
INTRODUCTION
Adducin is a ubiquitously expressed membrane-skeletal protein, which was firstly extracted from human erythrocyte in 1986 [1]. In 1991, Joshi and Gilligan found two adducin subunits named α adducin (ADD1) and β adducin (ADD2) [2], respectively. In 1995, Dong et al., reported the existence of γ adducin (ADD3) [3]. Adducin can be phosphorylated by PKA, PKC, and Rho-kinase. Besides, it can also bind to Ca2+/Calmodulin to exert functions. There is no doubt that abnormal phospho-adducin will disturb its function in cell proliferation, motility and signaling. Previous studies reported that adducin showed an abnormal level of expression or unusual phosphorylation in some tumors, suggesting that adducin may play a role in the evolution and progression in tumors.
Structure of adducin
Adducin is believed to self-associate into heterodimers and heterotetramers, which are composed of either α/β or α/γ. Heterodimers are the main modality. Adducin is localized at spectrin-actin junctions. α adducin and γ adducin are found in most tissues, whereas β adducin is abundant only in erythrocytes and brain.
The adducin family consists of three closely related genes: ADD1, ADD2, and ADD3. ADD1 has been mapped by positional cloning to human chromosome 4p16.3 [4]. Human ADD2 is localized at chromosome 2p13-p14 [5], whereas ADD3 is on chromosome 10q24.2-24.3 [6]. The sequences of α adducin and β adducin share 66% similarity at amino acid level, while γ adducin displays 60-70% sequence similarity with either α or β adducin.
It is demonstrated that all the three adducin proteins contain an N-terminal globular head domain, a neck domain and a C-terminal protease-sensitive tail domain. At the end of tail domain, there is a 22-residue MARCKS-related domain that has high homology to myristoylated alanine-rich C kinase substrate (MARCKS) protein. The MARCKS-related domain has clusters of lysine residues and is highly conserved among those three adducin subunits [1–3, 7–11].
Function of adducin
Adducin is important for formation and stabilization of membrane cytoskeleton
Adducin preferentially binds to spectrin-actin at the tail domain. It can not only recruit spectrin to spectrin-actin to promote forming complicated meshwork but also bundle actin filaments [1, 9, 12-16]. Previous studies indicated that there were two linkages between membrane skeleton and lipid bilayer: band 3-ankyrin-β-spectrin and glycophorin C-protein 4.1-β-spectrin. William demonstrated that adducin formed a bridge of band-3-adducin-spectrin to consolidate the stabilization of the membrane [17–19]. Additionally, adducin can inhibit capping the fast-growing ends of actin filaments as an actin capping protein. In this way, adducin could prevent addition or loss of actin subunits and make it easier to bundle actin filaments [15, 16].
Adducin is involved in cell signaling and ionic transportation
Adducin is the substrate of PKA, PKC, Rho-kinase and it can be phosphorylated by those kinases to get involved in cellular signal transduction. Moreover, adducin has been demonstrated to exert influence on ionic transport. In particular, adducin is related to Na+ transportation, such as Na+-H+ exchange, Na+-Li+ reverse transfer and Na+-K+-Cl- co-transport [9, 20, 21].
Adducin is essential for mitosis
Phosphorylation of ADD1 at Ser12 and Ser335 by cyclin-dependent kinase 1 (CDK1) enables ADD1 to bind to myosin-X (Myo10) and associate with mitotic spindles. The depletion of ADD1 results in distorted, elongated, and multipolar spindles, accompanied by aberrant chromosomal alignment [22, 23]. This phenomenon indicated that adducin played a crucial role in mitosis. Some studies revealed that ADD2 regulated structural stability and segregation of the chromosome as ADD2 bound to the chromosome in the mitotic metaphase. Furthermore, it is also found that adducin participated in meiosis of murine oocyte, suggesting that adducin may be indispensable in embryogenesis [24].
Adducin regulates cell motility
Phospho-adducin by Rho-kinase regulates membrane ruffling and cell motility [11]. ADD1 is dephosphorylated and mostly bound to spectrin in the membrane skeleton of the resting platelet at sites where actin filaments attach to the ends of spectrin molecules. After platelet activation, adducin that phosphorylated by PKC at Ser726 releases from the membrane skeleton concomitant with its dissociation from spectrin and actin, leading to the exposure of barbed actin filament ends which contributes to the actin assembly reactions in lamellar and filopodia [25–27]. PKCδ and phospho-adducin by PKCδ promote cell dissemination and migration [28]. Taken together, adducin is believed to play a role in regulating cell movement.
Adducin plays a critical role in cell-cell adhesion
It has been long recognized that ADD1 and ADD3 are enriched at intercellular junctions in cultured epithelial cells and simple mucosal epithelia in vivo [29]. Recent studies revealed that adducin promoted recruitment of β-spectrin so as to strengthen epithelial adherens junctions and tight junctions and antagonize stimulus-induced junctional disassembly. This novel function of adducin is important for remodeling of epithelial junctions and restoration of epithelial barrier [30].
Activation mechanism of adducin
Post-translational modification of proteins is of vital importance in human body. Most common post-translational modifications include phosphorylation, ubiquitination, glycosylation, methylation and acetylation. However, few studies have reported that adducin could be modified by other ways except phosphorylation. Phosphorylation of adducin at specific sites by PKA, PKC and Rho-kinase is indispensable (Figure 1). One study mentioned that adducin was phosphorylated in the hepatocyte growth factor (HGF)/c-MET signaling pathway but not in detail [31]. It has also been demonstrated that adducin could bind to Ca2+/Calmodulin at the domain of MARCKs to exert functions. Following are four main activation processes.
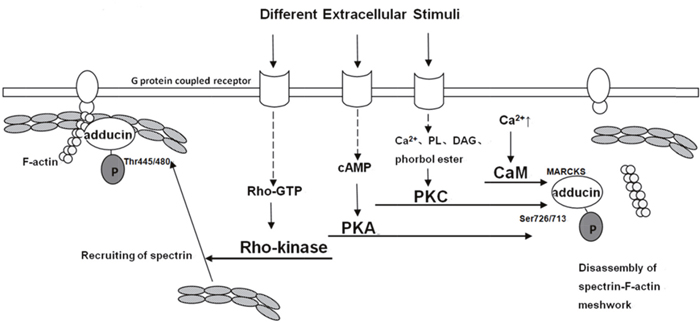
Figure 1: Model for different pathways of adducin phosphorylation [11, 26–36]. A spectrin-F-actin meshwork is linked to the membrane by association with membrane accessory proteins such as band 4.1. Upon the activation of Rho-kinase by Rho during the action of extracellular stimuli, α-adducin is phosphorylated at Thr445 and Thr480. Phosphorylated α-adducin binds to F-actin and then facilitates the recruitment of spectrin to F-actin. This may promote the formation of a spectrin-F-actin meshwork beneath membrane. On the other hand, phosphorylated α-adducin at Ser726 by PKA or PKC, and at MARCKS through Ca2+/CaM inhibits the activity of adducin in recruiting spectrin to F-actin and functioning as an actin capping protein.
Ca2+/Calmodulin pathway
The MARCKs-related domain of adducin has calcium-dependent binding of calmodulin. Different stimuli increase the concentration of intracellular calcium to trigger adducin bind to Ca2+/Calmodulin and hinder recruiting spectrin to spectrin-actin and capping the fast-growing ends of actin filaments [10, 30]. Calmodulin binding of native adducin and recombinant adducin polypeptides are inhibited by phosphorylation with PKC and by PKA. Other activities of adducin in association with spectrin-actin complexes are not affected by phosphorylation with PKC, which may indicate that the primary impact of phosphorylation by PKC is through modulation of the effects of calmodulin. Several observations showed that phosphorylation by PKC resulted in an almost complete loss of calmodulin binding for ADD2 and isolated peptide while only 50% reduction of binding to ADD1, suggesting that calmodulin preferentially binds to ADD2 when intracellular calcium is low while under higher levels of calcium it occupies binding sites of ADD1 as well as ADD2 [10].
cAMP-PKA pathway
Change in intracellular cAMP concentration is the main feature of this pathway. The production of cAMP, accelerated by activated adenylatecyclase (AC) contributes to the activation of PKA. Therefore, adducin, one downstream of PKA, is phosphorylated at Ser726 and Ser713 and it can also be targeted at Ser408, Ser436 and Ser481. Phospho-adducin inhibits the affinity of adducin and spectrin-actin and restrains from recruiting spectrin to actin [10, 11, 32].
PKC pathway
PKC isozymes is comprised of serine/threonine protein kinases that differ in cofactor requirement. For instance, the classical PKCs (α,β,γ) require a phospholipid (PL), diacylglycerol (DAG) or phorbol ester, and Ca2+ for optimal activation. The activation of PKCs (δ,ε,η,θ) is DAG, PL or phorbol ester dependent, but Ca2+ independent [29, 32, 33]. The phosphorylation of adducin at Ser 726 and Ser713 by PKC diminishes its interaction with actin and spectrin, hinders the recruitment of spectrin and exposes the barbed ends of actin filaments in motile cells [10, 26, 27, 32]. Recent studies elucidated that adducin was mainly targeted at Ser726 by PKCδ isozyme while PKCα may target at other sites except for Ser716 and Ser726 which remains unknown and requires further studies [28].
Rho/Rho-kinase pathway
Rho-kinase (ROCK) plays an important role in mitosis, muscle contraction, regulation of cytoskeleton, tumorigenesis and metastasis. Extracellular signals and some cytosolic proteins can affect the activity of ROCK. Extracellular stimuli (e.g. α-receptor agonist norepinephrine) aim at G protein-linked receptor and consequently activate Rho into Rho-GTP. The lateral binds the Rho binding domain of ROCK and leads to the exposure of ROCK’s catalytic active center. Therefore, ADD1 is phosphorylated at Thr445 and Thr480 and phospho-adducin promotes its binding to F-actin and facilitates the recruitment of spectrin to F-actin to enhance the formation of a spectrin-F-actin meshwork beneath plasma membrane for membrane ruffling. It also regulates adducin to function as an actin capping protein [11, 35, 36]. However, previous studies haven’t classified the role of Rho/Rho-kinase pathway in ADD2 or ADD3 phosphorylation.
Advances in the study of adducin in tumorigenesis and metastasis
Phosphorylation is one of the most common post-translational modifications, whose target is mainly serine, threonine and tyrosine residues. The state of the downstream has changed between phosphorylation and dephosphorylation, bringing about changes in subcellular localization, life span and interaction with other proteins. As a result, phosphorylation is involved in cell growth, differentiation and apoptosis. Abnormal phosphorylation results in instability of proteins and unusual activity, thus contributing to abnormal cell proliferation, dysdifferentiation and apoptosis suppression in etiopathology. Abundant studies revealed that abnormal phosphorylation was related to tumorigenesis and the hereditary variation on kinase, phosphatase or substrate impacts human susceptibility to tumors.
Different activation pathways have different binding sites. As mentioned above, PKA and PKC target at Ser726 and Ser713 of adducin, while Rho-kinase works at Thr445 and Thr480. Abnormal activation of adducin is bound to have influence on cell proliferation, cell motility and signaling. For example, when S716 mutation occurs, ADD1 can be adhered to α nucleoprotein and then be transferred into the nucleus which lead to abnormality of cell communication and cell-cell adhesion [37].
So far, limited studies have shown changes in adducin levels and/or phosphorylation during cancer development and the effect on the motility and metastatic potentials of cancer cells. Different tumor types may share diverse changes in adducin level (Table 1). Specifically, in renal carcinoma, total adducin levels decreased while Ser-660-phosphorylated adducin levels increased, possibly due to enhanced protein kinase C δ activity in these tumors [38]. Adducin and PKCδ levels were increased in mammary tumor cell lines [39, 40]. ADD1 and ADD2 transcripts were found to be upregulated in basal cell carcinoma and squamous cell carcinoma when compared with the regular levels in normal keratinocytes [41]. In contrast, ADD3 was found to be downregulated in migrating glioma cells. Concerning that few studies have reported ADD2 in tumors, we mainly make a brief overview on ADD1 and ADD3 in tumorigenesis and metastasis from the following subparts.
Table 1: Differences of adducin in different tumors
tumor type |
changes in adducin |
authors and references |
|---|---|---|
ovarian cancer |
down-regulation of ADD1 |
Syed V [41] |
non-cardia gastric cancer |
ADD1 rs4963 C→G mutation |
Wang MH [43] |
colorectal cancer |
ADD1 rs4963 C→G mutation |
Shen N [44] |
lung cancer |
up-regulation of ADD1 |
Jen J [45] |
small cell lung cancer |
up-regulation of ADD1 and ADD3 |
Ma PC [31] |
murine breast tumor |
up-regulation of ADD3-Ia |
Duretre M [46] |
non small cell lung cancer |
down-regulation of ADD3-Ib |
|
colorectal cancer |
up-regulation of ADD3-Ia/ADD3-Ib |
Tao M [49] |
glioblastoma |
down-regulation of ADD3 |
|
T-lymphoblastic leukemia |
ADD3 fused with NUP98 |
Lahortiga L [52] |
Note:ADD3-Ia or ADD3 (+14):encoded by variant 1 that expresses exon14; ADD3-Ib or ADD3 (-14):encoded by variant 2 and variant 3 that lose exon14
ADD1 in tumors
It is worth noticing that adducin seems to function oppositely in different tumors. From the following mentioned studies, we can see that α adducin may serve as a tumor suppressor in some tumors (e.g. ovarian tumor) but a promoter in tumors like lung cancer. However, few studies have mentioned the exact mechanism of how α adducin works in tumor development.
ADD1 in ovarian cancer
By transcriptional profiling and stringent data mining criteria, one research identified that ADD1 expression were up-regulated by estradiol-17β (E2) in human ovarian surface epithelial but down-regulated by estrogen in ovarian cancer (OVCA) cells. It was also found that ADD1 overexpression in OVCA cells was associated with reduced cell proliferation, soft agar, colony formation and invasion, suggesting adducin functions as a tumor suppressor in ovarian cancer cells. Such a tumor suppressive function could be mediated via promotion of cell differentiation and suppression of mobility in the cancer cells. However, specific mechanism still remains unclear [42].
ADD1 in non-cardia gastric cancer and colorectal cancer
Recently, a missense variant at the codon of ADD1’s phosphorylation site, rs4963 (Ser586Cys), is reported to modify the risk of non-cardia gastric cancer and colorectal cancer (CRC). The ADD1-rs4963 C allele is at the codon of TCT encoding serine, whereas the G allele is at the codon of TGT encoding cysteine. When C→G mutation occurs, the serine residue changes into a cysteine residue, which cannot be phosphorylated due to lack of hydroxyl groups. Wang et al., conducted a case-control study with 1998 cases and 2008 controls and found that ADD1-rs4963 might be associated with susceptibility to non-cardia gastric cancer [43]. Likewise, Shen et al., also performed a case-control study with a total of 1054 CRC cases and 1128 matched controls and also found that ADD1-rs4963 might modify the risk of CRC [44]. It is speculated that the G risk allele of ADD1-rs4963 might affect the phosphorylation of ADD1 and disturb its activity, including cell proliferation and communication, resulting in the development of non-cardia cancer and CRC [43, 44].
ADD1 in lung cancer
One study found that HGF and c-MET were overexpressed in small cell lung cancer (SCLC) and ADD1 was one of the downstream of HGF/c-MET signaling pathway [31]. Jen et al., observed that Zinc finger protein 322A (ZNF322A) was overexpressed in Asian and Caucasian lung cancer patients. Quantitative proteomic analysis revealed potential ZNF322A-regulated downstream targets, including ADD1, cyclin D1 (CCND1) and p53. They found that ZNF322A formed a complex with c-Jun and cooperatively activated ADD1 and CCND1 but repressed p53 gene transcription. Reconstitution experiments indicated that CCND1 and p53 were important to ZNF322A-mediated promotion of cell proliferation, whereas ADD1 was necessary for ZNF322A-mediated cell migration and invasion, suggesting ADD1 was involved in tumorigenesis and metastasis of lung cancer [45].
ADD3 in tumors
Apart from ADD1, some studies have reported the connection of ADD3 with tumors. ADD3 gene can be cut into three spliceosomes named variant 1, variant 2 and variant 3 and encode two kinds of proteins: one is ADD3-Ia encoded by variant 1 that expresses exon14 (ADD3(+14)); the other is ADD3-Ib encoded by variant 2 and variant 3 that lose exon14 (ADD3(-14)). Dutertrel observed that ADD3 (+14) was overexpressed in murine breast tumor of high degree of metastasis [46]. Similarly, ADD3 (-14) has been found to be down-regulated in non-small cell lung cancer compared with normal lung tissue [47, 48]. Tao’s experiment showed that the expression levels of ADD3 and ADD3-Ib decreased in CRC tissues as compared with the normal mucosa while ADD3-Ia/ADD3-Ib ratio increased in CRC tissue. In contrast, reduced expression of ADD3 and its splicing isoforms have been observed in colorectal polyps, suggesting that changes in ADD3 and its isoforms expression in polyps are the early stage of CRC, leading to the report of the change of ADD3-Ia/ADD3-Ib ratio useless. As polyps developed into CRC, ADD3 (+14) increased to normal level and ADD3-Ia/ADD3-Ib ratio changed from normal level to a higher level, which conformed to the development of CRC. Besides, the expression changes of ADD3 and its splicing isoforms in the CRC may be relevant to its invasion ability [49].
Besides, Rani et al., found a lower level of ADD3 in metastatic glioblastoma cells. Their study demonstrated that miR-145 acted as a tumor-suppressor in glioblastoma since it apparently reduced proliferation, adhesion and invasion of glioblastoma cells, apparently by suppressing the activity of ADD3 and oncogenic protein Sox9 [50, 51]. In SCLC, ADD3 was increased due to overexpression of HGF and c-MET [31]. Recently, ADD3 gene has been reported for the first time as a translocation partner of the nucleoprotein 98 gene (NUP98) in acute T-lymphoblastic leukemia [52]. However, none of those studies has mentioned how ADD3 exerts its function.
Taken together, we conclude that adducin has the following main functions relevant to tumor. Firstly, adducin promotes the ability of tumor cellular proliferation, migration and invasion. Secondly, adducin may function as a tumor suppressor in some tumors, such as in ovarian epithelial tumor and glioblastoma. Besides, adducin has different splicing isoforms that are differentially expressed in different stages of tumor progression. It is proved that differences exist in the phosphorylation sites of adducin in different tumors. For instance, phospho-adducin at Ser660 is at a high level in renal tumor. However, previous studies haven’t revealed the mechanism of adducin in tumorigenesis. Considering the mechanism of adducin activation and its biological effects, we suggest that imbalance of phospho-adducin and dephospho-adducin, and overexpression or down-regulation of adducin lead to instability of membrane skeleton, abnormal formation of chromosomes, loss of cell-cell adhesion or reinforced adhesion and unusual membrane ruffling, which ultimately result in abnormalities in differentiation, proliferation, migration and invasion. However, detailed mechanism requires further studies.
Prospects
Tumor is caused by abnormal cell proliferation from mutual interaction between heredity and environment. It is able to invade para-tissues and migrate to tissues far away from primary lesion through direct dissemination, lymph node metastasis, blood metastasis and implantation metastasis. However, the mechanism of tumorigenesis and metastasis in most tumors remains unknown. So far, adducin has not been reported to be modified by other ways except phosphorylation. Whether adducin has other modifications like ubiquitination and methylation remains unclear. It also interests us how other modifications may affect the phosphorylation of adducin and act on tumorigenesis. The splicing isoforms of adducin might be used as a biological marker for prophylaxis of tumor and a therapeutic target. Considering diverse phosphorylation sites of adducin in different tumors, whether there exists a phosphorylation site that expresses similarly in different tumors so it can be a targeted point to hinder the development of tumors. Additionally, given that adducin is not only involved in formation of the membrane cytoskeleton and cell motility and can also function as a tumor suppressor, the ability of adducin to control malignant biological behaviors worth studying. Nevertheless, all these hypotheses need further studies.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Gardner. K, Bennett V. A new erythrocyte membrane-associated protein with calmodulin binding activity. Identification and purification. J Biol Chem.1986; 261:1339–48.
2. Joshi R, Gilligan DM, Otto E, McLaughlin T, Bennett V. Primary structure and domain organization of human alpha and beta adducin. J Cell Biol.1991; 115:665–75.
3. Dong L, Chapline C, Mousseau B, Fowler L, Ramsay K, Stevens JL, Jaken S. 35H, a sequence isolated as a protein kinase C binding protein, is a novel member of the adducin family. J Biol Chem. 1995; 270:25534–40.
4. Lin B, Nasir J, McDonald H, Graham R, Rommens JM, Goldberg YP, Hayden MR. Genomic organization of the human alpha-adducin gene and its alternately spliced isoforms. Genomics. 1995; 25:93–9.
5. Gilligan DM, Lieman J, Bennett V. Assignment of the human beta-adducin gene (ADD2) to 2p13–p14 by in situ hybridization. Genomics. 1995; 28:610–2.
6. Citterio L, Azzani T, Duga S, Bianchi G. Genomic organization of the human gamma adducin gene. Biochem Biophys Res Commun. 1999; 266:110–4.
7. Joshi R, Bennett V. Mapping the domain structure of human erythrocyte adducin. J Biol Chem. 1990; 265:13130-6.
8. Hughes CA, Bennett V. Adducin: a physical model with implication for function in assembly of spectrin actin complexes. J Biol Chem. 1995; 270:18990–6.
9. Matsuoka Y, Li X, Bennett V. Adducin: structure, function and regulation. Cell Mol Life Sci. 2000; 57:884-95.
10. Matsuoka Y, Hughes CA, Bennett V. Adducin regulation: definition of the calmodulin-binding domain and sites of phosphorylation by protein kinases A and C. J Biol Chem. 1996; 271:25157-66.
11. Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsuura Y, Kaibuchi K. Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol. 1999; 145:347-61.
12. Mische SM, Mooseker MS, Morrow JS. Erythrocyte adducin: a calmodulin-regulated actin-bundling protein that stimulates spectrin-actin binding. J Cell Biol. 1987; 105:2837–45.
13. Kuhlman PA, Hughes CA, Bennett V, Fowler VM. A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J Biol Chem. 1996; 271:7986–91.
14. Li X, Bennett V. Identification of the spectrin subunit and domains required for formation of spectrin/ adducin/actin complexes. J Biol Chem. 1996; 271:15695-702.
15. Li X, Matsuoka Y, Bennett V. Adducin preferentially recruits spectrin to the fast growing ends of actin filaments in a complex requiring the MARCKS-related domain and a newly defined oligomerization domain. J Biol Chem.1998; 273:19329–38.
16. Kuhlman PA, Fowler VM. Purification and characterization of an alpha 1 beta 2 isoform of CapZ from human erythrocytes: cytosolic location and inability to bind to Mg2+ ghosts suggest that erythrocyte actin filaments are capped by adducin. Biochemistry. 1997; 36:13461–72.
17. Chang SH, Low PS. Regulation of the glycophorin C-protein 4.1 membrane-to-skeleton bridge and evaluation of its contribution to erythrocyte membrane stability. J Biol Chem. 2001; 276:22223-30.
18. Stefanovic M, Markham NO, Parry EM, Garrett-Beal LJ, Cline AP, Gallagher PG, Low PS, Bodine DM. An 11-amino acid β-hairpin loop in the cytoplasmic domain of band 3 is responsible for ankyrin binding in mouse erythrocytes. Proc Natl Acad Sci. 2007; 104:13972-7.
19. Aong WA, Franco T, Chu H, Weis TL, Devlin EE, Bodine DM, An X, Mohandas N, Low PS. Adducin forms a bridge between the erythrocyte membrane and its cytoskeleton and regulates membrane cohesion. Blood. 2009; 114:1904-12.
20. Warnock DG. Sticky business: cytoskeleton and Na + transport. J Clin Invest. 1996; 97:2691.
21. Ferrandi M, Salardi S, Tripodi G, Barassi P, Rivera R, Manunta P, Goldshleger R, Ferrari P, Bianchi G, Karlish SJ. Evidence for an interaction between adducin and Na +-K +-ATPase: relation to genetic hypertension. Am J Physiol. 1999; 277:H1338-49.
22. Chan PC, Hsu RY, Liu CW, Lai CC, Chen HC. Adducin-1 is essential for mitotic spindle assembly through its interaction with myosin-X. J Cell Biol. 2014; 204:19-28.
23. Woolner S, O’Brien LL, Wiese C, Bement WM. Myosin-10 and actin filaments are essential for mitotic spindle function. J Cell Biol. 2008; 182:77–88.
24. Muro AF, Marro ML, Gajović S, Porro F, Luzzatto L, Baralle FE. Mild spherocytic hereditary elliptocytosis and altered levels of α- and γ-adducins in β-adducin-deficient mice. Blood. 2000; 95:3978-85.
25. Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Mohandas N, Peters LL. Targeted disruption of the beta-adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci. 1999; 96:10717–22.
26. Gilligan DM, Sarid R, Weese J. Adducin in platelets: activation-induced phosphorylation by PKC and proteolysis by calpain. Blood. 2002; 99:2418–26.
27. Barkalow KL, Italiano JE Jr, Chou DE, Matsuoka Y, Bennett V, Hartwig JH. Alpha-adducin dissouciates from F-actin and spectrin during platelet activation. J Cell Biol. 2003; 161:557-70.
28. Chen CL, Hsieh YT, Chen HC. Phosphorylation of adducin by protein kinase C delta promotes cell motility. J Cell Sci. 2007; 120:1157–67.
29. Kaiser HW, O'Keefe E, Bennett V. Adducin: Ca++-dependent association with sites of cell-cell contact. J Cell Biol. 1989; 109:557–69.
30. Naydenove NG, Ivanov AI. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol Biol Cell. 2010; 21:3506-17.
31. Ma PC, Tretiakova MS, Nallasura V, Jagadeeswaran R, Husain AN, Salgia R. Downstream signaling and specific inhibition of c-MET/HGF pathway in small cell lung cancer: implications for tumor invasion. Br J Cancer. 2007; 97:368-77.
32. Matsuoka Y, Li X, Bennett V. Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J Cell Biol. 1998; 142:485–97.
33. Govekar RB, Zingde SM. Protein kinase C isoforms in human erythrocytes. Ann Hematol. 2001; 80:531–4.
34. George A, Pushkaran S, Li L, An X, Zheng Y, Mohandas N, Joiner CH, Kalfa TA. Altered phosphorylation of cytoskeleton proteins in sickle red blood cells:the role of protein kinase C, racGTPases, and reactive oxygen species. Blood Cells Mol Dis. 2010; 45:41-5.
35. Duan WG, Yuan ST, Liao H, Yan M, Zhang LY. [Advances in the study of Rho kinase and its inhibitors]. [Article in Chinese]. Yao Xue Xue Bao. 2007; 42:1013-22.
36. Kimura K, Fukata Y, Matsuoka Y, Bennett V, Matsuura Y, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of the association of adducin with actin filaments by rho-associated kinase (Rho-kinase) and myosin phosphatase. J Biol Chem. 1998; 273:5542–8.
37. Chen CL, Lin YP, Lai YC, Chen HC. α-Adducin translocates to the nucleus upon loss of cell-cell adhesions. Traffic. 2011; 12:1327-40.
38. Fowler L, Everitt J, Stevens JL, Jaken S. Redistribution and enhanced protein kinase C-mediated phosphorylation of alpha-and gamma-adducin during renal tumor progression. Cell Growth Differ. 1998; 9:405–13.
39. Kiley SC, Clark KJ, Duddy SK, Welch DR, Jaken S. Increased protein kinase C delta in mammary tumor cells: relationship to transformation and metastatic progression. Oncogene. 1999; 18:6748-57.
40. Kiley SC, Clark KJ, Goodnough M, Welch DR, Jaken S. Protein kinase C delta involvement in mammary tumor cell metastasis. Cancer Res. 1999; 59:3230-8.
41. Bowen SL, Bloor BK, Leigh IM, Waseem A. Adducin expression in cutaneous and oral lesions: alpha- and beta-adducin transcripts down-regulate with keratinocyte differentiation in stratified epithelia. J Pathol. 2003; 201:119-26.
42. Syed V, Zhang X, Lau KM, Cheng R, Mukherjee K, Ho SM. Profiling estrogen-regulated gene expression changes in normal and malignant human ovarian surface epithelial cells. Oncogene. 2005; 24:8128-43.
43. Wang MH, Chang J, Yu KD, Tan W, Lin DX. [A missense SNP in the codon of ADD1 phosphorylation site associated with non-cardia gastric cancer susceptibility in a Chinese population]. [Article in Chinese]. Zhonghua zhong liu za zhi. 2013; 35:311-4. doi: 10.3760/cma.j.issn.0253-3766.2013.04.016.
44. Shen N, Liu C, Li J, Chen X, Yang Y, Zhu Y, Gong Y, Gong J, Zhong R, Cheng L, Miao X. A phosphorylation-related variant ADD1-rs4963 modifies the risk of colorectal cancer. PLoS ONE. 2015; 10:e0121485.
45. Jen J, Lin LL, Chen HT, Liao SY, Tang YA, Su WC, Salgia R, Huang HC, Juan HF, Wang YC. Oncoprotein ZNF322A transcriptionally deregulates alpha-adducin, cyclin D1 and p53 to promote tumor growth and metastasis in lung cancer. Oncogene. 2016; 35:2357-69.
46. Dutertre M, Lacroix-Triki M, Driouch K, de la Grange P, Gratadou L, Beck S, Millevol S, Tazi J, Lidereau R, Vagner S, Auboeuf D. Exon-based clustering of murine breast tumor transcriptomes reveals alternative exons whose expression is associated with metastasis. Cancer Res. 2010; 70:896-905.
47. Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009; 656:85-106.
48. Langer W, Sohler F, Leder G, Beckmann G, Seidel H, Grone J, Hummel M, Sommer A. Exon array analysis using re-defined probe sets results in reliable identification of alternatively spliced genes in non-small cell lung cancer. BMC Genomic. 2010; 11:676.
49. Tao M, Huang LX, Cai PW, Jin L, Wu WB, Zeng CQ, Huang Y, Wu YA. [Differential expression of ADD3 splicing isoforms between colorectal cancer and normal mucosa tissues]. [Article in Chinese]. Chin J Pathophysiol. 2016; 32:451-7. doi: 10.3969/j.issn.1000-4718.2016.03.011.
50. Mariani L, Beaudry C, McDonough WS, Hoelzinger DB, Kaczmarek E, Ponce F, Coons SW, Giese A, Seiler RW, Berens ME. Death-associated protein 3 (Dap3) is overexpressed in invasive glioblastoma cells in vivo and in glioma cell lines with induced motility phenotype in vivo. Clin Cancer Res. 2001; 7:2480-9.
51. Rani SB, Rathod SS, Karthik S, Kaur N, Muzumdar D, Shiras AS. MiR-145 functions as a tumor-suppressive RNA by targeting Sox9 and adducin 3 in human glioma cells. Neuro Oncol. 2013; 15:1302-16.
52. Lahortiga I, Vizmanos JL, Agirre X, Vázquez I, Cigudosa JC, Larrayoz MJ, Sala F, Gorosquieta A, Perez-Equiza K, Calasanz MJ, Odero MD. NUP98 is fused to adducin 3 in a patient with T-cell acute lymphoblastic leukemia and myeloid markers, with a new translocation t(10;11)(q25;p15). Cancer Res. 2003; 63:3079-83.