
INTRODUCTION
The kinesin spindle protein Eg5 (also known as KSP or KIF11) is an important drug target in antimitotic cancer therapy. In early stages of mitosis, Eg5 contributes to centrosome separation through regulation of spindle elongation, which is essential for the formation of the bipolar spindle and segregation of sister chromatids [1, 2]. Successful blockage of Eg5 induces a hallmark phenotype known as the monopolar spindle, and results in cellular mitotic arrest and apoptosis [2, 3]. Overexpression of Eg5 leads to significant instability of the genome in mouse models and carcinogenesis [4]. The generally negligible expression of Eg5 in non-proliferating adult tissues results in a lower toxicity profile of Eg5-targeted therapy as compared to traditional anti-mitotic therapies. As Eg5 is strongly expressed in specific tissues such as blood and lymphatic vascular endothelial cells and is responsible for tumor angiogenesis, inhibition of Eg5 may have both an anti-proliferative effect as well as an anti-angiogenesis effect [5]. Loop L5 of Eg5 is considered to be an important component of the druggable Eg5 allosteric pocket. Among kinesin spindle protein family members this loop is the greatest in length, and thus better accommodates small molecule modulators, therefore providing an ideal structural basis for Eg5 inhibitor specificity [6, 7].
Numerous Eg5 inhibitors have been designed to target this allosteric pocket, and agents are currently undergoing clinical trials [8–15]. ARRY-520, developed by Array BioPharma, is at the forefront of Eg5 inhibitor research and development. The phase III clinical trial of this compound was successfully completed in August, 2015 (data from Thomson Reuters CortellisTM). Another promising Eg5 inhibitor candidate 4SC-205 which is ongoing under Phase I trial can be administrated orally for the treatment of solid tumor and hematologic neoplasia (ClinicalTrials.gov Identifier: NCT01065025). Neoplasm was stabilized in 67% of patients for 100 days with this agent, and median survival time was prolonged. In multiple clinical studies, Eg5 inhibitor treatment has shown favorable characteristics such as high tolerance, good pharmacokinetic/pharmacodynamic properties and low CNS toxicity [14, 16–18]. At the same time, other Eg5 inhibitor scaffolds such as S-trityl-L-cysteine (STLC) and its analogues [19, 20], benzimidazoles [21], thioxoimidazolidines [22], phenothiazanes [23], thiophenes [24], and biphenyls [25] are also in development.
Virtual screening is widely applied for identification of novel targeted drugs. This strategy is generally divided into structure based virtual screening (SBVS) and ligand based virtual screening (LBVS). Although these may appear to be two independent approaches to drug design, in practice they can be utilized together in a mutually advantageous way. Q.D. You pioneered use of LBVS and structure based drug design (SBDD) for discovery of novel Eg5 inhibitors, and this led to the discovery and identification of 1,4-dihydroquinolin-4-ones and 1,2,3,4-tetrahydroquinazolin-4-ones [26, 27]. The first successful application of virtual screening in this field was conducted by F. Kozielski, who combined pharmacophore modeling and molecular docking to identify three novel Eg5 inhibitors bearing quinazoline and thioxoimidazolidine scaffolds [22].
Inspired by these previous adaptations of computer aided drug design (CADD) in the field of Eg5 inhibitor discovery, we further optimized the workflow to search for a new scaffold with antitumor activity. In the first round of virtual screening for potential Eg5 inhibitors, a pharmacophore model (Supplementary Figure 1A, 1B) was combined with in silico docking (Supplementary Figure 1C) to identify the compound 7170 which had a moderate degree of antitumor activity (Supplementary Figure 1D). However, this compound does not break away from the triphenylmethyl scaffold of S-Trityl-L-cysteine (Table 1). In view of this scaffold’s limited potential for further development, we decided to conduct a second round of virtual screening by scaffold hopping. As the strict shape constraints of the pharmacophore model were likely to limit the ability of virtual screening to break away from the original scaffold, ROCS and EON from OpenEye was selected to perform a 3D similarity search. Multiple studies were used ROCS and EON for successful 3D similarity searches [28], offering an abundant source of material for refining our virtual screening protocol and optimizing the success rate [29, 30].
Table 1: EC50s (μM) of 3 compounds in enzyme and cell based assays
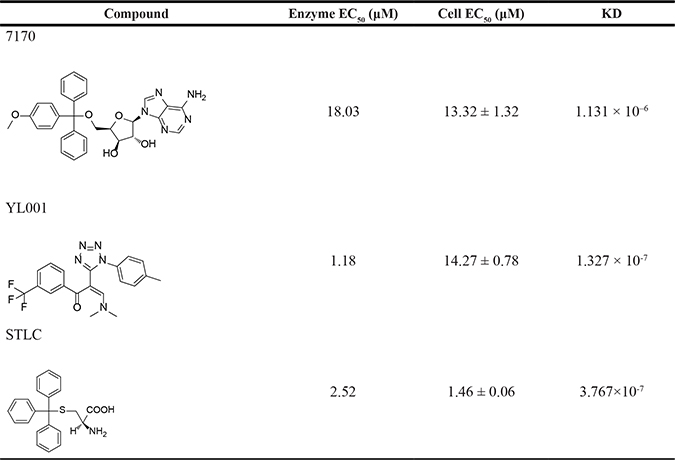
Enzymatic EC50s were assessed in Eg5 ATPase activity and cellular EC50s were evaluated on HeLa cell line. KD values were calculated with SPR Biacore T200. STLC was used as a control.
The 3D similarity search in the second round of virtual screening resulted in successful scaffold hopping, as the 1,5-disubstituted tetrazole compound YL001 is structurally unique in the field of Eg5 inhibitor research. This agent induces the Eg5-specific monopolar spindle phenotype and displays a strong antiproliferative activity over a broad range of cancer cell lines, including the taxol-resistant A2780 cell line and the 6TG-resistant 4T1 cell line. In a melanoma xenograft mouse model, YL001 inhibited tumor growth by 60% and prolonged median survival time by more than 50%. These findings demonstrated YL001 has the potential for Eg5 targeted therapy.
RESULTS
Discovery of YL001 by virtual screening
The workflow for virtual screening is shown in Figure 1A. The co-crystallized conformation of five STLC analogues (including STLC) with strong bioactivity (PDB ID: 2XAE, 3KEN, 4A50, 4A51 and 4BBG) were extracted, aligned and utilized to generate queries for the similarity search. These queries were subsequently validated through ROCS using the Database of Useful Decoys Enhanced (DUD-E) Eg5 inhibitor subset. The best query (query5), which featured ligands from 3KEN, 4A50 and 4BBG, and the best ROCS scoring method (ranked by shape tanimoto only) were selected due to their extraordinary performance in validation results (Supplementary Figure 2B–2D, 2F). Parameters for molecule conformation generation (OMEGA, OpenEye) were optimized and single-molecule queries for EON were subsequently validated and selected. Ligands of 4A51 and 4BBG were selected as single-molecule queries for EON rescoring, as they both showed enhanced performance compared to the Non-EON group and random selection group (Supplementary Figure 2E). Molecules from the Specs database (204,000 compounds) were filtered with FILTER (OpenEye) to exclude molecules with an unfavorable druggability profile; prepared with OMEGA using optimized parameters; screened, scored and filtered through ROCS and EON; docked into the allosteric pockets of three individual Eg5 crystal structures (PDB ID: 4A50, 4A51 and 4BBG; Figure 1E) to inspect the calculated binding affinity and the binding mode; clustered by structure, and manually selected. In the virtual screening, YL001 showed good molecular shape similarity with query5, with a shape tanimoto score of 0.70 (Figure 1C). This molecule also ranked first in EON electrostatic shape comparison. In Figure 1D, it is evident that the N,N-dimethylamine group closely mimics the amino group from the query, while the trifluoromethyl group simulates the function of the alkyl group. Docking studies were conducted with AutoDockVina to determine whether the superpositioned conformation was consistent with the docking results [31]. For three different receptors (4A50, 4A51 and 4BBG) the average binding affinity (n = 3) for the best binding conformations of YL001 were –8.9, –9.4 and –9.2 kcal/mol respectively. A hydrogen bond was found between the protonated N,N-dimethylamine group and Glu116, and the trifluoromethyl group fitted into the subpocket where the alkyl group of the original ligands was located, adequately filling the pocket as in the superpositioned conformation. This validated the ROCS and EON results (Figure 1B, 1E). After completion of the workflow, 23 molecules were purchased from Specs for evaluation in further assays.
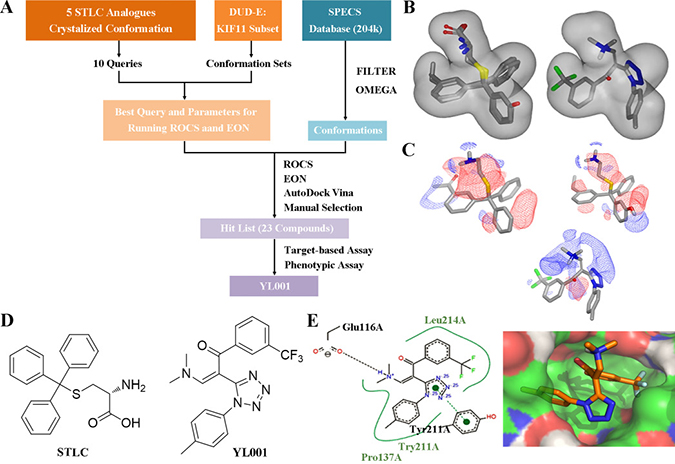
Figure 1: Identification of novel Eg5 inhibitors with 3D similarity search based virtual screening. (A) Virtual screening workflow. (B) Molecular shape comparison of query5 (left) and YL001 (right); grey shape contours in both figures are query5 shape contours. (C) Molecular surface electrostatic map showing the ligand of 4A51 (left), the ligand of 4BBG (right) and YL001 (below): positive charge (blue grid), negative charge (red grid). (D) Structure of STLC (left) and YL001 (right). (E) Docking pose of YL001 in the allosteric pocket of the receptor (PDB ID: 4A51). 2D interaction plot (left): hydrogen bonds (black dashes), pi-pi stacking interaction (green dashes). Surface plot (right): carbon (green), nitrogen (blue), oxygen (red), polar hydrogen (white).
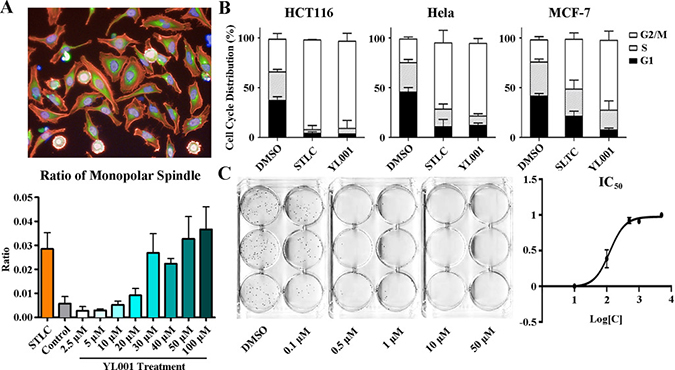
Figure 2: Phenotypic analysis of cells treated with YL001. (A) HeLa cells were treated with 50 μM YL001 or STLC (control) for 8 hours and fixed and stained for high content screening. Actin (red staining); tubulin (green staining); DNA (blue staining). The bar graph below shows the ratio of monopolar spindle cells to all cells treated with YL001 at different concentrations. (B) Cell cycle analysis of cells treated with YL001 in various cancer cell lines. YL001 efficiently arrested cells in mitosis and caused cell death. (C) HeLa cells were treated with gradient concentrations of YL001. The graph to the right shows inhibition with different treatment. YL001 exhibited antitumor activity by suppressing tumor growth in the colony formation assay.
Validation of YL001 as a highly selective antitumor agent targeted on Eg5
All 23 compounds selected by virtual screening were investigated with a novel comprehensive validation strategy to directly pick hits. This strategy combined enzymatic screening and SPR (as target-based screening) with cytotoxic and monopolar spindle screening (as phenotypic screening with high content imaging), allowing us to take advantage of both phenotypic and target-based screening, as well as to validate the compounds with strong anti-Eg5 activity (Supplementary Table 1). YL001 was selected using this strategy, and showed an EC50 of 1.18 μM on enzymatic assay, as well as an EC50 of 14.27 μM in HeLa cells with a monopolar spindle phenotype. Moreover, it bound to the Eg5 motor domain tightly, with a KD of 1.327×10–7 M as detected by SPR (Table 1). YL001 exhibited a KD constant which was two-fold stronger than the positive control STLC (3.767 × 10–7 M), and an order of magnitude stronger than compound 7170 which was identified in the first round of virtual screening (1.131 × 10–6 M). Through use of double validation with phenotypic and target-based screening, YL001 was identified as an Eg5 inhibitor with significant in vitro antitumor activity without obvious cytotoxicity against normal cells (Supplementary Table 2).
Activity and selectivity are two critical properties for small molecule enzyme inhibitors. Selectivity was a concern since YL001 has an α,β-unsaturated carbonyl bond which may react with endogenous nucleophiles via Michael addition and lead to cross-reaction with proteins in vivo. However, we found that YL001 is not active enough to react with highly active nucleophiles. YL001 was incubated with cysteine and lysine for 24 hours at 37°C and no new products were detected by HRMS. To further quantitatively analyze the selectivity of YL001, we assessed YL001 activity in a comprehensive panel of 468 typical, atypical and mutant human kinases at a concentration of 1 μM [32]. As expected, with a selectivity score of 0.00, YL001 displayed no activity at a concentration of 1 μM (taking into consideration the ~1% system false positive rate) (Supplementary Figure 3). These results confirm YL001 is not a covalent inhibitor, but a highly selective Eg5 inhibitor.
YL001 induces a monopolar spindle phenotype, resulting in mitotic arrest and cell death
YL001 strongly inhibited cell cycle progression. Exposure of cultured cells to YL001 produced the hallmark phenotype of Eg5 inhibition with an accumulation of cells in G2/M phase. High content imaging was performed on HeLa cells over a series of gradient concentrations of YL001, which induced a monopolar spindle phenotype in which cells showed a rosette of condensed chromosomes attached to a radial array of microtubules, wrapped in a round actin skeleton. As analyzed with Columbus, treatment with this compound led to monopolar spindles with an EC50 of 15.30 μM, demonstrating that normal mitotic spindle configuration is disrupted (Figure 2A).
Cytoflow was conducted on multiple cancer cell lines. As expected, all cell lines tested (HCT 116, HeLa and MCF7) showed arrest at G2/M, especially HCT 116. After 8 hours treatment with 50 μM YL001, the percentage of cells with 4N DNA content surged to over 80% (Figure 2B). To further observe the effect of YL001 induced cell cycle arrest, colony formation using different concentrations of the compound was carried out to measure the IC50. HeLa cells were exposed to YL001 for 24 hours and maintained in culture for 10 days without further treatment. The number of clones following YL001 treatment was significantly less than the number following STLC treatment. YL001 inhibited colony formation with an IC50 of 124 nM, demonstrating that in regard to antitumor activity, YL001 is stronger than STLC (IC50 = 1.25 μM) by an order of magnitude (Figure 2C).
YL001 suppresses tumor growth and prolongs median survival by suppressing Eg5 function in a B16 melanoma xenograft model
YL001 activity was tested in multiple human and rodent cancer cell lines to evaluate its spectrum of antitumor activity. YL001 showed broad-spectrum antitumor effects with EC50 values predominantly in the range of 5–20 μM (Table 2). CNE-2Z (nasopharyngeal cancer) and K562 (leukemia) cells were especially sensitive to treatment. HT1080 with an EC50 of over 70 μM was the only cell line which was not sensitive to treatment among the 15 cell lines tested. According to BioGPS data [33], in the four cell lines with available Eg5 gene expression level data, Eg5 expression ranked as HeLa > MCF-7 > U87 > HT1080. This shows the efficiency of YL001 activity is positively correlated with Eg5 gene expression. In addition, Eg5 inhibitors would be expected to be effective in cancers which are resistant to tubulin-targeting agents [34], where resistance results from mutation of beta-tubulin or alteration in the expression of tubulin isoforms [35, 36]. As expected, YL001 has shown activity in the taxol resistant cell line A2780 and the 6-Thiaguanine resistant cell line 4T1, which suggests YL001 has significant potential for therapy of drug-resistant tumors.
Table 2: Anti-proliferative activity EC50s of YL001 against a broad range of human and rodent cancer cell lines
Cell Lines |
Organism |
Type |
Source |
EC50 of YL001/ μM |
|---|---|---|---|---|
K562 |
Human |
Lymphoid |
Leukemia |
9.76 ± 0.52 |
CNE-2Z |
Human |
Epithelial |
Nasopharyngeal cancer |
5.94 ± 0.02 |
A549 |
Human |
Epithelial |
Lung cancer |
22.77 ± 0.72 |
Huh7 |
Human |
Epithelial |
Liver cancer |
14.59 ± 0.82 |
HeLa |
Human |
Epithelial |
Cervical cancer |
14.27 ± 0.78 |
B16 |
Mouse |
Epithelial |
Melanoma |
13.91 ± 5.03 |
HCT116 |
Human |
Epithelial |
Colon cancer |
9.04 ± 2.19 |
U87 |
Human |
Epithelial |
Brain cancer |
26.20 ± 0.12 |
DU145 |
Human |
Epithelial |
Prostate cancer |
26.46 ± 0.27 |
MCF-7 |
Human |
Epithelial |
Breast cancer |
14.90 ± 2.57 |
786-O |
Human |
Epithelial |
Renal cancer |
14.69 ± 0.62 |
AGS |
Human |
Epithelial |
Stomach cancer |
20.20 ± 0.01 |
HT1080 |
Human |
Epithelial |
Fibrosarcoma |
76.25 ± 0.77 |
A2780/taxol |
Human |
Epithelial |
Ovarian cancer |
20.14 ± 0.68 |
4T1/6TG |
Mouse |
Epithelial |
Breast cancer |
20.03 ± 0.50 |
Thirteen human cancer cell lines and two rodent cancer cell lines were used, including a human resistant cell line and a mouse resistant cell line. A2780/taxol is a taxol resistant A2780 cell line and 4T1/6TG is a 6TG resistant 4T1 cell line. Cellular activity of these three compounds showed positive correlation with Eg5 gene expression (HeLa > MCF-7 > U87 > HT1080).
The strong broad-spectrum antitumor activity displayed by YL001 against cultured cells prompted further evaluation of in vivo activity of YL001 in a B16 rodent melanoma xenograft model. After testing a range of YL001 doses in healthy B6 mice without tumor, we estimated the maximal therapeutic dose to be 200 mg/kg taking into consideration the solubility of YL001. Doses of 200 mg/kg were administrated daily for 10 days to B6 mice with tumor xenografts of the highly malignant melanoma B16. Animals with this xenograft in general exhibit a low survival rate and poor response to chemotherapy. However, i.p. treatment with YL001 significantly suppressed tumor growth with a minimum T/C ratio of 0.40 (P < 0.05 for tumor volume compared to controls) (Figure 3A) and an absence of toxicity (P > 0.05 for body weight loss compared to controls) (Figure 3B). Median survival results (Figure 3C) showed prolongation of the treatment group’s survival time by 50% over that of the control (Table 3). YL001 thus has excellent antitumor activity in this xenograft model, and no adverse effects were observed.
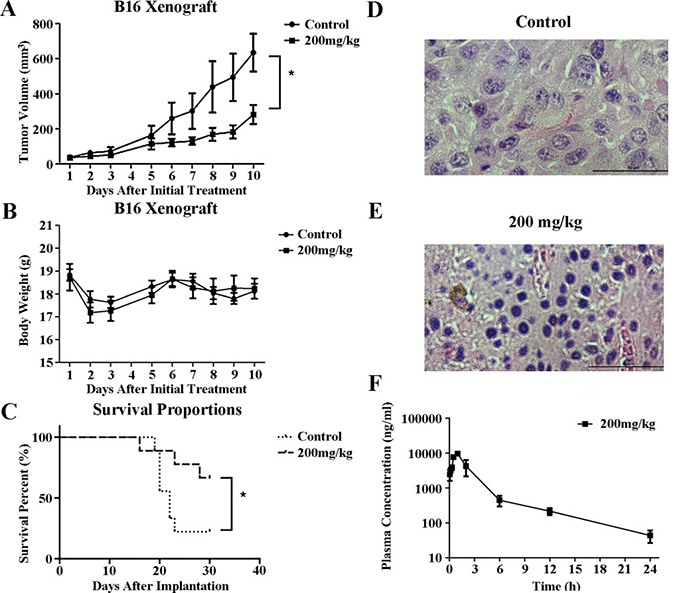
Figure 3: Antitumor efficacy of YL001 in a xenograft tumor model. (A) Tumor volume versus time after initial treatment with YL001 in B16 tumor-bearing mice. Columns, mean (n = 6); bars, SD. *P < 0.05. (B) Body weight versus days after initial treatment with YL001 in B16 tumor-bearing mice. Columns, mean (n = 6); bars, SD. (C) Percent survival versus time after implantation in B16 tumor-bearing mice treated with YL001 (n = 9). *P < 0.05. (D) H&E staining of a tumor from the control group. (E) H&E staining of a tumor from the YL001 treated group. (F) Pharmacokinetic study of YL001. Plasma concentration versus time after initial administration with YL001 in HeLa tumor-bearing Balb/c nude mice. Columns, mean (n = 3); bars, SD.
Table 3: Pharmacokinetic/pharmacodynamic studies of YL001
Control |
YL001 200 mg/kg |
|
|---|---|---|
Medium survival time/day |
22 |
> 33 |
Half-life in plasma/hour |
– |
5.3 |
Median survival time is shown in days. Half-life in plasma is presented in hours.
To determine whether YL001 inhibited Eg5 function in the B16 xenograft model, we examined the phenotype of mitotic cells in tumor tissues. Histochemisty analysis of tumor tissue demonstrated that YL001 induces mitotic arrest accompanied by formation of monopolar spindles through inhibition of Eg5 (Figure 3D, 3E), and has antitumor activity in xenograft tumor models (Figure 3A, 3C).
We also assessed the half-life of YL001 in the plasma in HeLa xenograft nude mice. The results (Figure 3F) show YL001 has a favorable pharmacokinetic property with t1/2 = 5.3 hours. These results indicate that YL001 has favorable pharmacokinetic and pharmacodynamic characteristics which warrant further investigation of this agent as an antitumor agent.
YL001 causes mitotic failure leading to cell death through activation of caspase-3
High content imaging in the YL001 treated group of cells showed cell fragments including many apoptotic bodies reflecting apoptosis, which leads to cell death by the activation of caspases. Caspase-3 is the most important of these caspases and plays a central role in transduction of apoptotic signals as an effector, and its activation leads to protein degradation.
Western blot was used for analysis of cleaved PARP1, which is a product of Caspase-3 that serves as a marker for intrinsic apoptosis. After 24 hours treatment with YL001, PARP1 cleavage increased dramatically (Figure 4A, lane 2 vs. lane 5), and the accumulation of cleaved PARP1 persisted for 48 hours (Figure 4A, lane 3 vs. lane 6). This shows YL001-induced cell death is due to apoptosis caused by Caspase-3 activation [34]. In addition, the marker of phosphor histone H3, which is specifically phosphorylated in mitosis, was evaluated with western blot. Immunostaining with phospho-specific antibody revealed that with YL001 treatment, the cells are evidently arrested in mitosis (Figure 4A, lane 5–6), leading eventually to apoptosis.
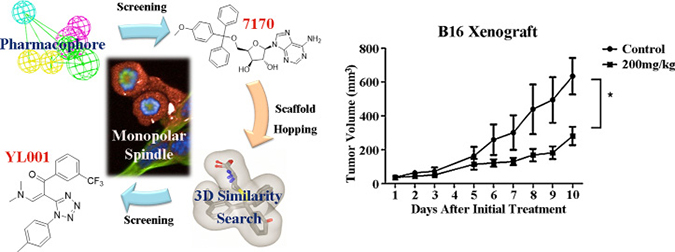
Graphical Abstract: Discovery of Eg5 inhibitor YL001 as a novel antitumor agent. Left, two rounds of virtual screening results in the discovery of YL001 as an Eg5 inhibitor which can lead to monopolar spindle phenotype. Right, YL001 exhibits potent antitumor activity in the B16 xenograft mouse model.
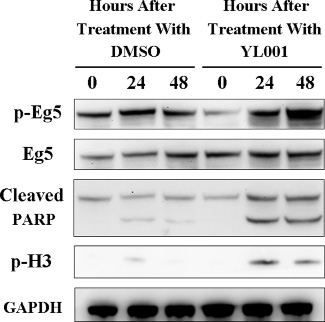
Figure 4: YL001 triggers caspase-3 activation and elevates Eg5 phosphorylation at T926. HeLa cells were treated with 50 μM of YL001 and DMSO (as a control) for a given length of time and cell lysate was tested for T926 phosphorylation of Eg5, total Eg5, cleaved PARP, S10 phosphorylation of histone H3 and GAPDH (control).
It came as a surprise to find comparison of each specific time point showed YL001 treatment consistently elevated phosphorylation of Eg5 at T926 (Figure 4A, lane 2–3 vs. lane 5–6). Eg5 phosphorylation is essential for its functional association with microtubules [2, 37, 38], but Eg5 hyperphosphorylation impairs normal Eg5 distribution and its function in spindle assembly [39]. These results demonstrate YL001 not only impedes Eg5 ATPase function as shown in Result 2.2, but also induces Eg5 hyperphosphorylation at T926, leading to mitotic catastrophe and subsequent apoptosis.
DISCUSSION
In this study, we have successfully combined virtual screening, target-based screening and phenotypic screening as a novel strategy to discover a novel scaffold Eg5 inhibitor with strong anti-neoplastic activity economically and efficiently. This novel strategy may offer guidance for discovery of other new targeted drugs.
Query is the core of a 3D similarity search in virtual screening. STLC was a structurally simple lead compound with distinctive antitumor activity [19, 20]. As numerous previous studies had discussed its SAR and co-crystallization structures with Eg5 [40–43], it was clear that STLC analogues were suitable for building our query, and supporting effective screening for novel modulators of the target as well. Analysis of co-crystal structures of STLC with Eg5 showed that the crystallized bioactive conformation of these STLC analogues superimposed well within the Eg5 allosteric pocket. We therefore supposed that use of a highly congruent superimposed bioactive conformation as a shape query would not only provide greater space for accommodating the molecular overlay, but would also circumvent the limitations of single molecule queries, and thus would be more compatible with discovery of new scaffolds.
Over the course of development of this compound, its chemical scaffold underwent a substantial structural change from the triphenylmethyl scaffold (STLC) to the final novel 1,5-disubstituted tetrazole (YL001). This would have been difficult to achieve using conventional drug design approaches such as bioisosteric fragment replacement. However, this virtual screening workflow yielded successful scaffold hopping. Similarity search based methodology generally outperforms molecular docking in efficiency, and 3D-similarity search achieves scaffold hopping albeit at the cost of a decrease in the success rate when compared to traditional 2D-similarity searches and molecular docking [44–46]. Thus, considering the small pool size of the compounds tested, the success rate (one hit out of 23 molecules tested, which is 4.3%) was satisfactory.
In phenotypic screening, an effective high content imaging method was introduced to identify potential hit compounds. In current practice, screening based on phenotype plays a more critical role in the early stages of drug discovery. Prior to the 1980s, most drugs were discovered with use of phenotypic screening in vitro or in vivo. However with the development of clone technology, phenotypic screening in drug discovery was gradually de-emphasized under comparison with the then emerging high-throughput, target-specific in vitro assays [47]. During the heyday of targeted cancer therapy at the beginning of 21st century, numerous potential drug targets were discovered, and screening assays were constructed based on these targets. However, this approach yielded many clinical candidates but resulted in few new marketed drugs [48].This was attributed to the fact that target-based drug screening did not take disease pathogenesis into account, and targeted drugs still incur the risk of inducing adaptive drug resistance [49]. We surmised that by combining phenotypic and target-based screening, advantages of both methods could be maximized and disadvantages could be minimized. This provided a novel platform for drug discovery which is economical and effective, and is adaptable to other research systems.
As expected, introduction of YL001 resulted in failure of mitosis by inhibiting ATPase function and accumulation of T926 phosphorylated Eg5, thus facilitating apoptosis by activating caspase-3. Although the mechanism of its activity has not been fully worked out, our results are sufficient to show YL001 has promise as an Eg5 inhibitor for cancer therapy.
MATERIALS AND METHODS
Virtual screening
Virtual screening round one procedure
A HypoGen pharmacophore model was generated by Discovery Studio 2.5 modeled on the study of Liu et al. [27]. Compounds from the PKU database were prepared and screened through this pharmacophore model, which contains four features including an aromatic ring, a hydrophobic, a hydrogen bond acceptor and a donor. The Eg5-ispinesib complex crystal structure (PDB code 4AP0) was used for in silico docking via GOLD 3.0. All compounds tested interacted with the Eg5 allosteric pocket. 100 top scored compounds in both two models were kept in the best-binding conformations for virtual analysis. Eventually six low weight molecules were obtained for the further validation.
Virtual screening round two procedures
Query validation and parameter optimization with OMEGA, ROCS and EON
Five STLC-typed ligands with strong binding capacity in their crystalized and bioactive conformations were selected (PDB ID: 2XAE, 3KEN, 4A50, 4A51 and 4BBG). The ligand 4BBG was modified to remove the unwanted aromatic tautomer state, and was then aligned with Discovery Studio V2.5 (Accelrys) using the default settings of the molecular overlay module. Five Queries were generated using aligned molecules by vROCS 3.1.2 (OpenEye) with the following parameters: maximum molecules per model = 5, models to keep = 5, merge color atoms = True. Four of five output queries were multi-ligand queries, and one query was a single-compound query. ROCS query validation was validated with the KIF11 subset of DUD-E selected to assess the performance of different queries, scoring functions and running modes [50]. Conformations of the validation set were generated by OMEGA 2.4.6 (OpenEye) using default parameters with the FLIPPER module turned on. All of the single-compound queries acquired directly from the original crystalized conformations were assessed as well. Query validation data is recorded in Supplementary Figure 1. Parameters of OMEGA were also subsequently optimized on query5 using the same validation method. Ranges of all relevant parameters (“ewindow” from 5, 10 to 15 kcal/mol and “rms” from 0.3, 0.4, 0.5 to 0.6 angstroms respectively) were chosen according to several important references. For maximum efficiency, we had the parameter “maxconfs” set to 100. The top 1% of the ROCS output hit list (ranked by shape tanimoto) for the optimized method was used as an input for EON (2.1.0, OpenEye) query validation. Since EON only accepts single-molecule queries, only the single-compound queries acquired directly from the original crystalized conformation were evaluated using the default parameters. The output was ranked by ET_Combo (Supplementary Figure 1F).
Database preparations
The Specs database (~204,000 compounds) was used as the screening library. All compounds were first treated with Pipeline Pilot v7.5 (Accelrys) to generate 3D coordination, strip salt, minimize molecule energy, and standardize the chemical table coding. FILTER 2.1.1 from OpenEye was then applied to filter unfavorable compounds using the recommended filter criteria documented in filter_blockbuster.txt provided by OpenEye. Conformations were generated by OMEGA 2.4.6 using the optimized parameters with the FLIPPER module turned on.
Virtual screening
All conformations were compared to query5 using ROCS 3.1.2 on molecular shape and ranked by shape tanimoto. The first 5,000 molecules were kept and re-evaluated by EON 2.1.0 to preserve the first 500 molecules with the highest ET_Combo score. Two separate hit lists produced by using the two different EON queries were merged with Pipeline Pilot. Molecules with the following properties were discarded: ADMET Solubility Level = 0, rotatable bonds > 9 (both properties calculated by Pipeline Pilot), which left 389 molecules for post screening analysis.
Pose screening analysis
All 389 molecules were docked into the allosteric pocket of three different receptors (PDB ID: 4A50, 4A51 and 4BBG) by AutoDockVina (1.1.2, Scripps) with default settings. Best-binding conformations were manually inspected in Open-Source PyMOL v1.3.x (Schrödinger). Hydrogen bond and ligand embedding ratios were speculatively assigned and calculated by the third-party PyMOL scripts. In the three separate docked poses corresponding to each receptor, at least two were expected to be successfully docked into the allosteric pocket to be considered a reliable pose, and the average embedding ratio of those poses was expected to be larger than 0.6. Moreover, at least two reliable poses were expected to have no less than one hydrogen bond interaction with the receptor, and the average hydrogen bond interaction count of these reliable poses was larger than one. In addition, the average binding affinity for these reliable poses was expected to be stronger than -9 kcal/mol. After docking analysis, 77 molecules were kept for further study. These molecules were clustered using the Ligand Cluster protocol provided by Pipeline Pilot, and the results were modified manually for better results. No more than two molecules were selected per cluster. Finally, 24 molecules were confirmed, and 23 of these were purchased from Specs (one of them was unavailable) to be evaluated in further assays. The 2D binding plot was generated by the PoseView web service [51].
Organic synthesis
Solvents and reagents were obtained from commercial suppliers and were used without further purification. Reactions were monitored with thin layer chromatography (TLC) on GF254 silica gel plates (Anhui LiangChen Silicon Material Co. Ltd.). The spots were visualized using an ultraviolet light. Flash chromatography was carried out on silica gel zcx-II (48–75 μm, Branch of Qingdao Haiyang Chemical Co. Ltd.) using the indicated solvents (PE, boiling range 60–90°C). 1H NMR spectra and 13C NMR spectra were recorded on a Bruker AVANCE III 400 (400 MHz) spectrometer in D-reagents with tetramethylsilane (TMS) as an internal reference. NMR chemical shifts are reported as values (ppm) relative to internal TMS. Splitting patterns were designated as: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet. Melting points were measured as uncorrected values. High resolution mass spectrometry (HRMS) was measured with an LTQ ObitrapElite spectrometer using ionspray methodology. High-performance liquid chromatography (HPLC) was carried out using a ZORBAX SB-C18 column 5 μm, 4.6 × 250mm.
Typical synthesis procedure for the target compound YL001 ((Z)-3-(dimethylamino)-2-(1-(p-tolyl)-1H-tetrazol-5-yl)-1-(3-(trifluoromethyl)phenyl)prop-2-en-1-one)
Step 1: A solution of 4.0 mL (30 mmol, 1 eq.) 4›-methyl-acetophenone was added to N,N-Dimethylformamide dimethyl acetal 8.0 mL (60 mmol, 2 eq.) and this mixture was stirred at 115°C under reflux for 30 hours. The reaction mixture was then cooled to room temperature, and red crystals were precipitated. The product was collected through suction filtration, washed with PE (15 mL × 3) and dried in vacuo. The product (4.532 g, 23.9 mmol), consisting of orange crystals, was pure enough to be used directly in the next reaction. The yield was 79.7%.
Step 2: To a solution of the product from step 1 (3.730 g, 19.7 mmol, 1 eq.) in 13.1 mL cold anhydrous dichloromethane (DCM) stirred at 0°C, a solution of POCl3 (1.84 mL, 19.7 mmol, 1 eq.) in 4.9 mL cold anhydrous DCM was added dropwise. This mixture was then stirred at room temperature for 20 minutes and 0°C for another 10 minutes. The intermediate was collected through suction filtration, and washed with 10 mL cold anhydrous DCM. While filtering the intermediate, 8.276 g (60.4 mmol, 3.1 eq.) of NaClO4•H2O was dissolved in 16.4 mL water and stirred at 0°C. The intermediate was added to this solution and stirred vigorously at 0°Cfor 30 minutes. The product was then collected by suction filtration, washed with a solution of 1.642 g (11.7 mmol, 0.6 eq.) NaClO4•H2O in 16.4 mL water, and dried in vacuo. The product (6.795 g) was as light yellow solid, was pure enough to be used directly in the next reaction. Yield was considered as 100%.
Step 3: In a bottle with two necks 2.945 g (45.3 mmol, 2.3 eq.) of NaN3 and 50 mL methanol were stirred at 60°C under reflux. Product from step 2 was added portion wise, and gas was allowed to release completely before adding the next portion. After product from step 2 was completely added, the mixture was stirred for another 30 minutes at 60°C under reflux. The mixture was then cooled to room temperature. Excess distilled water was poured into the mixture while stirring to let the product precipitate. The product was filtered by suction filtration, washed with diluted water (10 mL × 2), and dried in vacuo. The product (2.300 g, 10 mmol) was as pink solid, and was pure enough to be used directly in a next reaction. Yield was 50.5%.
Step 4: To a solution of the product from step 3 (229 mg, 1.0 mmol, 1 eq.) in 5 mL anhydrous toluene, triethylamine 0.17 mL (1.2 mmol, 1.2 eq.) was added and stirred at 0°C for 10 minutes. 3-(Trifluoromethyl)benzoyl chloride (1.2 mmol, 1.2 eq.) was added dropwise, and stirred for another 30 minutes at 0°C. The mixture was stirred and heated to 105°C under reflux for 8 hours. The mixture was then cooled to room temperature, and 10 mL EA and 50 mL of water were added to the mixture and extracted. The water phase was extracted with EA (10 mL × 3), and the organic phases were combined and washed with brine (50 mL × 3). The organic phase was dried with anhydrous Na2SO4. After the solvent was removed under vacuum, the residue was separated with silica gel column chromatography (PE: EA = 1: 1 to 1: 2 to 100% EA) which yielded the crude product. The crude product was heated and recrystallized in methanol, the crystal product was washed with anhydrous ether three times, and dried in vacuo. Final product, YL001, was as light yellow crystal, mp. 122.2 - 122.4°C. 1H NMR (400 MHz, CDCl3) δ 7.90 (s, 1H), 7.52 (d, J = 7.9 Hz, 1H), 7.22 (t, J = 7.8 Hz, 1H), 7.07 (m, 4H), 6.91 (d, J = 7.9 Hz, 2H), 3.34 (s, 3H), 2.75 (s, 3H), 2.36 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.09, 156.83, 151.38, 140.26, 139.88, 131.62, 130.95, 130.63, 130.03, 128.22, 126.86, 125.04, 124.86, 123.51, 122.76, 122.15, 91.34, 77.37, 77.05, 76.73, 48.01, 40.87, 21.01. HRMS calcd. for C20H18F3N5O+ [M+H]+ 402.15362, found 402.15337, ppm 1.96.
Biology
Cancer cell lines
Cancer cell lines (CNE-2Z, A549, Huh7, HeLa, B16, K562, MCF-7, HCT116, U87, DU145, 786-O, AGS, HT1080, A2780/taxol, 4T1/6TG) used for both in vitro antitumor and mechanism studies, and the in vivo xenograft mouse model were all obtained from the China Infrastructure of Cell Line Resources and American Type Culture Collection. Cells were cultured in DMEM (Macgene) or RPMI 1640 (Macgene) medium supplemented with 10% FBS in a 37°C incubator with 5% (v/v) CO2.
Compounds and reagents
STLC was from Alladin (1121390) (Shanghai, China). The compounds of interest identified by virtual screening were purchased from Specs. YL001 was first purchased from Specs, then synthesized in our laboratory. All compounds were prepared as 10 mM DMSO stock, and were sequentially diluted in growth medium. Cells were treated with compounds by medium exchange.
Antibodies
Cell lysates from cultured cells were prepared and analyzed by immunoblotting on nitrocellulose membranes. The following antibodies were used in this study: Mouse total anti-Eg5 T926 (620502) anti-Eg5 total (627802) was purchased from Biolegend. Rabbit anti-cleaved PARP (D64E10) (CS-5625P) and anti phospho histone H3 (ser10) (CST3377) antibody was from Cell Signaling Technologies. Mouse anti-GAPDH antibody was from Zhongshan Golden Bridge Biotechnology (Beijing, China). Anti-rabbit IgG and anti-mouse IgG coupled horseradish peroxidase (HRP) were from Sigma-Aldrich (St. Louis, MO, USA).
Inhibition of Eg5 ATPase Activity
All experiments were performed as previously described [52]. Data were analyzed using GraphPad Prism 5 and Microsoft Excel to obtain EC50 values. STLC was used as a positive control. Each inhibitory concentration was measured in triplicate, and averaged data points ± SD are shown.
SPR Assay
Interactions of the Eg5 motor domain and compounds of interest were analyzed using the Biacore T200 system (GE Healthcare, Uppsala, Sweden) at 25°C. Eg5 motor protein obtained as previously described (51) was immobilized on a sensor chip (CM5) using an amine coupling kit (GE Healthcare, Buckinghamshire, UK). Final Eg5 immobilization levels were typically ~23000 RU. Subsequently, compounds were injected as analytes at various concentrations, using PBS-P (10 mM phosphate buffer with 2.7 mM KCl and 137 mM NaCl, 0.05% Surfactant P20, pH 4.5, GE Healthcare, Uppsala, Sweden) as a running buffer. For binding studies, analytes were applied at corresponding concentrations in running buffer at a flow rate of 30 μL/minute with a contact time of 60 seconds and a dissociation time of 60 seconds. Chip platforms were washed with running buffer and 50% DMSO. Data were analyzed with Biacore evaluation software (T200 Version 1.0) by curve fitting using a steady state 1:1 binding model and a kinetic model.
Nucleophilic addition assay
0.5 mmol YL001 was solved in a solution of THF/H2O (v:v = 1:1) and stirred with nucleophiles (0.5 mmol L-cysteine or L-lysine) at 37°C for 24 hours. Products were then validated by HRMS.
Kinase profiling
Kinase profiling was conducted by the DiscoverX as KINOMEscan profiling service for testing the inhibition of YL001 of interest against 468 kinases [53]. Selectivity score S(35) was defined as follows:
S(35) = (number of non-mutant kinases with% Control < 35)/(number of non-mutant kinases tested).
Antiproliferation assays
Cytotoxic activities of the compounds of interest were investigated with human cancer cell lines using the Alamar Blue assay. Cells (5 × 103 cells/well) were seeded into 96-well plates (Corning, 3603). Various sample concentrations (compounds of interest) were added to each well in duplicate, and incubated at 37°C with 5% CO2 for two days such that cells were in the exponential growth phase at the time of drug addition. Ten microliters of the Alamar Blue Solution (Invitrogen, Alamar Blue) were added to each well and incubated at 37°C for up to 2 hours in a humidified, 5% CO2 atmosphere. After incubation, florescence was measured using a microplate reader (Tecan) at an excitation wavelength of 540 nm and emission of 590 nm. Each inhibitory concentration was measured in triplicate, and averaged data points ± SD are shown. Statistical analysis was carried out with GraphPad Prism 5 and the fifty-percent effective concentration is expressed as EC50.
Immunofluorescence microscopy
HeLa cells (5 × 103 cells/well) were seeded in 96-well plates (Corning, 3603). Drugs were diluted appropriately in medium from 10 mM stocks in 100% DMSO and added to the cells. Following 8 hours incubation with drugs, cells were fixed with 4% paraformaldehyde (20 minutes at room temperature), permeabilized with PBST (0.5% Triton X-100 in PBS) for 15 minutes, and washed before blocking. Cells were blocked with 4% BSA for at least 30 minutes at room temperature and stained with anti-β-tubulin monoclonal antibodies (Sigma) at 400-fold dilution for 1 hour with Alexafluor 488-conjugated goat anti-mouse secondary antibodies (Invitrogen) at 300-fold dilution for 1 hour. DNA was detected with 4’,6-diamidino-2-phenylindole dihydrochloride (DAPI). Actin was detected with Phalloidin. High content screening (Perkin Elmer Operetta) was used to photograph stained cells at 40× magnification, and Columbus software was used for statistical analysis of the percentage of mitotic cells with monopolar spindles present in treated cells over the total number of cells in mitosis counted after 8 hours incubation with drugs.
Flow cytometry
HeLa cells (1 × 106 cells per well) were plated in 6-well plates (Nunc, 140675). 10 mM stock was diluted into an appropriate final concentration solution with growth medium. After 12 hours, cells were incubated with compounds for 12 hours in 37°C with 5% CO2, using DMSO as a negative control and 50 μM STLC as a positive control. Medium was removed and PBS was used for washing twice. Cells were then digested with trypsin into a single-cell solution and fixed with methanol overnight at 4°C. Fixative was removed and cells were washed twice with PBS. 25 μL 20×RNaseA was added followed by incubation for 30 minutes at 37°C. 5 μL of PI was added followed by incubation for 30 minutes at 4°C. Cytoflow was carried out by BD FACSVerse (BD Biosciences, US).
Colony formation
HeLa cells (5 × 102 cells/well) were seeded into 6-well plates and were treated with a series of gradient concentrations of YL001 for 24 hours, followed by washing and continual culture for 10 days. DMSO was used as a negative control, and STLC was used as a positive control. Cells were fixed with ethanol for more than 30 minutes and dyed with crystal violet for more than 30 minutes. Clones were counted with OpenCFU [54].
In vivo tumor xenograft studies
B16 cells (1 × 106 cells) were inoculated s.c. into 6-week old C57BL/6 mice. Tumor volume was calculated as: V = 1/2× L (long axis)×W2 (short axis). When the volume of tumor was between 50 to 100 mm3, YL001 was administered i.p. daily on days 1 to 10 at 200 mg/kg. Doses and schedules were determined by tolerance studies. Mice were kept under specific pathogen-free conditions, and all animal experiments were approved by the Committee on Animal Research at our institute.
Vehicle (10% (v%) N,N-Dimethylacetamide (DMAC) in 200 μL olive oil per 20 g mouse) was administered daily i.p. as a control on days 1 to 10. Drug efficiency was expressed as the ratio of the mean experimental V/V0 value to that of the control group [treated versus control (T/C) ratio], where V is the tumor volume on the day of evaluation and V0 is the tumor volume on the day of initial treatment with the drug. Drug efficiency was also evaluated by median survival. All statistical analysis was performed using Graphpad Prism 5.
Histochemistry
On day 10 after the first treatment, all mice were sacrificed. Tumors from two mice in each group were fixed with formalin and analyzed histologically using H&E staining.
Pharmacokinetic study
A HPLC-MS/MS method was developed for single-dose pharmacokinetic (PK) study of YL001 in HeLa xenograft BALB/c nude mice. YL001 was administered intraperitoneally at a dose of 200 mg/kg. Blood samples were then collected by enucleating eyeballs at 0.083, 0.167, 0.333, 0.5, 1, 2, 6, 12, 24 hours in heparinized polypropylene tubes. Three mice were sacrificed at each time point. Samples were centrifuged immediately at 4000 r/min for 20 min to separate plasma. The final sample for HPLC-MS/MS detection by QTRAP 6500 (AB Sciex, USA) was a mixture (plasma: H2O: methanol = 1:1:8). All individual data were pooled together to further perform PK analysis. DAS 2.1.2 were used to estimate PK parameters by non-compartmental analysis.
Abbreviations
KSP, Kinesin Spindle Protein; SAR, Structure-Activity Relationship; CADD, computer aided drug design; STLC, S-Trityl-L-cysteine; DUD-E, Database of Useful Decoys: Enhanced; SPR, Surface Plasmon Resonance; KD, Dissociation Constant; TLC, Thin Layer Chromatography; PPI, Protein-protein interaction;HRMS, High Resolution Mass Spectrometry; HPLC, High-Performance Liquid Chromatography; H&E, Hematoxylin and eosin; DMAC, N,N-Dimethylacetamide.
ACKNOWLEDGMENTS
We thank Luhua Lai for kindly providing CADD instruction and Sulong Xiao for suggestions of organic synthesis. We thank Zhiyuan Hou for the help of in vivo assays, Yu Liu for technical support on cytoflow, and Xuyang Zhao and Jutuo Zhou for mass spectrum technical assistance.
CONFLICTS OF INTEREST
There are no conflicts of interest on the part of any of the authors.
GRANT SUPPORT
This work was supported by grants to Y. Yin, including the National Natural Scientific Foundation of China (31420103905, 81430056, and 81372491), and the Lam Chung Nin Foundation for Systems Biomedicine.
REFERENCES
1. Le Guellec R, Paris J, Couturier A, Roghi C, Philippe M. Cloning by differential screening of a Xenopus cDNA that encodes a kinesin-related protein. Mol Cell Biol. 1991; 11:3395–98.
2. Blangy A, Lane HA, d’Hérin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995; 83:1159–69.
3. Mayer TU, Kapoor TM, Haggarty SJ, King RW, Schreiber SL, Mitchison TJ. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science. 1999; 286:971–74.
4. Castillo A, Morse HC 3rd, Godfrey VL, Naeem R, Justice MJ. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res. 2007; 67:10138–47.
5. Exertier P, Javerzat S, Wang B, Franco M, Herbert J, Platonova N, Winandy M, Pujol N, Nivelles O, Ormenese S, Godard V, Becker J, Bicknell R, et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget. 2013; 4:2302–16. doi: 10.18632/oncotarget.1490.
6. Kapitein LC, Peterman EJ, Kwok BH, Kim JH, Kapoor TM, Schmidt CF. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005; 435:114–18.
7. Vale RD, Milligan RA. The way things move: looking under the hood of molecular motor proteins. Science. 2000; 288:88–95.
8. Mross K, Scharr D, Richly H, Bauer S, Krauss B, Krauss R, Mais A, Hauns B, Hentsch B, Braz E. First-in-Man Study of 4SC-205 (AEGIS), a novel oral inhibitor of Eg5 kinesin spindle protein. J Clin Oncol. 2014; 32:2564–2564.
9. Hollebecque A, Deutsch E, Massard C, Gomez-Roca C, Bahleda R, Ribrag V, Bourgier C, Lazar V, Lacroix L, Gazzah A, Varga A, de Baere T, Beier F, et al. A phase I, dose-escalation study of the Eg5-inhibitor EMD 534085 in patients with advanced solid tumors or lymphoma. Invest New Drugs. 2013; 31:1530–38.
10. Rosen L, Chen LC, Iyengar T, Goldman J, Lahr S, Chen CR, France DS, Chan TC, Schwartz B, Abbadessa G, Von Hoff DD. ARQ 621, a novel potent and selective inhibitor of Eg5: preclinical data and early results from a clinical phase 1 study. Cancer Res. 2010; 70:2750.
11. Wakui H, Yamamoto N, Kitazono S, Mizugaki H, Nakamichi S, Fujiwara Y, Nokihara H, Yamada Y, Suzuki K, Kanda H, Akinaga S, Tamura T. A phase 1 and dose-finding study of LY2523355 (litronesib), an Eg5 inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol. 2014; 74:15–23.
12. Shah JJ, Zonder J, Bensinger WI, Cohen AD, Kaufman JL, Nooka AK, Weber DM, Hilder B, Rush SA, Ptaszynski A. Prolonged survival and improved response rates with ARRY-520 in relapsed/refractory multiple myeloma (RRMM) patients with low α-1 acid glycoprotein (AAG) levels: results from a phase 2 study. Blood. 2013; 122:285.
13. Khoury HJ, Garcia-Manero G, Borthakur G, Kadia T, Foudray MC, Arellano M, Langston A, Bethelmie-Bryan B, Rush S, Litwiler K, Karan S, Simmons H, Marcus AI, et al. A phase 1 dose-escalation study of ARRY-520, a kinesin spindle protein inhibitor, in patients with advanced myeloid leukemias. Cancer. 2012; 118:3556–64.
14. LoRusso PM, Goncalves PH, Casetta L, Carter JA, Litwiler K, Roseberry D, Rush S, Schreiber J, Simmons HM, Ptaszynski M, Sausville EA. First-in-human phase 1 study of filanesib (ARRY-520), a kinesin spindle protein inhibitor, in patients with advanced solid tumors. Invest New Drugs. 2015; 33:440–49.
15. Lonial S, Cohen A, Zonder J, Bensinger W, Kaufman JL, Orlowski RZ, Harvey RD, Alexanian R, Thomas S, Weber D. The novel KSP inhibitor ARRY-520 demonstrates single-agent activity in refractory myeloma: results from a Phase 2 trial in patients with relapsed/refractory multiple myeloma (MM). Blood. 2011; 118:1266.
16. Owens B. Kinesin inhibitor marches toward first-in-class pivotal trial. Nat Med. 2013; 19:1550.
17. Lee CW, Bélanger K, Rao SC, Petrella TM, Tozer RG, Wood L, Savage KJ, Eisenhauer EA, Synold TW, Wainman N, Seymour L. A phase II study of ispinesib (SB-715992) in patients with metastatic or recurrent malignant melanoma: a National Cancer Institute of Canada Clinical Trials Group trial. Invest New Drugs. 2008; 26:249–55.
18. Holen K, DiPaola R, Liu G, Tan AR, Wilding G, Hsu K, Agrawal N, Chen C, Xue L, Rosenberg E, Stein M. A phase I trial of MK-0731, a kinesin spindle protein (KSP) inhibitor, in patients with solid tumors. Invest New Drugs. 2012; 30:1088–95.
19. Zee-Cheng KY, Cheng CC. Experimental antileukemic agents. Preparation and structure-activity study of S-tritylcysteine and related compounds. J Med Chem. 1970; 13:414–18.
20. Kessel D, Smith G, Blahnik J. Effects of S-(trityl)-L-cysteine and its analogs on cell surface properties of leukemia L1210 cells. Biochem Pharmacol. 1976; 25:1893–97.
21. Lahue BR, Ma Y, Shipps GW Jr, Seghezzi W, Herbst R. Substituted benzimidazoles: A novel chemotype for small molecule hKSP inhibitors. Bioorg Med Chem Lett. 2009; 19:3405–09.
22. Nagarajan S, Skoufias DA, Kozielski F, Pae AN. Receptor-ligand interaction-based virtual screening for novel Eg5/kinesin spindle protein inhibitors. J Med Chem. 2012; 55:2561–73.
23. Okumura H, Nakazawa J, Tsuganezawa K, Usui T, Osada H, Matsumoto T, Tanaka A, Yokoyama S. Phenothiazine and carbazole-related compounds inhibit mitotic kinesin Eg5 and trigger apoptosis in transformed culture cells. Toxicol Lett. 2006; 166:44–52.
24. Pinkerton AB, Lee TT, Hoffman TZ, Wang Y, Kahraman M, Cook TG, Severance D, Gahman TC, Noble SA, Shiau AK, Davis RL. Synthesis and SAR of thiophene containing kinesin spindle protein (KSP) inhibitors. Bioorg Med Chem Lett. 2007; 17:3562–69.
25. Holland JP, Kang A, Cohrs S, Selivanova SV, Milicevic Sephton S, Betzel T, Frey D, Wieser M, Jaussi R, Kammerer RA, Schibli R, Fischer E. Synthesis and evaluation of biphenyl compounds as kinesin spindle protein inhibitors. Chem Biodivers. 2013; 10:538–55.
26. Jiang C, Yang L, Wu WT, Guo QL, You QD. De novo design, synthesis and biological evaluation of 1,4-dihydroquinolin-4-ones and 1,2,3,4-tetrahydroquinazolin-4-ones as potent kinesin spindle protein (KSP) inhibitors. Bioorg Med Chem. 2011; 19:5612–27.
27. Liu F, You QD, Chen YD. Pharmacophore identification of KSP inhibitors. Bioorg Med Chem Lett. 2007; 17:722–26.
28. Rush TS 3rd, Grant JA, Mosyak L, Nicholls A. A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction. J Med Chem. 2005; 48:1489–95.
29. Naylor E, Arredouani A, Vasudevan SR, Lewis AM, Parkesh R, Mizote A, Rosen D, Thomas JM, Izumi M, Ganesan A, Galione A, Churchill GC. Identification of a chemical probe for NAADP by virtual screening. Nat Chem Biol. 2009; 5:220–26.
30. Hevener KE, Mehboob S, Su PC, Truong K, Boci T, Deng J, Ghassemi M, Cook JL, Johnson ME. Discovery of a novel and potent class of F. tularensis enoyl-reductase (FabI) inhibitors by molecular shape and electrostatic matching. J Med Chem. 2012; 55:268–79.
31. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010; 31:455–61.
32. Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011; 29:1046–51.
33. Jackson JR, Gilmartin A, Dhanak D, Knight S, Parrish C, Luo L, Sutton D, Caulder E, Diamond M, Giardiniere M. A second generation KSP inhibitor, SB-743921, is a highly potent and active therapeutic in preclinical models of cancer. AACR. 2006.
34. Tao W, South VJ, Zhang Y, Davide JP, Farrell L, Kohl NE, Sepp-Lorenzino L, Lobell RB. Induction of apoptosis by an inhibitor of the mitotic kinesin KSP requires both activation of the spindle assembly checkpoint and mitotic slippage. Cancer Cell. 2005; 8:49–59.
35. Monzó M, Rosell R, Sánchez JJ, Lee JS, O’Brate A, González-Larriba JL, Alberola V, Lorenzo JC, Núñez L, Ro JY, Martín C. Paclitaxel resistance in non-small-cell lung cancer associated with beta-tubulin gene mutations. J Clin Oncol. 1999; 17:1786–93.
36. Hasegawa S, Miyoshi Y, Egawa C, Ishitobi M, Taguchi T, Tamaki Y, Monden M, Noguchi S. Prediction of response to docetaxel by quantitative analysis of class I and III β-tubulin isotype mRNA expression in human breast cancers. Clin Cancer Res. 2003; 9:2992–97.
37. Cahu J, Olichon A, Hentrich C, Schek H, Drinjakovic J, Zhang C, Doherty-Kirby A, Lajoie G, Surrey T. Phosphorylation by Cdk1 increases the binding of Eg5 to microtubules in vitro and in Xenopus egg extract spindles. PLoS One. 2008; 3:e3936.
38. Blangy A, Arnaud L, Nigg EA. Phosphorylation by p34cdc2 protein kinase regulates binding of the kinesin-related motor HsEg5 to the dynactin subunit p150. J Biol Chem. 1997; 272:19418–24.
39. Wang J, Liu H. [Application of Nitrile in Drug Design]. [Article in Chinese]. Youji Huaxue. 2012; 32:1643–52.
40. Good JA, Wang F, Rath O, Kaan HY, Talapatra SK, Podgórski D, MacKay SP, Kozielski F. Optimized S-trityl-L-cysteine-based inhibitors of kinesin spindle protein with potent in vivo antitumor activity in lung cancer xenograft models. J Med Chem. 2013; 56:1878–93.
41. Kaan HY, Weiss J, Menger D, Ulaganathan V, Tkocz K, Laggner C, Popowycz F, Joseph B, Kozielski F. Structure-activity relationship and multidrug resistance study of new S-trityl-L-cysteine derivatives as inhibitors of Eg5. J Med Chem. 2011; 54:1576–86.
42. Wang F, Good JA, Rath O, Kaan HY, Sutcliffe OB, Mackay SP, Kozielski F. Triphenylbutanamines: kinesin spindle protein inhibitors with in vivo antitumor activity. J Med Chem. 2012; 55:1511–25.
43. Abualhasan MN, Good JA, Wittayanarakul K, Anthony NG, Berretta G, Rath O, Kozielski F, Sutcliffe OB, Mackay SP. Doing the methylene shuffle—further insights into the inhibition of mitotic kinesin Eg5 with S-trityl L-cysteine. Eur J Med Chem. 2012; 54:483–98.
44. Hawkins PC, Skillman AG, Nicholls A. Comparison of shape-matching and docking as virtual screening tools. J Med Chem. 2007; 50:74–82.
45. Venkatraman V, Pérez-Nueno VI, Mavridis L, Ritchie DW. Comprehensive comparison of ligand-based virtual screening tools against the DUD data set reveals limitations of current 3D methods. J Chem Inf Model. 2010; 50:2079–93.
46. Hu G, Kuang G, Xiao W, Li W, Liu G, Tang Y. Performance evaluation of 2D fingerprint and 3D shape similarity methods in virtual screening. J Chem Inf Model. 2012; 52:1103–13.
47. Johnson RK, McCabe FL, Caulder E, Inlow-Porter L, Whitacre M, Winkler JD, Bergnes G, Feng B, Morgans D, Wood K. SB-715992, a potent and selective inhibitor of the mitotic kinesin KSP, demonstrates broad-spectrum activity in advanced murine tumors and human tumor xenografts. Proc Am Assoc Cancer Res. 2002; 43:269.
48. Yokoyama H, Sawada J, Katoh S, Matsuno K, Ogo N, Ishikawa Y, Hashimoto H, Fujii S, Asai A. Structural basis of new allosteric inhibition in Kinesin spindle protein Eg5. ACS Chem Biol. 2015; 10:1128–36.
49. Strickley RG. Solubilizing excipients in oral and injectable formulations. Pharm Res. 2004; 21:201–30.
50. Mysinger MM, Carchia M, Irwin JJ, Shoichet BK. Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J Med Chem. 2012; 55:6582–94.
51. Stierand K, Rarey M. Drawing the PDB: Protein-Ligand Complexes in Two Dimensions. ACS Med Chem Lett. 2010; 1:540–45.
52. Kozielski F, DeBonis S, Skoufias DA. Screening for inhibitors of microtubule-associated motor proteins. Methods Mol Med. 2007; 137:189–207.
54. Geissmann Q. OpenCFU, a new free and open-source software to count cell colonies and other circular objects. PLoS One. 2013; 8:e54072.