
INTRODUCTION
Dyslipidemia, including hypertriglyceridemia, hypo–high density lipoprotein (HDL)–cholesterolemia, and hyper–low density lipoprotein (LDL)–cholesterolemia, is a multifactorial disorder that results from an interaction between genetic background and environmental factors [1, 2]. Given that dyslipidemia is an important risk factor for coronary artery disease, ischemic stroke [3], and colorectal cancer [4], its personalized prevention is an important public health goal.
Genome-wide association studies (GWASs) [5–8] and gene-centric meta-analysis [9] have implicated various genes and loci as determinants of blood lipid levels or in predisposition to dyslipidemia in European-ancestry populations. Genetic variants associated with lipid profiles have been extensively investigated, with one recent study identifying 157 such loci, including 62 variants not previously reported [10]. Recent GWASs [11, 12] or studies based on exome [13] or whole-genome [14] sequencing in European-ancestry populations also identified low-frequency or rare variants related to circulating lipid levels. Although several polymorphisms have been shown to be related to blood lipid profiles in the Japanese population [15], genetic variants—including low-frequency or rare variants—that influence circulating lipid levels or contribute to genetic susceptibility to dyslipidemia in Japanese remain to be identified definitively.
We have now performed exome-wide association studies (EWASs) to identify single nucleotide polymorphisms (SNPs)—in particular, low-frequency or rare coding variants with moderate to large effect sizes—that influence the serum concentrations of triglycerides, HDL-cholesterol, or LDL-cholesterol or confer susceptibility to dyslipidemia in Japanese. We used Illumina arrays that provide coverage of functional SNPs in entire exons including such variants.
RESULTS
EWASs for serum concentrations of triglycerides, HDL-cholesterol, or LDL-cholesterol
We applied linear regression analysis to examine the relation of genotypes for 41,371 SNPs to the serum triglyceride concentration in 13,414 subjects, for 41,225 SNPs to the serum HDL-cholesterol concentration in 14,119 subjects, or for 41,347 SNPs to the serum LDL-cholesterol concentration in 13,577 subjects. Manhattan plots for the EWASs are shown in Supplementary Figure 1. After Bonferroni’s correction, 46, 104, or 40 SNPs were significantly (P < 1.21 × 10–6) associated with the serum concentrations of triglycerides (Table 1), HDL-cholesterol (Supplementary Table 1), or LDL-cholesterol (Table 2), respectively.
Table 1: The 46 SNPs significantly (P < 1.21 × 10–6) associated with serum triglyceride concentration in the EWAS
Gene |
dbSNP |
Nucleotide |
Chromosome: position |
MAF (%) |
P (genotype) |
|---|---|---|---|---|---|
APOA5 |
rs2075291 |
C/A (G185C) |
11: 116790676 |
7.3 |
2.98 × 10–65 |
rs7350481 |
C/T |
11: 116715567 |
27.7 |
3.10 × 10–38 |
|
APOA5 |
rs2266788 |
T/C |
11: 116789970 |
26.2 |
1.03 × 10–35 |
BUD13 |
rs10790162 |
G/A |
11: 116768388 |
26.3 |
1.11 × 10–35 |
ZPR1 |
rs964184 |
C/G |
11: 116778201 |
26.3 |
2.17 × 10–35 |
rs9326246 |
G/C |
11: 116741017 |
26.5 |
6.24 × 10–34 |
|
ZPR1 |
rs2075290 |
T/C |
11: 116782580 |
26.7 |
4.87 × 10–33 |
MTFR2 |
rs143974258 |
G/A (R360*) |
6: 136231355 |
3.3 |
2.10 × 10–19 |
APOA4 |
rs5104 |
T/C (N147S) |
11: 116821618 |
35.7 |
3.57 × 10–14 |
C21orf59 |
rs76974938 |
C/T (D67N) |
21: 32609946 |
2.4 |
4.74 × 10–14 |
LPL |
rs328 |
C/G (S474*) |
8: 19962213 |
12.9 |
8.93 × 10–13 |
rs10096633 |
C/T |
8: 19973410 |
12.7 |
1.25 × 10–12 |
|
rs17482753 |
G/T |
8: 19975135 |
12.6 |
1.25 × 10–12 |
|
rs12678919 |
A/G |
8: 19986711 |
12.6 |
1.36 × 10–12 |
|
rs10503669 |
C/A |
8: 19990179 |
12.6 |
1.40 × 10–12 |
|
SIK3 |
rs2075292 |
G/T |
11: 116861796 |
42.7 |
1.58 × 10–12 |
GCKR |
rs1260326 |
T/C (L446P) |
2: 27508073 |
43.6 |
1.62 × 10–12 |
SIK3 |
rs10047462 |
G/T |
11: 116851325 |
47.6 |
1.78 × 10–12 |
GCKR |
rs780093 |
A/G |
2: 27519736 |
43.0 |
8.98 × 10–12 |
rs1260333 |
T/C |
2: 27525757 |
42.9 |
1.11 × 10–11 |
|
rs7016880 |
G/C |
8: 20019235 |
12.0 |
2.64 × 10–11 |
|
TNC |
rs138406927 |
C/T (A1096T) |
9: 115064848 |
2.1 |
2.66 × 10–10 |
LAIR2 |
rs34429135 |
T/A (F115Y) |
19: 54508164 |
2.5 |
1.24 × 10–9 |
PAFAH1B2 |
rs7112513 |
G/A |
11: 117166645 |
45.3 |
2.21 × 10–9 |
PAFAH1B2 |
rs4936367 |
A/G (V151M) |
11: 117166645 |
45.3 |
2.24 × 10–9 |
rs12269901 |
G/C |
11: 117103213 |
36.6 |
2.33 × 10–9 |
|
C2orf16 |
rs1919128 |
G/A (V774I) |
2: 27578892 |
42.2 |
2.45 × 10–9 |
OR4F6 |
rs141569282 |
G/A (A117T) |
15: 101806068 |
1.7 |
8.08 × 10–9 |
rs2954038 |
A/C |
8: 125495147 |
30.3 |
1.20 × 10–8 |
|
rs2197089 |
C/T |
8: 19968862 |
27.6 |
3.31 × 10–8 |
|
C2orf16 |
rs1919127 |
C/T (A685V) |
2: 27578626 |
42.2 |
4.30 × 10–8 |
LOC101929011 |
rs1240773 |
G/T |
11: 116649810 |
29.0 |
5.77 × 10–8 |
LPL |
rs15285 |
G/A |
8: 19967156 |
19.2 |
6.16× 10–8 |
LPL |
rs13702 |
A/G |
8: 19966981 |
19.2 |
6.17 × 10–8 |
rs2954026 |
G/T |
8: 125472284 |
33.1 |
8.37 × 10–8 |
|
LPL |
rs326 |
A/G |
8: 19961928 |
19.4 |
8.74 × 10–8 |
COL6A5 |
rs200982668 |
G/A (E2501K) |
3: 130470894 |
1.3 |
8.94 × 10–8 |
rs1264429 |
A/G |
6: 30597324 |
13.4 |
9.04 × 10–8 |
|
rs1441756 |
T/G |
8: 20010875 |
19.0 |
1.29 × 10–7 |
|
rs2083637 |
T/C |
8: 20007664 |
19.0 |
1.53 × 10–7 |
|
rs2954033 |
G/A |
8: 125481504 |
33.3 |
1.55 × 10–7 |
|
LPL |
rs301 |
T/C |
8: 19959423 |
19.3 |
1.62× 10–7 |
MUC17 |
rs78010183 |
A/T (T1305S) |
7: 101035329 |
1.8 |
5.96 × 10–7 |
MARCH1 |
rs61734696 |
G/T (Q137K) |
4: 164197303 |
1.2 |
7.44 × 10–7 |
rs4938303 |
T/C |
11: 116714271 |
43.0 |
8.13 × 10–7 |
|
MRVI1 |
rs4909945 |
C/T (V11I) |
11: 10652192 |
0.3 |
1.03× 10–6 |
The relation of genotypes of SNPs to the serum concentration of triglycerides was examined by linear regression analysis. aMajor allele/minor allele.
Table 2: The 40 SNPs significantly (P < 1.21 × 10–6) associated with serum LDL-cholesterol concentration in the EWAS
Gene |
dbSNP |
Nucleotide (amino acid) |
Chromosome: position |
MAF (%) |
P (genotype) |
|---|---|---|---|---|---|
APOE |
rs7412 |
C/T (R176C) |
19: 44908822 |
4.3 |
6.42 × 10–51 |
APOC1 |
rs445925 |
C/T |
19: 44912383 |
6.6 |
4.19 × 10–18 |
APOB |
rs13306206 |
G/A (P955S) |
2: 21019859 |
3.2 |
3.34 × 10–16 |
PCSK9 |
rs151193009 |
C/T (R93C) |
1: 55043912 |
1.1 |
3.35 × 10–14 |
APOE |
rs769449 |
G/A |
19: 44906745 |
7.7 |
3.58 × 10–11 |
PSRC1 |
rs599839 |
A/G |
1: 109279544 |
7.9 |
1.51 × 10–10 |
CELSR2 |
rs629301 |
A/C |
1: 109275684 |
7.8 |
1.96 × 10–10 |
APOB |
rs13306194 |
G/A (R532W) |
2: 21029662 |
12.1 |
3.05 × 10–10 |
CELSR2 |
rs12740374 |
G/T |
1: 109274968 |
7.7 |
4.45 × 10–10 |
rs602633 |
C/A |
1: 109278889 |
7.6 |
4.80 × 10–10 |
|
CELSR2 |
rs646776 |
A/G |
1: 109275908 |
7.7 |
4.98× 10–10 |
ABO |
rs1053878 |
G/A (P156L) |
9: 133256264 |
22.8 |
1.63 × 10–9 |
rs651007 |
G/A |
9: 133278431 |
27.9 |
1.12 × 10–8 |
|
rs579459 |
T/C |
9: 133278724 |
27.9 |
1.18 × 10–8 |
|
rs635634 |
G/A |
9: 133279427 |
27.8 |
1.28 × 10–8 |
|
rs507666 |
G/A |
9: 136149399 |
27.8 |
1.36 × 10–8 |
|
MUC22 |
rs117024916 |
A/G (T71A) |
6: 31025642 |
9.9 |
7.55 × 10–8 |
VARS |
rs11751198 |
G/A |
6: 31785749 |
9.5 |
1.67 × 10–7 |
CCHCR1 |
rs147733073 |
C/G (H486Q) |
6: 31145462 |
10.2 |
1.94 × 10–7 |
rs2853969 |
C/T |
6: 31388797 |
9.7 |
2.02 × 10–7 |
|
MSH5 |
rs11754464 |
C/T |
6: 31755958 |
9.5 |
2.88 × 10–7 |
VARS |
rs5030798 |
C/T (V1055I) |
6: 31779733 |
9.5 |
3.60 × 10–7 |
PRRC2A |
rs11538264 |
G/A (V1774M) |
6: 31635412 |
9.5 |
3.71 × 10–7 |
FAM65B |
rs150142878 |
C/T (R371Q) |
6: 24847657 |
5.6 |
3.72 × 10–7 |
HSPA1B |
rs6457452 |
C/T |
6: 31827773 |
9.7 |
3.85 × 10–7 |
LY6G6C |
rs117894946 |
G/C (G75A) |
6: 31719250 |
9.5 |
4.04 × 10–7 |
C6orf48 |
rs11968400 |
C/T |
6: 31836952 |
9.7 |
4.13× 10–7 |
rs12210887 |
G/T |
6: 31847946 |
9.7 |
4.61 × 10–7 |
|
KIAA0319 |
rs4576240 |
G/T (P142T) |
6: 24596250 |
5.5 |
4.85 × 10–7 |
rs2596574 |
G/A |
6: 31366397 |
9.7 |
5.67 × 10–7 |
|
ZSCAN31 |
rs6922302 |
C/G (P128A) |
6: 28327533 |
9.6 |
6.40 × 10–7 |
NEU1 |
rs13118 |
T/A |
6: 31859509 |
9.7 |
6.46 × 10–7 |
ZSCAN26 |
rs76463649 |
A/G (N15S) |
6: 28271963 |
9.6 |
6.71 × 10–7 |
LY6G6F |
rs17200983 |
C/A (P34Q) |
6: 31707506 |
9.5 |
6.89 × 10–7 |
rs3129029 |
A/C |
6: 29694666 |
23.0 |
7.50 × 10–7 |
|
LY6G6F |
rs9267546 |
G/A |
6: 31705659 |
9.8 |
9.05 × 10–7 |
LY6G6F |
rs9267547 |
G/A (A107T) |
6: 31707724 |
10.0 |
9.88 × 10–7 |
TNXB |
rs140770834 |
C/G (L2271V) |
6: 32064851 |
8.8 |
1.14 × 10–6 |
TNXB |
rs11751545 |
A/C |
6: 32073266 |
8.8 |
1.14 × 10–6 |
ABCF1 |
rs4148249 |
C/A |
6: 30590413 |
10.1 |
1.17 × 10–6 |
The relation of genotypes of SNPs to the serum concentration of LDL-cholesterol was examined by linear regression analysis. aMajor allele/minor allele.
EWASs for hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia
We performed the EWAS for hypertriglyceridemia with 13,414 subjects (4742 cases, 8672 controls), that for hypo–HDL-cholesterolemia with 14,119 subjects (2646 cases, 11,473 controls), and that for hyper–LDL-cholesterolemia with 13,577 subjects (4489 cases, 9088 controls). Characteristics of the subjects are shown in Table 3. In the study of hypertriglyceridemia, age, the frequency of men, body mass index (BMI), and the prevalence of smoking, hypertension, diabetes mellitus, chronic kidney disease, and hyperuricemia as well as the serum concentrations of triglycerides and LDL-cholesterol and the ratio of LDL-cholesterol to HDL-cholesterol were greater, whereas the serum concentration of HDL-cholesterol was lower, in subjects with hypertriglyceridemia than in controls. In the study of hypo–HDL-cholesterolemia, age, the frequency of men, BMI, and the prevalence of smoking, hypertension, diabetes mellitus, chronic kidney disease, and hyperuricemia as well as the serum concentration of triglycerides and the ratio of LDL-cholesterol to HDL-cholesterol were greater, whereas the serum concentration of HDL-cholesterol was lower, in subjects with hypo–HDL-cholesterolemia than in controls. In the study of hyper–LDL-cholesterolemia, BMI, the prevalence of smoking, the serum concentrations of triglycerides and LDL-cholesterol, and the ratio of LDL-cholesterol to HDL-cholesterol were greater, whereas age, the prevalence of chronic kidney disease, and the serum concentration of HDL-cholesterol were lower, in subjects with hyper–LDL-cholesterolemia than in controls.
Table 3: Characteristics of the 14,337 study subjects
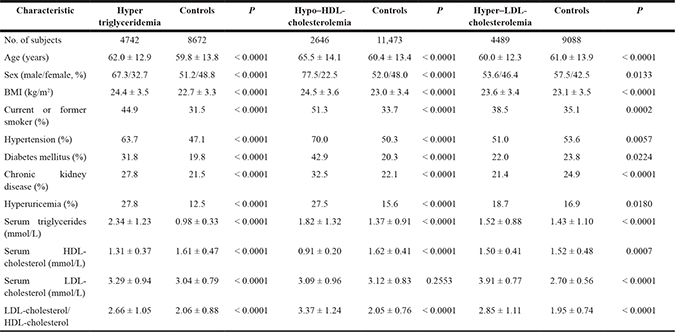
Quantitative data are means ± SD and were compared between subjects with hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia and corresponding controls with the unpaired Student’s t test. Categorical data were compared between two groups with Fisher’s exact test. Based on Bonferroni’s correction, a P value of < 0.0014 (0.05/36) was considered statistically significant.
We applied Fisher’s exact test to examine the relation of allele frequencies of 41,371 SNPs to hypertriglyceridemia, of 41,225 SNPs to hypo–HDL-cholesterolemia, or of 41,347 SNPs to hyper–LDL-cholesterolemia. Manhattan plots for the EWASs are shown in Supplementary Figure 2. After Bonferroni’s correction, 73, 87, or 114 SNPs were significantly (P < 1.21 × 10–6) associated with hypertriglyceridemia (Supplementary Table 2), hypo–HDL-cholesterolemia (Supplementary Table 3), or hyper–LDL-cholesterolemia (Supplementary Table 4), respectively. The genotype distributions of these SNPs were in Hardy-Weinberg equilibrium (P > 0.001) among the corresponding controls for hypertriglyceridemia (Supplementary Table 5), hypo–HDL-cholesterolemia (Supplementary Table 6), or hyper–LDL-cholesterolemia (Supplementary Table 7).
Multivariable logistic regression analysis of hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia
The relation of the 73, 87, or 114 SNPs identified by the EWASs for dyslipidemia to hypertriglyceridemia, hypo–HDL-cholesterolemia, and hyper–LDL-cholesterolemia, respectively, was examined further by multivariable logistic regression analysis with adjustment for age and sex. Two SNPs—rs10790162 (G/A) of BUD13 and rs7350481 (C/T) at 11q23.3—were significantly [P < 1.71 × 10–4 (0.05/292)] associated with hypertriglyceridemia (Table 4, Supplementary Table 8); three SNPs—rs146515657 [T/C (N650S)] of USP4, rs147317864 [C/T (A262T)] of TRABD2B, and rs12229654 (T/G) at 12q24.1—were significantly [P < 1.44 × 10–4 (0.05/348)] associated with hypo–HDL-cholesterolemia (Table 4, Supplementary Table 9); and nine SNPs—rs7771335 (A/G) at 6p22.1, rs76974938 [C/T (D67N)] of C21orf59, rs2071653 (C/T) of MOG, rs2853969 (C/T) at 6p21.3, rs2269704 (C/T) of PPP1R18, rs2269703 (G/A) of NRM, rs495089 (T/C) at 6p21.3, rs2269702 (A/G) of MDC1, and rs1233399 (C/T) at 6p22.1—were significantly [P < 1.10 × 10–4 (0.05/456)] associated with hyper–LDL-cholesterolemia (Table 4, Supplementary Table 10).
Table 4: Relation of SNPs to hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia as determined by multivariable logistic regression analysis
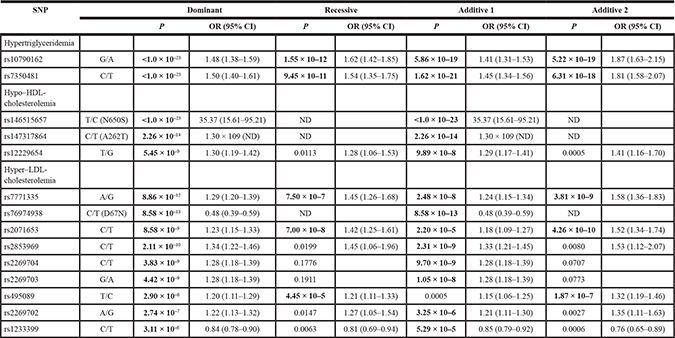
Multivariable logistic regression analysis was performed with adjustment for age and sex. Based on Bonferroni’s correction, P values of < 1.71 × 10–4 (0.05/292), of < 1.44 × 10–4 (0.05/348), or of < 1.10 × 10–4 (0.05/456) were considered statistically significant in the analysis of hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia, respectively, and are shown in bold. OR, odds ratio; CI, confidence interval; ND, not determined.
Linkage disequilibrium and haplotype analysis
Given that eight SNPs (rs7771335, rs2071653, rs2853969, rs2269704, rs2269703, rs495089, rs2269702, rs1233399) associated with hyper–LDL-cholesterolemia were all located at chromosome 6p22.1-p21.3, we examined linkage disequilibrium among these SNPs as well as the relation of their haplotypes to this condition. The eight SNPs were all in strong linkage disequilibrium (Supplementary Table 11). Haplotype analysis revealed that the haplotypes A (rs7771335)–C (rs2071653)–C (rs2853969)–C (rs2269704)–G (rs2269703)–T (rs495089)–A (rs2269702)–T (rs1233399) and G (rs7771335)–T (rs2071653)–T (rs2853969)–T (rs2269704)–A (rs2269703)–C (rs495089)–G (rs2269702)–C (rs1233399) were significantly (P < 4.31 × 10–4) associated with hyper–LDL-cholesterolemia, with the former haplotype being protective against and the latter representing a risk factor for this condition (Supplementary Table 12).
Relation of identified SNPs to serum triglyceride, HDL-cholesterol, or LDL-cholesterol levels
We examined the relation of genotypes of identified SNPs to serum triglyceride, HDL-cholesterol, or LDL-cholesterol levels by one-way analysis of variance. The 46 SNPs identified in the EWAS for serum triglyceride concentration, including the two SNPs also found to be associated with hypertriglyceridemia (rs10790162 of BUD13, rs7350481 at 11q23.3), were all significantly [P < 0.0011 (0.05/46)] associated with serum triglyceride level (Supplementary Table 13). The 104 SNPs identified in the EWAS for serum HDL-cholesterol concentration, including two of the three SNPs found to be associated with hypo–HDL-cholesterolemia (rs146515657 of USP4, rs12229654 at 12q24.1), were all significantly [P < 4.76 × 10–4 (0.05/105)] associated with the serum HDL-cholesterol level, as was the SNP associated with hypo–HDL-cholesterolemia alone (rs147317864 of TRABD2B) (Supplementary Table 14). The 40 SNPs identified in the EWAS for serum LDL-cholesterol concentration, including one SNP also found to be associated with hyper–LDL-cholesterolemia (rs2853969 at 6p21.3), were all significantly [P < 0.0010 (0.05/48)] associated with serum LDL-cholesterol level, as were five of the eight SNPs associated with hyper-LDL-cholesterolemia alone (rs7771335 at 6p22.1, rs2071653 of MOG, rs2269704 of PPP1R18, rs2269703 of NRM, rs2269702 of MDC1) (Supplementary Table 15).
Relation of SNPs identified in the present study to dyslipidemia-related phenotypes examined in previous GWASs
We examined the relation of genes, chromosomal loci, and SNPs identified in the present study to dyslipidemia-related phenotypes included in previous GWASs deposited in GWAS Catalog (http://www.ebi.ac.uk/gwas). Among the 24 loci associated with triglyceride-related traits in the present study, 13 loci—BUD13 [15], 11q23.3 [16], APOA5 [15, 16], ZPR1 [16], APOA4 [15, 17], LPL [15, 17], 8p21.3 [15, 18], SIK3 [19, 20], GCKR [16], 2p23 [17, 21], C2orf16 [22], 8q24.1 [23], and LOC101929011 [21]—were previously shown to be related to the circulating triglyceride level or hypertriglyceridemia (Supplementary Table 16). Among the 69 loci associated with HDL-cholesterol–related traits in the present study, 16 loci—12q24.1 [24], 16q13 [15, 16], CETP [15, 16], APOA5 [16], LIPC [16, 17], HECTD4 [24], LILRB2 [8], LPL [15, 16], 8p21.3 [15, 16], LOC101928635 [10, 17], BUD13 [16], ZPR1 [23], ABCA1 [10, 16], 11q23.3 [16], OAS3 [24], and CD36 [25]—were previously shown to be related to the blood HDL-cholesterol level or hypo–HDL-cholesterolemia (Supplementary Table 17). Among the 32 loci associated with LDL-cholesterol–related traits in the present study, nine loci—APOE [26], APOC1 [27], APOB [16, 17], PCSK9 [17], PSRC1 [12, 28], CELSR2 [10, 16], 1p13.3 [16], ABO [17], and 9q34.2 [8, 17]—were previously shown to be related to the circulating LDL-cholesterol concentration or hyper–LDL-cholesterolemia (Supplementary Table 18).
DISCUSSION
We found that two SNPs—rs10790162 of BUD13 and rs7350481 at 11q23.3—were significantly related to both the serum triglyceride concentration and hypertriglyceridemia; three SNPs—rs146515657 of USP4, rs147317864 of TRABD2B, and rs12229654 at 12q24.1—were significantly related to both the serum HDL-cholesterol concentration and hypo–HDL-cholesterolemia; and six SNPs—rs2853969 at 6p21.3, rs7771335 at 6p22.1, rs2071653 of MOG, rs2269704 of PPP1R18, rs2269703 of NRM, and rs2269702 of MDC1—were significantly related to both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia. Among these SNPs, rs146515657 of USP4 and rs147317864 of TRABD2B may be novel determinants of hypo–HDL-cholesterolemia, whereas rs2853969 at 6p21.3, rs7771335 at 6p22.1, rs2071653 of MOG, rs2269704 of PPP1R18, rs2269703 of NRM, and rs2269702 of MDC1 may be new determinants of hyper–LDL-cholesterolemia. We also found that 12, 61, 23, and 3 SNPs may be new determinants of the serum triglyceride, HDL-cholesterol, or LDL-cholesterol concentrations or of hyper–LDL-cholesterolemia, respectively.
SNPs associated with serum HDL-cholesterol or LDL-cholesterol levels
The ubiquitin specific peptidase 4 gene (USP4) is located at chromosome 3p21.31 (NCBI Gene, https://www.ncbi.nlm.nih.gov/gene) and is expressed in various tissues and organs (The Human Protein Atlas, http://www.proteinatlas.org). Ubiquitin-specific proteases promote posttranslational protein modification by reversing protein ubiquitination and thereby activates multiple biological processes including the cell cycle, DNA repair, and intracellular signaling [29]. USP4 accelerates the growth, invasion, and metastasis of colorectal cancer [29, 30]. We have now shown that rs146515657 [T/C (N650S)] of USP4 was significantly associated with both the serum HDL-cholesterol concentration and hypo–HDL-cholesterolemia, with the minor C allele being related to a lower serum level of HDL-cholesterol, although the molecular mechanism underlying this association remains unclear.
The TraB domain containing 2B gene (TRABD2B) is located at chromosome 1p33 (NCBI Gene) and is expressed ubiquitously (The Human Protein Atlas). TRABD2B encodes the metalloprotease TIKI2 that inhibits the Wnt/β-catenin signaling by cleavage of the amino terminus of Wnt protein [31]. The expression of TIKI2 was reduced in osteosarcoma specimens and the increased expression of TIKI2 inhibited the growth of osteosarcoma in vivo, suggesting TIKI2 suppresses the growth of osteosarcoma [32]. We have now shown that rs147317864 [C/T (A262T)] of TRABD2B was significantly associated with both the serum concentration of HDL-cholesterol and hypo–HDL-cholesterolemia, with the minor T allele being related to a reduced level of serum HDL-cholesterol, although the functional relevance underlying this association remains unclear.
The myelin oligodendrocyte glycoprotein gene (MOG) is located at chromosome 6p22.1 (NCBI Gene) and is highly expressed in brain (The Human Protein Atlas). The MOG protein is localized to the outer surface of the myelin sheath of neurons in the central nervous system and is a key antigen for autoimmune responses that result in inflammation and demyelination [33, 34]. Both B cell responses and antibodies to MOG have also been detected in patients with demyelinating diseases such as multiple sclerosis and acute disseminating encephalomyelitis [34]. We have now shown that rs2071653 (C/T) of MOG was significantly associated with both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia, with the minor T allele being related to an increased serum level of LDL-cholesterol, although the underlying molecular mechanism remains unknown.
The protein phosphatase 1 regulatory subunit 18 gene (PPP1R18) is located at chromosome 6p21.33 (NCBI Gene) and is expressed in various tissues (The Human Protein Atlas). Protein phosphatase 1 binds to regulatory subunits that target the enzyme to different intracellular locations to exert its activity toward specific substrates [35]. The PPP1R18 protein is a regulatory subunit that targets protein phosphatase 1 to the F-actin cytoskeleton [36]. We have now shown that rs2269704 (C/T) of PPP1R18 was significantly associated with both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia, with the minor T allele being related to an increased serum LDL-cholesterol level, although the molecular mechanism underpinning this association is unclear.
The nurim gene (NRM) is located at chromosome 6p21.33 (NCBI Gene) and is expressed ubiquitously (The Human Protein Atlas). The NRM protein contains transmembrane domains and resides within the inner nuclear membrane, where it is tightly bound to the nuclear envelope [37]. NRM is expressed in a broad range of cancers, with its expression level being correlated with tumor grade [38]. NRM deficiency was found to alter the shape of the nuclear envelope and to enhance ultraviolet light–induced apoptosis in HeLa cells, implicating NRM in suppression of apoptosis [38]. We have now shown that rs2269703 (G/A) of NRM was significantly associated with both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia, with the minor A allele being related to an increased serum LDL-cholesterol level, although the molecular mechanism is unknown.
The mediator of DNA damage checkpoint 1 gene (MDC1) is located at chromosome 6p21.33 (NCBI Gene) and is expressed in various tissues and organs (The Human Protein Atlas). MDC1 is a nuclear protein required for activation of the intra–S phase and G2-M phase checkpoints of the cell cycle in response to DNA damage [39]. We have now shown that rs2269702 (A/G) of MDC1 was significantly associated with both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia, with the minor G allele being related to an increased serum LDL-cholesterol level, although the molecular mechanism remains unclear.
We also found that rs2853969 (C/T) at 6p21.3 and rs7771335 (A/G) at 6p22.1 were significantly associated with both the serum LDL-cholesterol concentration and hyper–LDL-cholesterolemia, with the minor T and G alleles, respectively, being related to an increased serum level of LDL-cholesterol. Eight SNPs (rs7771335, rs2071653, rs2853969, rs2269704, rs2269703, rs495089, rs2269702, rs1233399) associated with hyper–LDL-cholesterolemia are all located at chromosomal region 6p22.1-p21.3 and were in strong linkage disequilibrium.
General considerations
In previous GWASs of blood lipid traits in East Asian populations [24], a minor allele frequency (MAF) and effect size of identified SNPs were 10% to 33% and –0.088 to –0.050 mg/dL for triglycerides, 12% to 15% and –0.035 to 0.043 mg/dL for HDL-cholesterol, and 26% and 2.203 mg/dL for LDL-cholesterol, respectively. In trans-ancestry GWASs for lipid profiles [10], the MAF and effect size of identified SNPs ranged from 9% to 49% and from –0.033 to 0.037 mg/dL for triglycerides, from 9% to 50% and from –0.051 to 0.034 mg/dL for HDL-cholesterol, and from 4% to 48% and from –0.051 to 0.103 mg/dL for LDL-cholesterol. In more recent GWASs that included low-frequency or rare variants, the MAF and effect size of identified SNPs ranged from 1.76% to 3.25% and from –30% to 21% for triglycerides, from 0.20% to 2.01% and from –3 to 17 mg/dL for HDL-cholesterol, and from 0.05% to 3.43% and from –40 to 71 mg/dL for LDL-cholesterol [11]; as well as from 1% to 47% and from –0.170 to 0.128 mmol/L for triglycerides, from 5% to 20% and from –0.141 to 0.044 mmol/L for HDL-cholesterol, and from 1% to 21.6% and from –0.049 to 0.648 mmol/L for LDL-cholesterol, respectively [12]. Studies based on exome and whole-genome sequencing identified SNPs with a MAF and effect size from 0.06% to 28.2% and from –98.0 to 51.5 mg/dL, respectively, for LDL-cholesterol [13]; as well as those with a MAF of 0.007% to 4.6% and effect size of –65.3 to 12.0% for triglycerides and –0.087 to 0.40 mmol/L for HDL-cholesterol [14].
In our study, among 46 SNPs associated with the serum triglyceride concentration, one SNP was a rare variant (MAF, 0.3%) with a large effect size (difference in serum triglyceride level among genotypes, 28.1%), eight SNPs were low-frequency variants (1.2–3.3%) with a moderate to large effect size (14.6–19.8%), and 37 SNPs were common variants (7.3–47.6%) with a small to large effect size (6.2–45.2%) (Supplementary Table 19). Among 104 SNPs associated with the serum HDL-cholesterol concentration, nine SNPs were rare variants (MAF, 0.2–0.3%) with a large effect size (difference in serum HDL-cholesterol level among genotypes, 17.3–29.3%), 32 SNPs were low-frequency variants (0.5–4.5%) with a moderate to large effect size (5.2–25.8%), and 63 SNPs were common variants (5.0–47.6%) with a small to large effect size (3.3–18.7%) (Supplementary Table 20). Among 40 SNPs associated with the serum LDL-cholesterol concentration, three SNPs were low-frequency variants (MAF, 1.1–4.3%) with a large effect size (difference in serum LDL-cholesterol level among genotypes, 16.8–24.8%) and 37 SNPs were common variants (6.6–27.9%) with a small to moderate effect size (2.8–9.2%) (Supplementary Table 21).
Study limitations
There are several limitations to the present study. (1) Our results were not replicated and will therefore require validation in independent subject panels or in other ethnic groups. (2) Subjects who had treatment for other diseases such as diabetes mellitus were included in the study. It was possible that such treatment affected lipid profiles of the subjects. (3) SNPs identified in our study might be in linkage disequilibrium with other polymorphisms in the nearby genes that are actually responsible for the observed associations. (4) Three SNPs associated with hyper–LDL-cholesterolemia were not significantly related to the serum LDL-cholesterol concentration, which may be attributable to the effects of medical treatment. (5) The functional relevance of the observed associations remains to be determined.
MATERIALS AND METHODS
Study subjects
A total of 14,337 subjects (8354 individuals with dyslipidemia, 5983 controls) was recruited as described previously [40].
Venous blood was collected in the early morning after the subjects had fasted overnight. Blood samples were centrifuged at 1600 × g for 15 min at 4°C, and serum was separated for subsequent analysis. Serum concentrations of triglycerides, HDL-cholesterol, and LDL-cholesterol were measured at the clinical laboratory of each hospital. The 4742 subjects with hypertriglyceridemia and 8672 controls had serum triglyceride concentrations of ≥ 1.69 mmol/L (range, 1.69 to 20.14 mmol/L) and < 1.69 mmol/L (0.14 to 1.68 mmol/L), respectively; the 2646 subjects with hypo–HDL-cholesterolemia and 11,473 controls had serum HDL-cholesterol concentrations of < 1.03 mmol/L (0.26 to 1.01 mmol/L) and ≥1.03 mmol/L (1.03 to 4.73 mmol/L), respectively; and the 4489 subjects with hyper–LDL-cholesterolemia and 9088 controls had serum LDL-cholesterol concentrations of ≥ 3.62 mmol/L (3.62 to 12.31 mmol/L) and < 3.62 mmol/L (0.26 to 3.59 mmol/L), respectively. Individuals with dyslipidemia had at least one of hypertriglyceridemia, hypo–HDL-cholesterolemia, and hyper–LDL-cholesterolemia, or were taking anti-dyslidemic medications. The 1300 subjects with both hypertriglyceridemia and hypo–HDL-cholesterolemia as well as 7844 controls overlapped between the corresponding studies, as did the 2002 subjects with both hypertriglyceridemia and hyper–LDL-cholesterolemia and 6326 controls as well as the 712 subjects with both hypo–HDL-cholesterolemia and hyper–LDL-cholesterolemia and 7776 controls. Individuals with single-gene disorders such as familial hypercholesterolemia or with endocrinologic or metabolic diseases that cause dyslipidemia were excluded from the study. Those taking medications that may cause secondary dyslipidemia were also excluded. Autopsy cases were excluded from controls.
The study protocol complied with the Declaration of Helsinki and was approved by the Committees on the Ethics of Human Research of Mie University Graduate School of Medicine, Tokyo Metropolitan Institute of Gerontology, Hirosaki University Graduate School of Medicine, and participating hospitals. Written informed consent was obtained from each participant or families of the deceased subjects.
EWASs
Methods for collection and extraction of genomic DNA samples were described previously [40]. EWASs for the serum concentrations of triglycerides (13,414 subjects), HDL-cholesterol (14,119 subjects), or LDL-cholesterol (13,577 subjects) or for hypertriglyceridemia (4742 cases, 8672 controls), hypo–HDL-cholesterolemia (2646 cases, 11,473 controls), or hyper-LDL-cholesterolemia (4489 cases, 9088 controls) were performed with HumanExome-12 v1.1 or v1.2 DNA Analysis BeadChip or Infinium Exome-24 v1.0 BeadChip arrays (Illumina, San Diego, CA). Detailed information of the exome arrays and methods of quality control were described previously [40]. Totals of 41,371, 41,225, and 41,347 SNPs passed quality control in the EWASs for hypertriglyceridemia, hypo–HDL-cholesterolemia, and hyper–LDL-cholesterolemia, respectively, and were included in the analysis.
Statistical analysis
The relation of SNP genotypes to the serum concentrations of triglycerides, HDL-cholesterol, or LDL-cholesterol in the EWASs was examined with linear regression analysis. For analysis of characteristics of the study subjects, quantitative and categorical data were compared between cases and controls with the unpaired Student’s t test and Fisher’s exact test, respectively. Allele frequencies were estimated by the gene counting method, and departure from Hardy-Weinberg equilibrium was identified with Fisher’s exact test. The relation of allele frequencies of SNPs to hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia in the EWASs was examined with Fisher’s exact test. To compensate for multiple comparisons of genotypes or allele frequencies with lipid concentrations or dyslipidemia, we applied Bonferroni’s correction for statistical significance of association. Given that 41,225 to 41,371 SNPs were analyzed, a P value of < 1.21 × 10–6 [0.05/(41,225 to 41,371] was considered statistically significant for the EWASs. Quantile-quantile plots for P values of genotypes or allele frequencies in the EWASs are shown in Supplementary Figures 3 and 4, respectively. The inflation factor (λ) was 1.05 for serum triglycerides, 0.97 for serum HDL-cholesterol, 1.06 for serum LDL-cholesterol, 1.20 for hypertriglyceridemia, 1.29 for hypo–HDL-cholesterolemia, and 1.20 for hyper–LDL-cholesterolemia. Multivariable logistic regression analysis was performed with hypertriglyceridemia, hypo–HDL-cholesterolemia, or hyper–LDL-cholesterolemia as a dependent variable and independent variables including age, sex (0, woman; 1, man), and genotype of each SNP. A detailed method of analysis was described previously [40]. Relations of genotypes of identified SNPs to serum concentrations of triglycerides, HDL-cholesterol, or LDL-cholesterol were examined by one-way analysis of variance. Bonferroni’s correction was also applied to other statistical analysis as indicated. Statistical tests were performed with JMP Genomics version 6.0 software (SAS Institute, Cary, NC).
CONCLUSIONS
The SNPs rs146515657 of USP4 and rs147317864 of TRABD2B may be novel determinants of hypo–HDL-cholesterolemia whereas rs2853969 at 6p21.3, rs7771335 at 6p22.1, rs2071653 of MOG, rs2269704 of PPP1R18, rs2269703 of NRM, and rs2269702 of MDC1 may be new determinants of hyper–LDL-cholesterolemia. In addition, 12, 61, 23, or 3 SNPs may be new determinants of the serum triglyceride, HDL-cholesterol, or LDL-cholesterol concentrations or of hyper–LDL-cholesterolemia, respectively. Determination of genotypes for these SNPs may prove informative for assessment of the genetic risk for dyslipidemia in Japanese.
Authors’ contributions
Y. Yamada contributed to conception and design of the study; to acquisition, analysis, and interpretation of the data; and to drafting of the manuscript. J. Sakuma, I. Takeuchi, and Y. Yasukochi contributed to analysis and interpretation of the data as well as to revision of the manuscript. K. Kato, M. Oguri, T. Fujimaki, H. Horibe, M. Muramatsu, M. Sawabe, Y. Fujiwara, Y. Taniguchi, S. Obuchi, H. Kawai, S. Shinkai, S. Mori, and T. Arai contributed to acquisition of the data and to revision of the manuscript. M. Tanaka contributed to acquisition, analysis, and interpretation of the data as well as to revision of the manuscript. All authors approved submission of the final version of the article for publication.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by CREST (grant number, JPMJCR1302), Japan Science and Technology Agency (to Y. Yamada, J. Sakuma, and I. Takeuchi) and by Japan Society for the Promotion of Science KAKENHI grants JP15H04772 (to Y. Yamada), JP25242062 (to M. Tanaka), and JP16H01872 (to M. Tanaka).
REFERENCES
1. Heller DA, DeFaire U, Pedersen N, Dahlén G, McClearn GE. Genetic and environmental influences on serum lipid levels in twins. N Engl J Med. 1993; 328:1150–1156.
2. Khera AV, Rader DJ. Discovery and validation of new molecular targets in treating dyslipidemia: the role of human genetics. Trends Cardiovasc Med. 2009; 19:195–201.
3. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, et al. Heart Disease and Stroke Statistics—2016 Update: A Report from the American Heart Association. Circulation. 2016; 133:e38–e360.
4. Agnoli C, Grioni S, Sieri S, Sacerdote C, Vineis P, Tumino R, Giurdanella MC, Pala V, Mattiello A, Chiodini P, Iacoviello L, De Curtis A, Cattaneo L, et al. Colorectal cancer risk and dyslipidemia: a case-cohort study nested in an Italian multicentre cohort. Cancer Epidemiol. 2014; 38:144–151.
5. Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, Wahlstrand B, Hedner T, Corella D, et al. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008; 40:189–197.
6. Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T, Voight BF, Bonnycastle LL, Jackson AU, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009; 41:56–65.
7. Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T, Martin NG, Pedersen NL, Kyvik KO, et al. Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009; 41:47–55.
8. Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010; 466:707–713.
9. Asselbergs FW, Guo Y, van Iperen EP, Sivapalaratnam S, Tragante V, Lanktree MB, Lange LA, Almoguera B, Appelman YE, Barnard J, Baumert J, Beitelshees AL, Bhangale TR, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet. 2012; 91:823–838.
10. Global Lipids Genetics Consortium, Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013; 45:1274–1283.
11. Peloso GM, Auer PL, Bis JC, Voorman A, Morrison AC, Stitziel NO, Brody JA, Khetarpal SA, Crosby JR, Fornage M, Isaacs A, Jakobsdottir J, Feitosa MF, et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet. 2014; 94:223–232.
12. Surakka I, Horikoshi M, Mägi R, Sarin AP, Mahajan A, Lagou V, Marullo L, Ferreira T, Miraglio B, Timonen S, Kettunen J, Pirinen M, Karjalainen J, et al. The impact of low-frequency and rare variants on lipid levels. Nat Genet. 2015; 47:589–597.
13. Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, Bizon C, Lange EM, Smith JD, Turner EH, Jun G, Kang HM, Peloso G, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014; 94:233–245.
14. Helgadottir A, Gretarsdottir S, Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A, Jonasdottir A, Kristjansson H, Sulem P, Oddsson A, Sveinbjornsson G, Steinthorsdottir V, Rafnar T, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016; 48:634–639.
15. Kurano M, Tsukamoto K, Kamitsuji S, Kamatani N, Hara M, Ishikawa T, Kim BJ, Moon S, Jin Kim Y, Teramoto T. Genome-wide association study of serum lipids confirms previously reported associations as well as new associations of common SNPs within PCSK7 gene with triglyceride. J Hum Genet. 2016; 61:427–433.
16. Below JE, Parra EJ, Gamazon ER, Torres J, Krithika S, Candille S, Lu Y, Manichakul A, Peralta-Romero J, Duan Q, Li Y, Morris AP, Gottesman O, et al. Meta-analysis of lipid-traits in Hispanics identifies novel loci, population-specific effects, and tissue-specific enrichment of eQTLs. Sci Rep. 2016; 6: 19429.
17. Lu X, Huang J, Mo Z, He J, Wang L, Yang X, Tan A, Chen S, Chen J, Gu CC, Chen J, Li Y, Zhao L, et al. Genetic susceptibility to lipid levels and lipid change over time and risk of incident hyperlipidemia in Chinese populations. Circ Cardiovasc Genet. 2016; 9:37–44.
18. Kamatani Y, Matsuda K, Okada Y, Kubo M, Hosono N, Daigo Y, Nakamura Y, Kamatani N. Genome-wide association study of hematological and biochemical traits in a Japanese population. Nat Genet. 2010; 42:210–215.
19. Ko A, Cantor RM, Weissglas-Volkov D, Nikkola E, Reddy PM, Sinsheimer JS, Pasaniuc B, Brown R, Alvarez M, Rodriguez A, Rodriguez-Guillen R, Bautista IC, Arellano-Campos O, et al. Amerindian-specific regions under positive selection harbour new lipid variants in Latinos. Nat Commun. 2014; 5: 3983.
20. Kooner JS, Chambers JC, Aguilar-Salinas CA, Hinds DA, Hyde CL, Warnes GR, Gómez Pérez FJ, Frazer KA, Elliott P, Scott J, Milos PM, Cox DR, Thompson JF. Genome-wide scan identifies variation in MLXIPL associated with plasma triglycerides. Nat Genet. 2008; 40:149–151.
21. Waterworth DM, Ricketts SL, Song K, Chen L, Zhao JH, Ripatti S, Aulchenko YS, Zhang W, Yuan X, Lim N, Luan J, Ashford S, Wheeler E, et al. Genetic variants influencing circulating lipid levels and risk of coronary artery disease. Arterioscler Thromb Vasc Biol. 2010; 30:2264–2276.
22. Weissglas-Volkov D, Aguilar-Salinas CA, Nikkola E, Deere KA, Cruz-Bautista I, Arellano-Campos O, Muñoz-Hernandez LL, Gomez-Munguia L, Ordoñez-Sánchez ML, Reddy PM, Lusis AJ, Matikainen N, Taskinen MR, et al. Genomic study in Mexicans identifies a new locus for triglycerides and refines European lipid loci. J Med Genet. 2013; 50:298–308.
23. Kraja AT, Vaidya D, Pankow JS, Goodarzi MO, Assimes TL, Kullo IJ, Sovio U, Mathias RA, Sun YV, Franceschini N, Absher D, Li G, Zhang Q, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011; 60:1329–1339.
24. Kim YJ, Go MJ, Hu C, Hong CB, Kim YK, Lee JY, Hwang JY, Oh JH, Kim DJ, Kim NH, Kim S, Hong EJ, Kim JH, et al. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat Genet. 2011; 43:990–995.
25. Coram MA, Duan Q, Hoffmann TJ, Thornton T, Knowles JW, Johnson NA, Ochs-Balcom HM, Donlon TA, Martin LW, Eaton CB, Robinson JG, Risch NJ, Zhu X, et al. Genome-wide characterization of shared and distinct genetic components that influence blood lipid levels in ethnically diverse human populations. Am J Hum Genet. 2013; 92:904–916.
26. Rasmussen-Torvik LJ, Pacheco JA, Wilke RA, Thompson WK, Ritchie MD, Kho AN, Muthalagu A, Hayes MG, Armstrong LL, Scheftner DA, Wilkins JT, Zuvich RL, Crosslin D, et al. High density GWAS for LDL cholesterol in African Americans using electronic medical records reveals a strong protective variant in APOE. Clin Transl Sci. 2012; 5:394–399.
27. Postmus I, Trompet S, Deshmukh HA, Barnes MR, Li X, Warren HR, Chasman DI, Zhou K, Arsenault BJ, Donnelly LA, Wiggins KL, Avery CL, Griffin P, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014; 5: 5068.
28. Sandhu MS, Waterworth DM, Debenham SL, Wheeler E, Papadakis K, Zhao JH, Song K, Yuan X, Johnson T, Ashford S, Inouye M, Luben R, Sims M, et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet. 2008; 371:483–491.
29. Fraile JM, Quesada V, RodriguezD, Freije JM, Lopez-Otin C. Deubiquitinases in cancer: new functions and therapeutic options. Oncogene 2012; 31:2373–88.
30. Clerici M, Luna-Vargas MP, Faesen AC, Sixma TK. The DUSP-Ubl domain of USP4 enhances its catalytic efficiency by promoting ubiquitin exchange. Nat Commun. 2014; 5: 5399.
31. Zhang X, MacDonald BT, Gao H, Shamashkin M, Coyle AJ, Martinez RV, He X. Characterization of Tiki, a New Family of Wnt-specific Metalloproteases. J Biol Chem. 2016; 291:2435–2443.
32. Li R, Liu J, Wu H, Liu L, Wang L, Zhang S. TIKI2 suppresses growth of osteosarcoma by targeting Wnt/β-catenin pathway. Mol Cell Biochem. 2014; 392:109–116.
33. Johns TG, Bernard CC. The structure and function of myelin oligodendrocyte glycoprotein. J Neurochem. 1999; 72:1–9.
34. Lee DH, Linker RA. The role of myelin oligodendrocyte glycoprotein in autoimmune demyelination: a target for multiple sclerosis therapy? Expert Opin Ther Targets. 2012; 16:451–462.
35. Aggen JB, Nairn AC, Chamberlin R. Regulation of protein phosphatase-1. Chem Biol. 2000; 7: R13–R23.
36. Kao SC, Chen CY, Wang SL, Yang JJ, Hung WC, Chen YC, Lai NS, Liu HT, Huang HL, Chen HC, Lin TH, Huang HB. Identification of phostensin, a PP1 F-actin cytoskeleton targeting subunit. Biochem Biophys Res Commun. 2007; 356:594–598.
37. Holmer L, Worman HJ. Inner nuclear membrane proteins: functions and targeting. Cell Mol Life Sci. 2001; 58:1741–1747.
38. Chen H, Chen K, Chen J, Cheng H, Zhou R. The integral nuclear membrane protein nurim plays a role in the suppression of apoptosis. Curr Mol Med. 2012; 12:1372–1382.
39. Jungmichel S, Stucki M. MDC1: the art of keeping things in focus. Chromosoma. 2010; 119:337–349.
40. Yamada Y, Sakuma J, Takeuchi I, Yasukochi Y, Kato K, Oguri M, Fujimaki T, Horibe H, Muramatsu M, Sawabe M, Fujiwara Y, Taniguchi Y, Obuchi S, et al. Identification of STXBP2 as a novel susceptibility locus for myocardial infarction in Japanese individuals by an exome-wide association study. Oncotarget. 2017; 8:33527–33535. doi: 10.18632/oncotarget.16536.