vimodrone, Milano, Italy). HPLC grade water (18.2 MΩ) was prepared by using a Millipore Milli-Q purification system (Merck-Millipore).
Cell culture, cell viability assay and apoptotic nuclei staining
HG3 cells, a lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection, were cultured in RPMI medium supplemented with 10%, fetal bovine serum, 1% L-glutamine and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2 [45].
Cell viability was assayed using neutral red viability test [83]. Cells were cultured at density of 0.25–0.5 × 106/ml in 48 multi-well plates and incubated (24 h) in a medium containing the indicated compounds. Cell viability assay was performed as described [36]. Briefly neutral red (0.066% v/v final concentration) was added and incubated for 2 h. Cells were collected and centrifuged at 400 × g for 5 minutes, washed once with PBS and incubated with lysis buffer (50 mM Tris-HCl pH 7.4; 150 mM NaCl; 5 mM dithiotreitol; 1% Triton-100) containing 1% acetic acid and 50% ethanol. Absorbance was spectrophotometrically measured at 540 nm (Synergy HT multi-well reader; Bio-Tek Instruments, Milano, Italy). The quantity of adsorbed dye was proportional to number of living cells and/or with an active metabolism.
Combination index (C.I.) values were calculated according to the Chou and Talalay mathematical model for drug interactions [84], as previously reported [31]. Dose–response curves, dose–effect analysis and C.I. for the combination treatment groups were generated using the equations reported by Chou and Talalay using the CompuSyn software (freely available at: www.combosyn.com).
Caspase assays
To determine caspase-3 enzymatic activity, 2 × 106/ml cells were treated as indicated for 6 h. Subsequently, cells were collected and centrifuged at 400 × g for 5 minutes, washed twice in PBS and suspended in lysis buffer (10 mM Hepes, pH 7.4; 2 mM ethylenediaminetetraacetic acid; 0.1% [3-(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate, 5 mM dithiothreitol, 1 mM phenylmethylsulfonylfluoride, 10 μg/ml pepstatin-A, 10 μg/ml apronitin, 20 μg/ml leupeptin). Cell extracts (10 μg) were added with the reaction buffer and the conjugated amino-4-trifluoromethyl coumarin (AFC) substrate: benzyloxycarbonyl-Asp (OMe)-Glu (OMe)-Val-Asp (OMe)-AFC (Z-DEVD-AFC). The samples were incubated at 37°C for 30 minutes. Upon proteolytic cleavage of the substrate by caspase-3, the free fluorochrome AFC was detected by a spectrofluorometer multiplate reader (Bio-Tek Instruments) with excitation 400 ± 20 nm and emission at 530 ± 20 nm. To quantify enzymatic activity, we determined an AFC standard curve. Caspase-3 specific activity was calculated as nmol of AFC produced per min per μg proteins at 37°C in the presence of saturating substrate concentration (50 μM) [36].
Annexin V assay
Phosphatidylserine exposure was measured using the binding of fluorescein-isothiocyanate-labeled (FITC) Annexin V to phosphatidylserine, as indicated in the manufacturer’s protocol (Milteny-Biotech, Milano, Italy). Briefly, cells (1 × 106/ml) were treated as indicated for 16–18 h (over-night). Cells were collected and centrifuged at 400 × g for 5 minutes, washed in PBS and suspended in binding buffer (10 mM Hepes, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2). Annexin V FITC (10 μL) and propidium iodide (25 μg/ml) were added to the cells for 10 min in the dark at room temperature and analyzed with flow cytometer (FACSCalibur; Becton Dickinson, Mountain View, CA, USA) equipped with argon laser (488 nm) and filtered at 530 and 585 nm for FITC and phycoerythrin respectively. Low fluorescence debris and necrotic cells, permeable to propidium iodide, were gated out before to analysis. Data were analyzed using CellQuest software (Becton Dickinson).
Immunoblot
After treatments, cells (generally 1.5 × 106) were suspended in lysis buffer containing 50 mM Tris/HCl, pH 7.4; 150 mM NaCl; 5 mM ethylenediaminetetraacetic acid; 1% Nonidet P-40; 0.5 mM dithiotreitol; 1 mM Na3VO4; 40 mM NaF; 1 mM Na4P2O7; 7.4 mg/ml 4-p-nitrophenyl phosphate; 10% glycerol; 100 μg/ml phenylmethylsulfonyl fluoride and the cocktail of protease inhibitors ‘complete’ (Roche Applied Science; Monza, Italy). Following measurement of protein concentration [85], total lysates (20–25 μg) were added with loading buffer (Bio-Rad Laboratories, Milano, Italy), boiled for 5 minutes and loaded on a 12% pre-cast gel (CRITERION XT; Bio-Rad Laboratories) using MOPS [3-(N-morfolin) propanosulfonic] buffer (1 M MOPS; 1 M Tris/Base; 69.3 mM SDS; 20.5 mM EDTA) or MES [2-(N-morpholino)ethanesulfonic acid] buffer (50 mM MES, 50 mM Tris/Base, 0.1% SDS, 1 mM EDTA) and a constant voltage (200 V). Proteins were blotted onto polyvinylidene difluoride (PVDF), membrane (Transfer Pack Bio-Rad Laboratories), using TRANS-Blot TURBO System (Bio-Rad laboratories), with a constant amperage (2.5 mA) for 7 min room temperature. The membranes were rinsed with T-TBS (0.1% Tween-20; 25 mM Tris; 137 mM NaCl; 2.69 mM KCl, pH 8) and blocked using 5% (w/v) non-fat dry milk resuspended in T-TBS for 1 h at room temperature, before incubation for 16 h at 4°C with specific antibodies. The primary antibodies used were anti-Mcl-1, anti-pAKT, anti pPTEN (Cell Signaling; Milano; Italy), anti-Caspase-3 (Genetex), anti-p85-α and -β regulatory subunits of class I PI3Ks (Santa Cruz Biotechnologies, Heidelberg, Germany), anti-Akt1/2/3 (Santa-Cruz Biotechnologies; kindly provided by Dr. Paola Ungaro), anti-PTEN (Upstate Biotechnology; kindly provided by Prof. Adriana Borriello), anti-CK2α subunit (Ab 276 which recognizes α–α′ subunits, [86]), anti-α-tubulin (Sigma-Aldrich). PVDF membranes were finally incubated with horseradish peroxidase linked secondary antibody against mouse or rabbit (GE Healthcare, Milano, Italy) and immunoblots developed using the ECL Plus Western Blotting Detection System Kit (Perkin-Elmer, Milano, Italy). Band intensities were quantified measuring optical density on Gel Doc 2000 Apparatus and Multi-Analyst Software (Bio-Rad Laboratories).
Kinase assays
PI3K activity was determined using a commercially available kit (Abcam, Cambridge, UK) by measuring the amount of radioactively labeled [γ-32 P] ATP incorporated into a lipid substrate, following separation using thin layer chromatography (TLC), as described in the manufacturer’s protocol. The enzymatic activity was determined using the pure enzyme, i.d. the PI3K recombinant catalytic subunit included in the kit, or the active enzyme immunoprecipitated from HG3 cells treated with quercetin as indicated in figure legends. The immunoprecipitation was performed using an antibody reacting against the p85-α and -β regulatory subunits of class I PI3Ks (Santa Cruz Biotechnology; cat. # sc-423) following a previously described protocol [87].
CK2 activity was determined on cell lysates deriving from DMSO (0.1%, v/v) and HG3 treated cells with different reagents (25 μM quercetin, 12.5 μM TBBz, as indicated in figure legends). The reaction mixture contained in a total volume of 50 μl, 50–90 μM [γ-32 P] ATP (1500–3000 cpm/pmol ATP) (Perkin-Elmer), 0.5 mM peptide substrate ETE (RRREEETEEE, amino acid sequence one letter code; Promega, Milan, Italy) in CK2 kinase buffer (50 mM MOPS, pH 7.0, 10 mM MgCl2, 10 mM NaCl, 60 mM β-glycerophosphate), as previously reported [40, 86]. Reactions were incubated for 30–40 min at 30°C in the presence of 5–10 μg of cell lysate and terminated by transferring the supernatants to P81 paper (Whatman; GE Healthcare) to determine phosphate incorporation on ETE peptide as described [88].
Measurement of intracellular levels of quercetin
To detect the intracellular levels of quercetin, HG3 cells (1.5–2.0 × 106) were incubated in the presence of different concentrations of quercetin (5–50 μM) in RPMI containing 10% FBS, 1% L-glutamine, and 1% penicillin/streptomycin. At the end of incubation (5–60 min), cells were harvested by centrifugation and washed two times in PBS. Cellular pellets were re-suspended three times with methanol and sonicated. Finally, supernatants were frozen at −80°C, until HPLC analysis was performed. Quercetin concentrations in extracts were determined by HPLC–UV analysis using a HP 1110 series HPLC (Agilent, Palo Alto, CA, USA) equipped with a binary pump (G-1312A) and a UV detector (G-1314A). Samples were analyzed using a reverse phase Hypersil BDS C18 column (250 mm 4.6 mm, 5μm) (Thermo, Bellefonte, PA, USA) at a flow rate of 1 ml min-1. Solvent A was 0,1% formic acid and solvent B was 0.1% formic acid in acetonitrile. The gradient for B was as follows: 30% for 5 min, from 30% to 95% in 20 min, from 95% to 100% in 2 min. The eluate was monitored at 380 nm. In order to confirm the identity of quercetin HPLC eluted peaks were analyzed by Electrospray Ionization multistage Ion Trap Mass Spectrometry (ESI-ITMSn) using a Finnigan LCQ DECA XP Max ion trap mass spectrometer (Thermo Finnigan, San Josè, CA, USA), equipped with Xcalibur® system manager data acquisition software (Thermo Finnigan, San José, CA, USA). Mass spectra were recorded from mass-to-charge ratio (m/z) 80 to 600 in negative ionization mode. The capillary voltage was set at −10 V, the spray voltage was at 3 kV and the tube lens offset was at −10 V. The capillary temperature was 275°C. Data were acquired in MS, MS/MS and MSn scanning mode. Quantification of quercetin was performed with external calibration curves generated by repeated injections of a fixed volume of quercetin standard over a concentration range of 0.01 a 0.2 μg μl−1, with five different concentrations and duplicate injections at each level. All samples were prepared and analyzed in duplicate. Experiments were performed three times in duplicates and expressed as ng quercetin/2 × 106 cells. Table 1 represents the mean of three experiments (± s.d).
DPBA staining
To qualitatively detect quercetin uptake in HG3 cells, we used DPBA (2-aminoethyl diphenylborinate; Sigma-Aldrich) staining, a reagent used in plant physiology that selectively binds flavonols emitting fluorescent when exited at 485 ± 20 nm (emission at 530 ± 20 nm) [89]. The method was slightly modified to qualitatively assess the intracellular presence of quercetin in human cell lines. Briefly, HG3 cells were plated at a cell density of 1 × 106/ml in a 12-wells plate and treated with 25 μM quercetin (5–60 min); subsequently, cells were centrifuged, washed with PBS, suspended in 1 ml of DPBA solution (2.5 mg/ml) dissolved in 10% formalin and finally incubated at 37°C in a humidified atmosphere containing 5% CO2 for 15 min. After incubation, cells were visualized using a fluorescent microscopy and photographed in phase contrast and in FITC filter with 400× magnification (Axiovert 200 Zeiss, Iena Germany).
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics (version 23.0.; IBM Corp). Results are expressed as mean ± standard deviation (s.d.). Differences between groups were tested using analysis of variance (ANOVA) with Bonferroni’s correction for multiple comparisons. The significance was set at p < 0.05 with specific values indicated in figure legends.
ACKNOWLEDGMENTS
We warmly thank Dr. Paola Ungaro (Istituto di Endocrinologia ed Oncologia Sperimentale ‘G. Salvatore’, Consiglio Nazionale delle Ricerche, Napoli, Italy) and Prof. Adriana Borriello (Department of Biochemistry, Biophysics and General Pathology, Università degli Studi della Campania Luigi Vanvitelli, Napoli, Italy) for the reagents (anti-Akt1/2/3 and anti-PTEN antibodies, respectively) kindly provided.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was partially supported by the following national and regional grants: (1) C.I.S.I.A. project “Innovazione e Sviluppo del Mezzogiorno - Conoscenze Integrate per Sostenibilita` ed Innovazione del Made in Italy Agroalimentare - Legge 191/2009” from the Italian Ministry of Economy and Finance to the National Research Council; (2) BenTeN project (Wellness from biotechnologies: New Processes and Products for Nutraceutical, Cosmeceutical and Human Nutrition), within the Biotechnology Network of Campania Region (Italy).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Fabbri G, Dalla-Favera R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat Rev Cancer. 2016; 16:145–62.
3. Zenz T, Mertens D, Kuppers R, Dohner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010; 10:37–50.
4. Kikushige Y, Ishikawa F, Miyamoto T, Shima T, Urata S, Yoshimoto G, Mori Y, Iino T, Yamauchi T, Eto T, Niiro H, Iwasaki H, Takenaka K, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011; 20:246–59.
5. Rawstron AC, Green MJ, Kuzmicki A, Kennedy B, Fenton JA, Evans PA, O’Connor SJ, Richards SJ, Morgan GJ, Jack AS, Hillmen P. Monoclonal B lymphocytes with the characteristics of “indolent” chronic lymphocytic leukemia are present in 3.5% of adults with normal blood counts. Blood. 2002; 100:635–9.
6. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, Buchbinder A, Budman D, Dittmar K, Kolitz J, Lichtman SM, Schulman P, Vinciguerra VP, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999; 94:1840–7.
7. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999; 94:1848–54.
8. Lobetti-Bodoni C, Bertoni F, Stussi G, Cavalli F, Zucca E. The changing paradigm of chronic lymphocytic leukemia management. Eur J Intern Med. 2013; 24:401–10.
9. Hamblin TJ, Orchard JA, Ibbotson RE, Davis Z, Thomas PW, Stevenson FK, Oscier DG. CD38 expression and immunoglobulin variable region mutations are independent prognostic variables in chronic lymphocytic leukemia, but CD38 expression may vary during the course of the disease. Blood. 2002; 99:1023–9.
10. Eichhorst B, Cramer P, Hallek M. Initial therapy of chronic lymphocytic leukemia. Semin Oncol. 2016; 43:241–50.
11. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008; 111:5446–56.
12. Keating MJ, O’Brien S, Albitar M, Lerner S, Plunkett W, Giles F, Andreeff M, Cortes J, Faderl S, Thomas D, Koller C, Wierda W, Detry MA, et al. Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. J Clin Oncol. 2005; 23:4079–88.
13. Tam CS, O’Brien S, Wierda W, Kantarjian H, Wen S, Do KA, Thomas DA, Cortes J, Lerner S, Keating MJ. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood. 2008; 112:975–80.
14. Dreger P, Corradini P, Kimby E, Michallet M, Milligan D, Schetelig J, Wiktor-Jedrzejczak W, Niederwieser D, Hallek M, Montserrat E. Indications for allogeneic stem cell transplantation in chronic lymphocytic leukemia: the EBMT transplant consensus. Leukemia. 2007; 21:12–7.
15. Billard C. Apoptosis inducers in chronic lymphocytic leukemia. Oncotarget. 2014; 5:309–25. doi: 10.18632/oncotarget.1480.
16. Pleyer L, Egle A, Hartmann TN, Greil R. Molecular and cellular mechanisms of CLL: novel therapeutic approaches. Nat Rev Clin Oncol. 2009; 6:405–18.
17. Tam CS, Seymour JF, Roberts AW. Progress in BCL2 inhibition for patients with chronic lymphocytic leukemia. Semin Oncol. 2016; 43:274–9.
18. Delbridge AR, Strasser A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015; 22:1071–80.
19. Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nature reviews Drug discovery. 2008; 7:989–1000.
20. Balakrishnan K, Gandhi V. Bcl-2 antagonists: a proof of concept for CLL therapy. Invest New Drugs. 2013; 31:1384–94.
21. Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009; 15:1126–32.
22. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005; 435:677–81.
23. Tromp JM, Geest CR, Breij EC, Elias JA, van Laar J, Luijks DM, Kater AP, Beaumont T, van Oers MH, Eldering E. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res. 2012; 18:487–98.
24. Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, Rybicki LA, Kalaycio M, Maciejewski JP, Houghton JA, Almasan A. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood. 2011; 118:3579–90.
25. Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012; 30:488–96.
26. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202–8.
27. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, Kipps TJ, Anderson MA, Brown JR, Gressick L, Wong S, Dunbar M, Zhu M, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. New Engl J Med. 2016; 374:311–22.
28. Green DR. A BH3 Mimetic for Killing Cancer Cells. Cell. 2016; 165:1560.
29. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA. 2005; 102:13944–9.
30. Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007; 67:782–91.
31. Russo M, Spagnuolo C, Volpe S, Tedesco I, Bilotto S, Russo GL. ABT-737 resistance in B-cells isolated from chronic lymphocytic leukemia patients and leukemia cell lines is overcome by the pleiotropic kinase inhibitor quercetin through Mcl-1 down-regulation. Biochem Pharmacol. 2013; 85:927–36.
32. Spagnuolo C, Cerella C, Russo M, Chateauvieux S, Diederich M, Russo GL. Quercetin downregulates Mcl-1 by acting on mRNA stability and protein degradation. Br J Cancer. 2011; 105:221–30.
33. Alford SE, Kothari A, Loeff FC, Eichhorn JM, Sakurikar N, Goselink HM, Saylors RL, Jedema I, Falkenburg JH, Chambers TC. BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res. 2015; 75:1366–75.
34. Jilg S, Reidel V, Muller-Thomas C, Konig J, Schauwecker J, Hockendorf U, Huberle C, Gorka O, Schmidt B, Burgkart R, Ruland J, Kolb HJ, Peschel C, et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia. 2016; 30:112–23.
35. Russo M, Nigro P, Rosiello R, D’Arienzo R, Russo GL. Quercetin enhances CD95- and TRAIL-induced apoptosis in leukemia cell lines. Leukemia. 2007; 21:1130–3.
36. Russo M, Palumbo R, Mupo A, Tosto M, Iacomino G, Scognamiglio A, Tedesco I, Galano G, Russo GL. Flavonoid quercetin sensitizes a CD95-resistant cell line to apoptosis by activating protein kinase Calpha. Oncogene. 2003; 22:3330–42.
37. Russo M, Palumbo R, Tedesco I, Mazzarella G, Russo P, Iacomino G, Russo GL. Quercetin and anti-CD95(Fas/Apo1) enhance apoptosis in HPB-ALL cell line. FEBS Lett. 1999; 462:322–8.
38. Russo M, Spagnuolo C, Volpe S, Mupo A, Tedesco I, Russo GL. Quercetin induced apoptosis in association with death receptors and fludarabine in cells isolated from chronic lymphocytic leukaemia patients. Br J Cancer. 2010; 103:642–8.
39. Russo GL, Russo M, Spagnuolo C. The pleiotropic flavonoid quercetin: from its metabolism to the inhibition of protein kinases in chronic lymphocytic leukemia. Food Funct. 2014; 5:2393–401.
40. Russo GL, Russo M, Spagnuolo C, Tedesco I, Bilotto S, Iannitti R, Palumbo R. Quercetin: a pleiotropic kinase inhibitor against cancer. Cancer Treat Res. 2014; 159:185–205.
41. Russo M, Spagnuolo C, Tedesco I, Bilotto S, Russo GL. The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochem Pharmacol. 2012; 83:6–15.
42. Spagnuolo C, Russo M, Bilotto S, Tedesco I, Laratta B, Russo GL. Dietary polyphenols in cancer prevention: the example of the flavonoid quercetin in leukemia. Ann N Y Acad Sci. 2012; 1259:95–103.
43. Block KI, Gyllenhaal C, Lowe L, Amedei A, Amin AR, Amin A, Aquilano K, Arbiser J, Arreola A, Arzumanyan A, Ashraf SS, Azmi AS, Benencia F, et al. Designing a broad-spectrum integrative approach for cancer prevention and treatment. Semin Cancer Biol. 2015; 35:S276–304.
44. Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, Yang H, Samadi AK, Russo GL, et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015; 35:S78–103.
45. Rosen A, Bergh AC, Gogok P, Evaldsson C, Myhrinder AL, Hellqvist E, Rasul A, Bjorkholm M, Jansson M, Mansouri L, Liu A, Teh BT, Rosenquist R, et al. Lymphoblastoid cell line with B1 cell characteristics established from a chronic lymphocytic leukemia clone by in vitro EBV infection. Oncoimmunology. 2012; 1:18–27.
46. Multi-author review. Protein kinase CK2 in health and disease. Cell Mol Life Sci. 2009; 66:1795–889.
47. Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003; 369:1–15.
48. Mandato E, Manni S, Zaffino F, Semenzato G, Piazza F. Targeting CK2-driven non-oncogene addiction in B-cell tumors. Oncogene. 2016; 35:6045–6052.
49. Piazza F, Manni S, Ruzzene M, Pinna LA, Gurrieri C, Semenzato G. Protein kinase CK2 in hematologic malignancies: reliance on a pivotal cell survival regulator by oncogenic signaling pathways. Leukemia. 2012; 26:1174–9.
50. Shehata M, Schnabl S, Demirtas D, Hilgarth M, Hubmann R, Ponath E, Badrnya S, Lehner C, Hoelbl A, Duechler M, Gaiger A, Zielinski C, Schwarzmeier JD, et al. Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3-K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood. 2010; 116:2513–21.
51. Barata JT. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv Enzyme Regul. 2011; 51:37–49.
52. Martins LR, Lucio P, Silva MC, Anderes KL, Gameiro P, Silva MG, Barata JT. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood. 2010; 116:2724–31.
53. Martins LR, Lucio P, Melao A, Antunes I, Cardoso BA, Stansfield R, Bertilaccio MT, Ghia P, Drygin D, Silva MG, Barata JT. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia. 2014; 28:179–82.
54. Prins RC, Burke RT, Tyner JW, Druker BJ, Loriaux MM, Spurgeon SE. CX-4945, a selective inhibitor of casein kinase-2 (CK2), exhibits anti-tumor activity in hematologic malignancies including enhanced activity in chronic lymphocytic leukemia when combined with fludarabine and inhibitors of the B-cell receptor pathway. Leukemia. 2013; 27:2094–6.
55. Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010; 70:10288–98.
56. Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood. 2008; 111:846–55.
57. Matter WF, Brown RF, Vlahos CJ. The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem Biophys Res Commun. 1992; 186:624–31.
58. Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000; 6:909–19.
59. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010; 11:329–41.
60. Barata JT. The impact of PTEN regulation by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv Enzyme Regul. 2011; 51:37–49.
61. Russo M, Spagnuolo C, Bilotto S, Tedesco I, Maiani G, Russo GL. Inhibition of protein kinase CK2 by quercetin enhances CD95-mediated apoptosis in a human thymus-derived T cell line. Food Res Int. 2014; 63:244–51.
62. Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J. 2008; 415:353–65.
63. Lessene G, Czabotar PE, Sleebs BE, Zobel K, Lowes KN, Adams JM, Baell JB, Colman PM, Deshayes K, Fairbrother WJ, Flygare JA, Gibbons P, Kersten WJ, et al. Structure-guided design of a selective BCL-X(L) inhibitor. Nat Chem Biol. 2013; 9:390–7.
64. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013; 12:1691–700.
65. Wang Z, Azmi AS, Ahmad A, Banerjee S, Wang S, Sarkar FH, Mohammad RM. TW-37, a small-molecule inhibitor of Bcl-2, inhibits cell growth and induces apoptosis in pancreatic cancer: involvement of Notch-1 signaling pathway. Cancer Res. 2009; 69:2757–65.
66. Eichhorst B, Cramer P, Hallek M. Initial therapy of chronic lymphocytic leukemia. Semin Oncol. 2016; 43:241–50.
67. Hallek M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am J Hematol. 2015; 90:446–60.
68. Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. New Engl J Med. 2014; 370:997–1007.
69. Vitiello M, Zullo A, Servillo L, Mancini FP, Borriello A, Giovane A, Della Ragione F, D’Onofrio N, Balestrieri ML. Multiple pathways of SIRT6 at the crossroads in the control of longevity, cancer, and cardiovascular diseases. Ageing Res Rev. 2017; 35:301–11.
70. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000; 351:95–105.
71. Agullo G, Gamet-Payrastre L, Manenti S, Viala C, Remesy C, Chap H, Payrastre B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem Pharmacol. 1997; 53:1649–57.
72. Lolli G, Cozza G, Mazzorana M, Tibaldi E, Cesaro L, Donella-Deana A, Meggio F, Venerando A, Franchin C, Sarno S, Battistutta R, Pinna LA. Inhibition of protein kinase CK2 by flavonoids and tyrphostins. A structural insight. Biochemistry. 2012; 51:6097–107.
73. Wang RE, Hunt CR, Chen J, Taylor JS. Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg Med Chem. 2011; 19:4710–20.
74. Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011; 117:591–4.
75. Inuzuka H, Fukushima H, Shaik S, Liu P, Lau AW, Wei W. Mcl-1 ubiquitination and destruction. Oncotarget. 2011; 2:239–44. doi: 10.18632/oncotarget.242.
76. Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001; 298:865–72.
77. Zaher M, Akrout I, Mirshahi M, Kolb JP, Billard C. Noxa upregulation is associated with apoptosis of chronic lymphocytic leukemia cells induced by hyperforin but not flavopiridol. Leukemia. 2009; 23:594–6.
78. Tromp JM, Geest CR, Breij EC, Elias JA, van Laar J, Luijks DM, Kater AP, Beaumont T, van Oers MH, Eldering E. Tipping the Noxa/Mcl-1 balance overcomes ABT-737 resistance in chronic lymphocytic leukemia. Clin Cancer Res. 2012; 18:487–98.
79. Primikyri A, Chatziathanasiadou MV, Karali E, Kostaras E, Mantzaris MD, Hatzimichael E, Shin JS, Chi SW, Briasoulis E, Kolettas E, Gerothanassis IP, Tzakos AG. Direct binding of Bcl-2 family proteins by quercetin triggers its pro-apoptotic activity. ACS Chem Biol. 2014; 9:2737–41.
80. Ferry DR, Smith A, Malkhandi J, Fyfe DW, deTakats PG, Anderson D, Baker J, Kerr DJ. Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res. 1996; 2:659–68.
81. Nam JS, Sharma AR, Nguyen LT, Chakraborty C, Sharma G, Lee SS. Application of Bioactive Quercetin in Oncotherapy: From Nutrition to Nanomedicine. Molecules. 2016; 21:E108.
82. Wang S, Su R, Nie S, Sun M, Zhang J, Wu D, Moustaid-Moussa N. Application of nanotechnology in improving bioavailability and bioactivity of diet-derived phytochemicals. J Nutr Biochem. 2014; 25:363–76.
83. Fautz R, Husein B, Hechenberger C. Application of the neutral red assay (NR assay) to monolayer cultures of primary hepatocytes: rapid colorimetric viability determination for the unscheduled DNA synthesis test (UDS). Mutat Res. 1991; 253:173–9.
84. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22:27–55.
85. Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976; 72:248–54.
86. Russo GL, Vandenberg MT, Yu IJ, Bae YS, Franza BR Jr, Marshak DR. Casein kinase II phosphorylates p34cdc2 kinase in G1 phase of the HeLa cell division cycle. J Biol Chem. 1992; 267:20317–25.
87. Havasi A, Li Z, Wang Z, Martin JL, Botla V, Ruchalski K, Schwartz JH, Borkan SC. Hsp27 inhibits Bax activation and apoptosis via a phosphatidylinositol 3-kinase-dependent mechanism. J Biol Chem. 2008; 283:12305–13.
88. Marshak DR, Carroll D. Synthetic peptide substrates for casein kinase II. Methods Enzymol. 1991; 200:134–56.
89. Endo A, Tatematsu K, Hanada K, Duermeyer L, Okamoto M, Yonekura-Sakakibara K, Saito K, Toyoda T, Kawakami N, Kamiya Y, Seki M, Nambara E. Tissue-specific transcriptome analysis reveals cell wall metabolism, flavonol biosynthesis and defense responses are activated in the endosperm of germinating Arabidopsis thaliana seeds. Plant Cell Physiol. 2012; 53:16–27.