
INTRODUCTION
Human ERK8 (alias MAPK15), located at 8q24.3, is the most recently identified member of the ERK family [1]. Over the past decades, despite the fact that in-depth investigations have been done on typical ERKs (ERK1 and 2), the functions of ERK8 remain relatively understudied, in which there are still a lot of unanswered questions for its cellular function. ERK8 resembles ERK1, ERK2, and ERK5 that possesses a Thr-Glu-Tyr motif in the activation loop [1]. ERK8 has been considered as an atypical MAPK partly because it contains the long carboxyl-terminal domain that regulating its activity, cellular localization and function [1–3]. Furthermore, ERK8 is regulated by auto-phosphorylation and no specific upstream activating kinase has been identified so far [1, 4, 5]. Earlier studies from several research groups have shown that ERK8 activity can be stimulated by serum, DNA damage, and human oncogenes such as RET/PTC3 (an active form of the RET proto-oncogene) [1, 6, 7]. Moreover, ERK8 has recently been shown to increase tumorigenesis in human colon cancer cells by activating c-jun and in gastric cancer cells by stabilizing c-Jun [8, 9]. Besides, ERK8 is involved in maintaining genome stability as well as autophagy [10, 11]. Yet, it is still unclear whether ERK8 acts as a proto-oncogene or tumor suppressor. However, it should be noted that protein expressed in one cell type might actually function differently in another, leading to diverse phenotypes. Therefore, no unified functions of ERK8 can be drawn conclusively at present.
Arsenic trioxide (As2O3), a traditional Chinese medicine, inhibits growth and promotes apoptosis in many different cancer cell types. It has been proven especially to be highly effective against hematologic malignancies, such as acute promyelocytic leukemia (APL) [12, 13]. Moreover, promising preclinical activity of As2O3 against malignancies other than APL was noted, such as myeloid leukemia, lymphoma, lymphocytic leukemia, and solid tumor cell lines of prostate, cervix, bladder, ovary, colon, stomach, and esophagus [12]. Lung cancers are malignant tumors with high incidences in China and worldwide [14] and characterized with high mortality because of the development of acquired resistance to chemotherapy [15]. Although recent studies have shed light on the potential of As2O3 against human lung cancers [16–21], however, there are still missing links to be explored.
In this study, we provide evidence to show that ERK8 is highly expressed in several human lung cancer cell lines. Remarkably, we report for the first time that As2O3 at physiologically relevant concentrations (effective in as low as ~5 μM) induces the phosphorylation of ERK8 and activated ERK8 subsequently promotes the phosphorylation and degradation of IκBα, which leads to the activation of NF-κB and lung cancer cell apoptosis. The pro-apoptotic role of ERK8 and NF-κB played in As2O3 cytotoxicity has been supported by the fact that short-hairpin RNA-specific knockdown of ERK8 or inhibition of NF-κB activity by NF-κB inhibitor in high ERK8-expressing human lung cancer H1299 cells blunted the As2O3-induced NF-κB activation and cytotoxicity towards these cells, indicating that both ERK8 and NF-κB are critical players in mediating the effects of As2O3. Taken together, our findings establish a novel regulatory circuit of NF-κB activation by ERK8 upon As2O3 treatment, and implicate the potential of As2O3 in targeting lung cancers with high ERK8 expression.
RESULTS
As2O3 induces the phosphorylation of ERK8
Our group has been investigating novel functions of oncokinases and their downstream substrates as potential signaling axis/molecular targets for cancer intervention. Here, we focused on novel functions and signaling cascade mediated by ERK8. Previously, ERK8 has been shown to be phosphorylated and activated by serum and growth factors such as epidermal growth factor (EGF) and increased tumorigenesis of human colon cancer [8]. However, other stimuli that would lead to its activation is unclear and the involvement of ERK8 in ROS stress/redox signaling is largely unexplored. Among the ROS-inducing drugs, we are particularly keen on As2O3 as it is a well-known anti-cancer therapeutic agent with promising efficacy on hematologic malignancies such as APL. To determine whether ERK8 can be activated by As2O3, we transfect Xpress-ERK8 in HEK293T cells and they were subsequently exposed to As2O3. As the phosphorylation of ERK8 at Thr175 and Tyr177 signifies its kinase activation status and therefore we detected the levels of p-ERK8 at Thr175 and Tyr177 upon As2O3 treatment in these cells by western blot analyses. Surprisingly, from the results, it can be seen that the phosphorylation of ERK8 is increased by As2O3 treatment in a dose- and time-dependent manner, suggesting that ERK8 can be activated by As2O3 and that ERK8 may mediate the cellular effects of As2O3 (Figure 1A–1C). This prompts us further to delineate the possible relationship of As2O3 and ERK8 and the downstream targets.
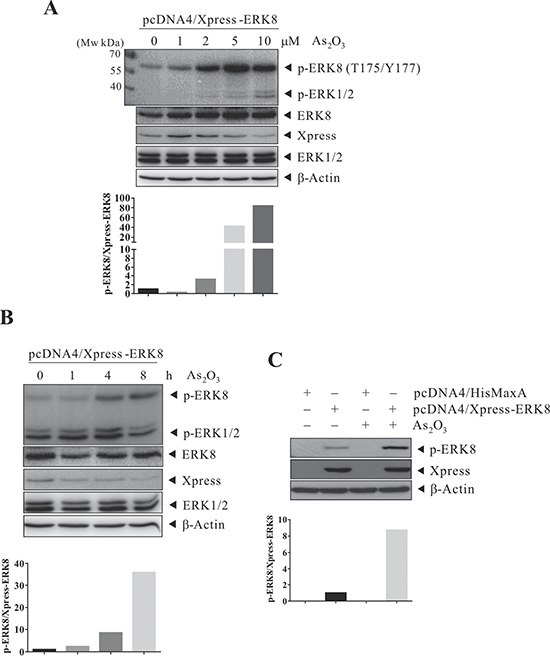
Figure 1: Activation of ERK8 by As2O3 in a dose and time-dependent manner in Xpress-ERK8 transfected cells. HEK293T cells were transfected with pcDNA4/HisMaxA control or pcDNA4/Xpress-ERK8 expression plasmids (6 μg). At 24 h post-transfection, they were cultured in serum-free medium for 24 h, after which they were sham exposed or exposed to As2O3 and monitored. (A) 1 to 10 μM As2O3 for 4 h; (B) 5 μM As2O3 for 1 to 8 h; and (C) 5 μM As2O3 for 4 h. Cells were lysed, and protein extracts were subjected to SDS-PAGE followed by immunobloting using antibodies against p-ERK8 (Thr175/Tyr177). After development, the membranes were stripped and reprobed with regular antibodies against ERK8, Xpress, ERK1/2, and β-actin to monitor the total level of ERK8, ERK1/2, and loading difference, respectively. The data are representative of three independent experiments.
IκBα as a novel substrate of ERK8
To date, knowledge about the cellular physiological protein substrate(s) of ERK8 is limited. Previous studies have shown that c-Jun is an ERK8 substrate and related to colon and gastric cancer development [8, 9]. For this reason, we sought to discover novel ERK8 substrates and we are particular keen on redox signaling pathways since we found that ERK8 can be activated by the oxidant As2O3. To identify novel substrates of ERK8, we first expressed and purified active His-tagged ERK8 and perform in vitro kinase assay using a list of potential substrates available in our laboratory. Remarkably, among these substrates, we found that IκBα can be robustly phosphorylated by ERK8 at Ser32 and Ser36 as determined by western blot analyses (Figure 2A and 2B), but not the ERK8 K42R kinase-dead mutant (Figure 2C).
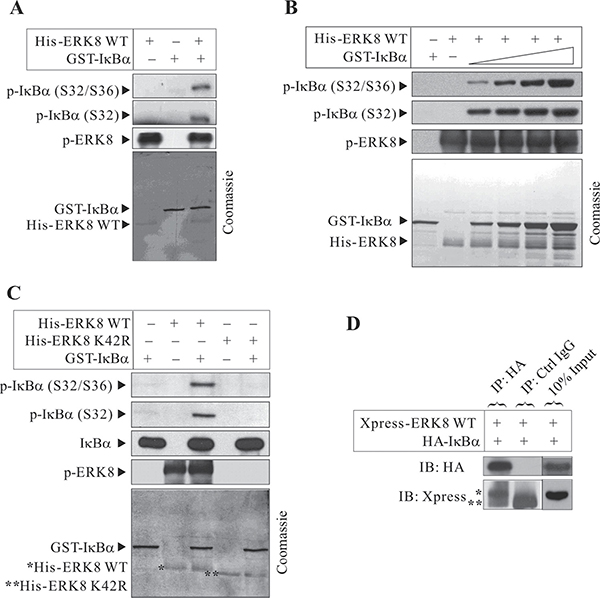
Figure 2: ERK8 phosphorylates and interacts with IκBα. In vitro kinase assays were conducted using (A) recombinant active His-ERK8 WT and GST-IκBα; (B) recombinant active His-ERK8 WT and increasing amounts of GST-IκBα; and (C) recombinant active (WT) or kinase dead (K42R) His-ERK8 and GST-IκBα as described in “Materials and methods”. Western blot analyses of IκBα phosphorylation after the kinase assays using phospho-specific antibody against p-IκBα (Ser32) or p-IκBα (Ser32/Ser36) were conducted. p-ERK8 levels signifies its kinase activity which could be detected in active His-ERK8 WT but not the ERK8 K42R mutant. (D) Xpress-ERK8 WT and HA-IκBα expression plasmids (3 μg of each) were transiently transfected into HEK293T cells. At 24 h post-transfection, they were cultured in serum-free medium for 24 h, after which they were exposed to 10 μM MG132 for 4 h. IκBα was immunoprecipitated (IP) with anti-HA or Ctrl IgG agarose beads followed by immunoblotting with anti-HA or anti-Xpress antibodies (*, Xpress-ERK8 WT; **, IgG heavy chain). For (A), (B), and (C), the corresponding gels are stained with Coomassie Blue to monitor equal protein loading (bottom panels of each). In (C), *, His-ERK8 WT; **, His-ERK8 K42R). The data are representative of three independent experiments.
To determine whether ERK8 and IκBα interact each other, plasmids encoding HA-IκBα and Xpress-ERK8 were cotransfected into HEK293T cells. IκBα proteins were immunoprecipitated with anti-HA agarose beads and subjected to western blot analyses using anti-Xpress antibody. ERK8 was confirmed to interact with IκBα ex vivo (Figure 2D). From the results, we reported for the first time that IκBα is a novel substrate of ERK8 and the likelihood of ERK8 in regulating NF-κB pathway.
ERK8 promotes the phosphorylation and degradation of IκBα
Among the redox sensitive transcription factors, NF-κB is regulated by its inhibitor IκBα which sequesters NF-κB from entering the nucleus. Upon stimulus, phosphorylation of IκBα at Ser32 and Ser36 would lead to the dissociation of IκBα from NF-κB and its subsequent ubiquitin-proteasomal degradation; as a result, NF-κB is translocated from the cytosol into nucleus and activates NF-κB-dependent gene expression [22]. To investigate the cellular physiological role of ERK8 in modulation of NF-κB pathway, first, we transfected Xpress-ERK8 into HEK293T cells and they were subsequently treated with 5 μM As2O3 for 4 h. From the western blot results, we observed that ectopically-expressed ERK8 can be activated by As2O3 treatment and the p-ERK8 levels positively correlate to the amount of the ERK8 plasmid transfected (Figure 3A). Moreover, if IκBα is a physiological substrate of ERK8 and ERK8 would catalyze the phosphorylation of IκBα at Ser32/Ser36 in cells, then ERK8 should also presumably promote the degradation of IκBα. As expected, when we transfect an increasing amount (1 to 6 μg) of Xpress-ERK8 expression plasmid into cells and subsequently dosed them with 5 μM As2O3, the expression level of endogenous IκBα declined accordingly (Figure 3A). To correlate if the decline of IκBα level is due to phosphorylation, we perform similar experiments and also monitor the level of p-IκBα at Ser32/Ser36. From the results, it can be seen that there is an obvious enhancement of phosphorylation of IκBα at Ser32/Ser36 in ERK8 transfected cells with As2O3 treatment (Figure 3B, lane 4). Although there is also elevation of the p-IκBα level in As2O3-treated cells (Figure 3B, lane 3), however, the protein level of IκBα declined more dramatically in Xpress-ERK8 transfected cells with As2O3 treatment (Figure 3B, lane 4). The ratio of p-IκBα/IκBα has the highest value in Xpress-ERK8 transfected cells with As2O3 treatment, reflecting that endogenous IκBα protein level is rapidly declined. To further substantiate the notion that the decline of IκBα protein level is due to its proteasomal degradation, we pretreated these cells with the proteasome inhibitor MG132 before exposure to As2O3. From the results, treatment of MG132 prior to As2O3 treatment prevented the decline of IκBα upon As2O3 treatment (compare lane 2 to lane 3 in Figure 3C). To determine whether IκBα degradation would lead to enhanced NF-κB activity, we performed NF-κB luciferase assay by cotransfecting the 5× κB firefly luciferase reporter with Xpress-ERK8 expression plasmid in NIH/3T3 cells and then challenged the cells with 5 μM As2O3 for 4 h. Our results indicate that As2O3 can synergize ERK8 in promotion of NF-κB activity (Figure 3D).
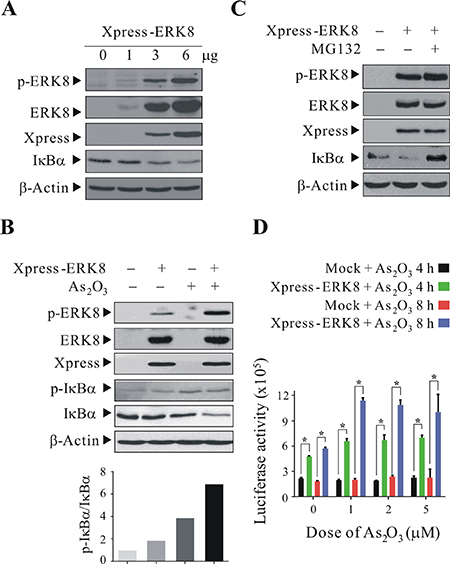
Figure 3: ERK8 promotes the phosphorylation and degradation of IκBα. HEK293T cells were transfected with Xpress-ERK8 expression plasmid. At 24 h post-transfection, they were cultured in serum-free medium for 24 h, after which they were exposed to 5 μM As2O3 for 4 h. (A) cells transfected with 1 to 6 μg Xpress-ERK8 expression plasmid; (B) cells transfected with 6 μg Xpress-ERK8 expression plasmid; (C) cells transfected with 6 μg Xpress-ERK8 expression plasmid, and prior to As2O3 treatment, cells were first pretreated with 10 μM MG132 for 4 h. Cells were lysed, and protein extracts were subjected to SDS-PAGE followed by immunobloting using antibodies against p-ERK8 and p-IκBα (Ser32/Ser36). After development, the membranes were stripped and reprobed with regular antibodies against ERK8, Xpress, IκBα, and β-actin to monitor the total level of ERK8, IκBα and loading difference, respectively. The intensities of the bands of p-IκBα and IκBα in (B) were quantified and expressed as relative ratios, setting 1 for control. (D) NF-κB luciferase assay using NIH/3T3 cells treated with As2O3. NIH/3T3 cells seeded at 6 cm dish were transient transfected with ERK8 expressing plasmid (6 μg) along with the 5× kB luciferase reporter plasmid (6 μg) and the Renilla luciferase reporter plasmid (150 ng). At 24 h post-transfection, they were seeded into 24 well-plate at 1 × 105 cells/well. After 24 h, they were sham-exposed or exposed to 1, 2, or 5 μM As2O3 for additional 4 and 8 h. NF-κB-luciferase activity was measured and normalized against Renilla luciferase activity as described in “Materials and methods”. *A significant difference of P < 0.05. The data are representative of three independent experiments.
ERK8 promotes the phosphorylation and degradation of IκBα, as well as nuclear translocation of NF-κB in human lung cancer cells
The results thus far suggested a potential paradigm of NF-κB activation by ERK8. However, whether similar phenomenon occurs in other cells would need to be verified. As our team has been focusing on lung-related disease, especially lung cancers; for this reason, we screened the basal levels of ERK8 in several human lung cancer cell lines available in our laboratory, including Calu-3, H1299, H358, and H460, by quantitative real-time RT-PCR and western blot analyses. From the results, it can be seen that ERK8 is expressed at low level in Calu-3 cells; however, among the lung cancer cells, ERK8 has a relatively high expression in H1299, H358, and H460 cells (Supplementary Figure 1A and 1B). To determine whether there is NF-κB activation in high ERK8 expressing cells, we checked on H1299 cells since the subcellular fractionation quality turns out to be the best among these cell lines. So, after we treated H1299 cells with 5 μM As2O3, we fractionated individual subcellular components and it was found that the levels of nuclear NF-κB in nuclear-soluble and chromatin-bound fractions increased as compared with untreated control (Figure 4A). Similar results were observed in H358 cells (data not shown). This indicates that there is an enhanced NF-κB chromatin-binding in As2O3-treated cells, and suggests that NF-κB might likely be involved in modulating the subsequent gene expressions. To further strengthen the relationship of As2O3-ERK8-IκBα-NF-κB cascade, we generate H1299 stable ERK8 knockdown cells and examine the levels of p-IκBα and IκBα upon As2O3 treatment. From our results, knocking down ERK8 in H1299 cells blunted the phosphorylation and degradation of IκBα (compare lane 3 to lane 4 in Figure 4B), and as a result sustained the ratio of p-IκBα/IκBα close to untreated controls. Furthermore, we also performed NF-κB luciferase assay by transfecting the 5× κB firefly luciferase reporter in these shMock and shERK8 cells and then challenged the cells with 5 μM As2O3. Our results indicate that the NF-κB luciferase activity is significantly suppressed in ERK8 knockdown cells as compared to shMock control (Figure 4C). In addition, the mRNA level of NF-κB downstream genes Fas [23, 24] was detected to further confirm the activation of NF-κB. Interestingly, the expression of Fas increased in shMock but not in stable shERK8 H1299 cells upon exposure to As2O3 (Figure 4D). These results suggest that ERK8 is critical in mediating the effect of As2O3 and the subsequent NF-κB activation in these cells.
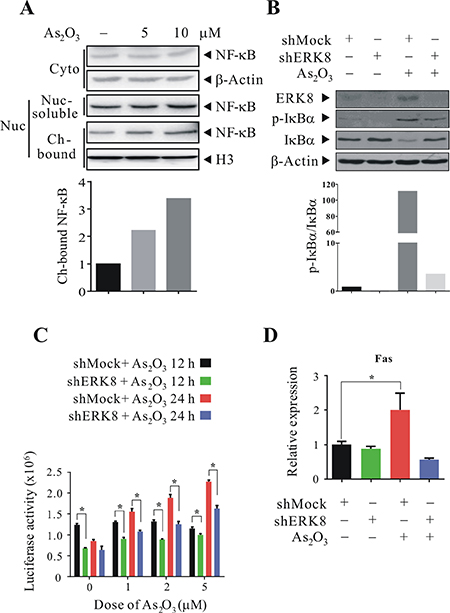
Figure 4: ERK8 promotes the phosphorylation and degradation of IκBα, as well as nuclear translocation of NF-κB in human lung cancer cells. (A) H1299 cells were treated with 5 μM As2O3 for 4 h; individual subcellular fractions (cytoplasmic, nuclear-soluble, and chromatin-bound) were isolated and subjected to western blot analysis using NF-κB p65 antibody. The same blot was stripped and reprobed with the monoclonal β-actin or histone H3 antibody to monitor the loading difference. The intensities of the bands of chromatin-bound NF-κB and histone H3 were quantified and expressed as relative ratios, setting 1 for control. (B) Stable mock control (shMock) and ERK8 knockdown (shERK8) H1299 cells were cultured in serum-free medium for 24 h, after which they were exposed to 5 μM As2O3 for 4 h. Cells were lysed, and protein extracts were subjected to SDS-PAGE followed by immunobloting using antibodies against ERK8 and p-IκBα (Ser32/Ser36). After development, the membranes were stripped and reprobed with regular antibodies against IκBα and β-actin to monitor the total level of IκBα and loading difference, respectively. The intensities of the bands of p-IκBα and IκBα were quantified and expressed as relative ratios, setting 1 for control. (C) NF-κB luciferase assay using shMock and shERK8 H1299 cells treated with As2O3. Stable shMock and shERK8 H1299 cells at 6 cm dish were transient transfected with the 5× κB luciferase reporter plasmid (6 μg) and the Renilla luciferase reporter plasmid (150 ng). At 24 h post-transfection, they were seeded into 24 well-plate at 1 × 105 cells/well. After 24 h, they were sham-exposed or exposed to 1, 2, or 5 μM As2O3 for additional 12 and 24 h. NF-κB-luciferase activity was measured and normalized against Renilla luciferase activity as described in “Materials and methods”. (D) Quantitative real-time RT-PCR to determine the mRNA level of NF-κB downstream gene Fas in stable shMock and shERK8 H1299 cells treated with 5 μM As2O3 after serum starvation for 24 h. β-Actin was used as an internal control. *A significant difference of P < 0.05. The data are representative of three independent experiments.
As2O3 promotes cell death in high ERK8-expressing lung cancers
From the above sections, we observed the activation of ERK8 by As2O3 treatment, with the concomitant phosphorylation and degradation of IκBα, as well as induction of NF-κB activity, but whether this would ultimately impact on cell growth or cell death regulation is unclear. For this reason, we sought to study the phenotype of cells mediated by this regulatory circuit. As mentioned earlier, since there is a relatively high ERK8 expression in H1299 cells, we would therefore want to know the outcome of As2O3 treatment to this high ERK8-expressing lung cancer cell line. H1299 cells were sham-exposed or treated with As2O3 (5 to 20 μM) and monitored, it can be seen that massive cytotoxicity occurred in a dose- and time-dependent manner in this cell line (Figure 5A). To check whether the observed cytotoxicity is due to the execution of apoptosis, we checked the expression levels of several important apoptosis mediators by western blot analysis. The induction/cleavage of BAX/CASP9/CASP3 can well be observed (Figure 5B), suggesting that As2O3 treatment indeed leads to apoptotic cell death. As a proof of concept, we perform similar experiments on shMock and shERK8 H1299 cells. This time, the cell viabilities were monitored by MTS assay post 48 h of As2O3 treatment. Notably, knocking down ERK8 in H1299 cells sustained the cell viability of shERK8 cells upon As2O3 treatment, but not in shMock cells (Figure 5C). Similar results were observed in H460 cells (data not shown). In addition, knocking down ERK8 in H1299 cells blunted the induction/cleavage of marker proteins involved in apoptosis (Figure 5D).
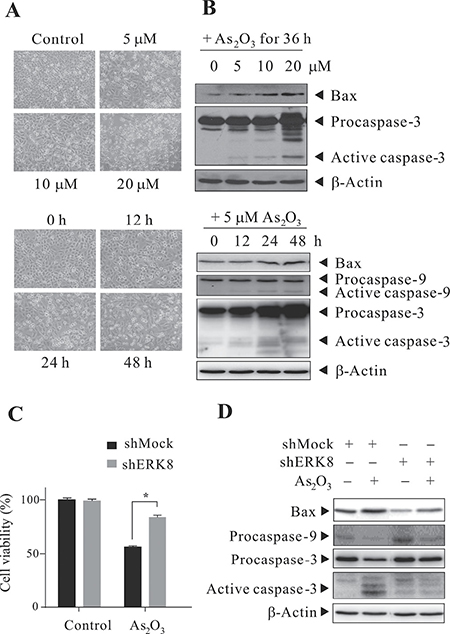
Figure 5: As2O3 promotes cell death in high ERK8-expressing lung cancers. Dose- and time-dependent cytotoxicity of As2O3 towards H1299 cells. H1299 cells were sham-exposed or exposed to 5-20 μM of As2O3 for 36 h or 5 μM As2O3 for 12-48 h. (A) cell morphology of H1299 cells under light microscope. (B) The corresponding western blot of (A), cells were lysed and protein extracts were subjected to western blot analyses using antibodies against Bax, Caspase-9, and Caspase-3. After development, the membranes were stripped and reprobed with β-actin to monitor the loading difference. (C) Stable shMock and shERK8 H1299 cells were sham-exposed or exposed to 5 μM As2O3 for 48 h, the percentage of cell viability was determined by MTS assay. *A significant difference of P < 0.05. (D) The corresponding western blot of (C) for the determination of marker proteins involved in apoptosis. The data are representative of three independent experiments.
NF-κB specific inhibitor, JSH-23, inhibits ERK8 promoted As2O3-induced lung cancer cell death
To further confirm that ERK8 promotes As2O3-induced lung cancer cell apoptosis mainly and specifically through IκBα associated NF-κB signaling, the NF-κB specific inhibitor (JSH-23) was employed. First, NF-κB luciferase assay was performed to detect the effectiveness of JSH-23 on the suppression of NF-κB activity. As we can see in Figure 6A, JSH-23 treatment significantly suppressed As2O3-induced NF-κB luciferase activity in H1299 cells. To correlate the sensitivity of H1299 cells to As2O3 is due to ERK8 mediated NF-κB activation, we performed cell viability assay in stable shMock and shERK8 H1299 cells. As expected, JSH-23 treatment significantly sustained the cell viability of As2O3-treated shMock H1299 cells (compare column 3 to column 4 in Figure 6B). In the previous section, since we showed that knocking down ERK8 significantly suppressed the NF-κB activity and sustained the cell viability of shERK8 H1299 cells upon As2O3 treatment (Figures 4C and 5C), therefore, no significant difference in As2O3 sensitivity was observed at all between JSH-23-untreated or JSH-23-treated shERK8 H1299 cells (compare column 7 to column 8 in Figure 6B). These data indicate that lowering the ERK8 level alone is sufficient and reminiscent to reducing the activity of NF-κB signaling, which ultimately diminished the cellular sensitivity to As2O3. Altogether, our data suggest that As2O3 is very effective in promoting cell death regulation in high ERK8-expressing lung cancer cells and we document for the first time on the interplay of As2O3-ERK8-IκBα-NF-κB signaling axis.
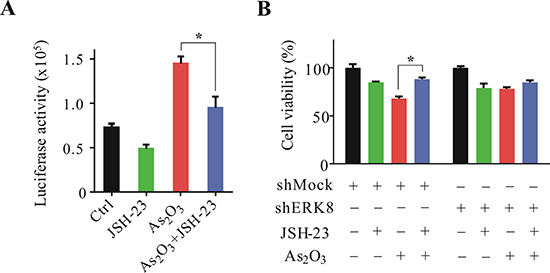
Figure 6: JSH-23 inhibits ERK8 promoted As2O3-induced lung cancer cell death. (A) NF-κB luciferase assay of H1299 cells in the presence or absence of JSH-23 (30 μM) post 12 h exposure to As2O3 (5 μM). The NF-κB luciferase activity was determined similarly as described in the previous sections. (B) Stable shMock and shERK8 H1299 cells were exposed to As2O3 (5 μM) in the presence or absence of JSH-23 (30 μM) for 36 h, the cell viability was determined by NBB assay. All the results were expressed as the mean ± SD of triplicate samples and the reproducibility was confirmed in three separate experiments. *A significant difference of P < 0.05.
DISCUSSION
Our group has been investigating novel functions of protein post-translational modifications. In the present study, we focused on novel substrates of ERK8. Previously, it has been reported that ERK8 is overexpressed in human colon cancers [8], suggesting that ERK8 might be a promising chemotherapeutic target. Here, we first wonder whether ERK8 may be overexpressed as well in lung cancers. For this reason, we screened the levels of ERK8 in several human lung cancer cell lines available in our lab, including Calu-3, H1299, H358, and H460, by quantitative real-time RT-PCR and western blot analyses. From the results, it can be seen that ERK8 is expressed at low level in Calu-3 cells. However, among the lung cancer cells, ERK8 has a relatively high expression in H1299, H358, and H460 cells (Supplementary Figure 1A and 1B).
The traditional Chinese medicine As2O3 is a well-known anti-cancer therapeutic agent with promising efficacy on hematologic malignancies such as APL. Interestingly, we found that As2O3 induced preferential cytotoxicity in several high ERK8-expressing lung cancer cells (H1299, H358, and H460) (Figure 5; Supplementary Figure 1C). Indeed, the pivotal role of ERK8 that it plays in mediating the effect of As2O3 has been demonstrated in our study, in which shRNA knockdown of ERK8 or inhibition of NF-κB activity by JSH-23 treatment blunted the As2O3-induced NF-κB activation and cell death in H1299 cells. Although the phosphorylation of ERK1/2 and p38 MAPK also increased slightly by As2O3 treatment (Figure 1A and 1B; Supplementary Figure 2), however, the magnitude of ERK8 phosphorylation is significantly higher as compared to those of ERK1/2 and p38 MAPK in all the respective time points and As2O3 dose examined, suggesting that ERK8 is dominantly activated by As2O3 in high ERK8 expressing cells (Figure 1A–1C; Supplementary Figure 2). Altogether, these data confirm that the interplay of ERK8-IκBα-NF-κB is critical for mediating the apoptotic signal exerted by As2O3 (Figure 7).
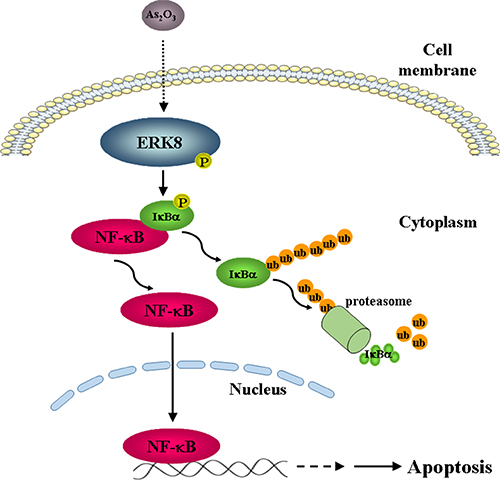
Figure 7: A model of As2O3 action based on our data is presented. As2O3 treatment induces the phosphorylation of ERK8 and activated ERK8 subsequently promotes the phosphorylation and degradation of IκBα, which leads to the activation of NF-κB and lung cancer cell apoptosis.
To date, besides IkappaB kinases and ribosomal S6 kinase 2 [25, 26], other potential kinases on the phosphorylation of IκBα at Ser32 and Ser36 is unknown/understudied. Moreover, knowledge about cellular physiological protein substrate(s) of ERK8 is limited. Previous studies have shown that c-Jun is an ERK8 substrate and related to colon and gastric cancer development [8, 9]. In here, we show that As2O3 can specifically promote the activation of ERK8 and the subsequent phosphorylation and degradation of IκBα, leading to NF-κB activation and inducing cell death in high ERK8 expressing human lung cancer cells. The reason of the Janus-faced role of ERK8 in promotion of cell growth and cell death may be very complex and cell type-dependent. Therefore, similar to JNK/c-Jun axis, depending on the types and magnitude of stimuli, which can either promote cell death or cell transformation; suggesting that although EGF (mitogenic stimulus) or As2O3 (apoptotic stimulus) can both activate the phosphorylation of ERK8 at Thr175/Tyr177, however, the relaying downstream targets are crucial in dictating the cell fate that ultimately lead to cell transformation or cell death. Nevertheless, further study is required to address these issues to find out the NF-κB dependent genes in response to As2O3 treatment (besides Fas as we have demonstrated here) that lead to cell death in high ERK8-expressing human lung cancer cells.
In summary, we demonstrate for the first time a regulatory paradigm of NF-κB activation by ERK8 upon As2O3 treatment and that IκBα is a newly identified cellular physiological substrate of ERK8. Given the important role of ERK8 in mediating the therapeutic efficacy of As2O3 against lung cancer cells, we suggest that As2O3 (besides its promising role in the treatment of hematologic malignancies) as a potential agent in the treatment of lung cancer patients with high ERK8 expression such that this might gain more selective killing of cancer cells. Finally, the work we report here might also implicate the need for ongoing research on the identification and understanding of novel signaling circuit(s) in response to As2O3 treatment in various cancer types, and hopefully shed light in providing strategies for anti-cancer management.
MATERIALS AND METHODS
Materials
Arsenic trioxide (As2O3) and MG132 were purchased from Sigma-Aldrich (St. Louis, MO). The NF-κB specific inhibitor JSH-23 (S7351) was purchased from Selleck (Shanghai, China). The Subcellular Protein Fractionation Kit for Cultured Cells was from Thermo Scientific (Rockford, IL). shRNAs were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ERK8 shRNA plasmid (h) (sc-77462-SH) is a target-specific lentiviral vector plasmid encoding a 19–25 nt (plus hairpin) shRNA to knock down gene expression. Control shRNA plasmid-A (sc-108060) encodes a scrambled shRNA sequence that will not lead to the specific degradation of any known cellular mRNA. All other general chemicals were purchased from GE Healthcare (Uppsala, Sweden), Qiagen (Valencia, CA) and Sigma-Aldrich. The pCMV4-3 HA/IκBα (21985) and pRL-SV40P (27163) plasmids were purchased from Addgene (Cambridge, MA). The 5× κB luciferase reporter plasmid has been described previously [27]. The Homo sapiens ERK8 gene coding sequence (NM_139021) expression plasmid pGEX-2T-GST-ERK8 wild-type (WT) and the corresponding kinase-dead mutant (K42R) were generously provided by Dr. Mark K. Abe from the Univesity of Chicago (Chicago, IL). Antibodies used for western blot were purchased from BBI Life Sciences (Shanghai, China), Santa Cruz Biotechnology, GeneTex (Irvine, CA), Cell Signaling Technology (Danvers, MA), Invitrogen (Waltham, MA) and Sigma-Aldrich, with the following dilutions: ERK8 (AB21813b; BBI Life Sciences), 1:1000; p-ERK1/2 Thr202/Tyr204 (4370; Cell Signaling Biotechnology, also being utilized for the detection of phosphorylated ERK8 at Thr175/Tyr177 as described previously), 1:1000 [8]; ERK1/2 (sc-292838; Santa Cruz Biotechnology), 1:1000; p-p38 MAPK (4511; Cell Signaling Technology), 1:1000; p38 MAPK (8690; Cell Signaling Technology), 1:1000; p-JNK (sc-293138; Santa Cruz Biotechnology), 1:1000; JNK (sc-7345; Santa Cruz Biotechnology), 1:1000; p-IκBα Ser32 (2859; Cell Signaling Biotechnology), 1:1000; p-IκBα Ser32/Ser36 (9246; Cell Signaling Biotechnology), 1:1000; IκBα (AB20138a; BBI Life Sciences), 1:1000; NF-κB p65 (sc-8008; Santa Cruz Biotechnology), 1:1000; Bax (GTX109683; GeneTex), 1:1000; Caspase-9 (GTX112888; GeneTex), 1:1000; Caspase-3 (GTX110543; GeneTex), 1:1000; HA-probe (sc-7392; Santa Cruz Biotechnology), 1:1000; Xpress (46-0528; Invitrogen), 1:5000; and β-Actin (A5441; Sigma-Aldrich), 1:10000.
Construction of ERK8 expression plasmids and the corresponding mutants
The ERK8 WT and ERK8 K42R were each inserted into the pET46 Ek/LIC plasmid and expressed as His-tagged fusions as described previously [8]. The ERK8 proteins were purified using Ni-NTA agarose (Qiagen). The ERK8 gene coding sequence was also amplified from pGEX-2T-GST-ERK8 WT by PCR. After restriction digestion, the ERK8 sequence was ligated to the BamHI/XbaI site of pcDNA4/HisMaxA to generate the pcDNA4/Xpress-ERK8. For preparation of GST-IκBα, the IκBα gene coding sequence was amplified from pCMV4-3 HA/IκB-alpha by PCR. After restriction digestion, the IκB-alpha sequence was ligated to the EcoRI/NotI site of pGEX-5X-1 to generate the pGEX-5X-1/GST- IκBα. The recombinant plasmids were confirmed by DNA sequencing (Beijing Genomics Institute, Shenzhen, Guangdong, China).
Cell culture and transfection
All the cell lines employed in this study, including human lung cancer cells (Calu-3, H1299, H358, and H460) as well as HEK293T, NIH/3T3, HL-60, HepG2, and HeLa cells, were purchased from and authenticated by the ATCC Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured following the supplier’s recommended conditions. Cell viability was measured by MTS or NBB assay as described previously [8, 28]. Transfection of the expression vectors was performed using Lipofectamine (Invitrogen) following the manufacturer’s suggested protocol. For generation of ERK8 knockdown cells, viral particles were packed with control or ERK8 shRNA. H1299 cells, grown to about 70% confluence, were infected with the above lentiviral shRNAs in the presence of 8 μg/ml polybrene for 24 h. Uninfected cells were eliminated by exposure to 2 μg/ml puromycin for 4 days before use.
Quantitative real-time RT-PCR
In brief, total RNA was extracted from cells using TRIzol Reagent (Thermo Fisher Scientific, 15596018). cDNA was synthesized using PrimeScript Reverse Transcriptase (Takara, 2680A) according to the manufacturer’s instructions. Quantitative real-time RT-PCR was performed with GoTaq qPCR Master Mix (A6001) from Promega (Fitchburg, WI) and gene-specific primers on Applied Biosystems 7500 Real-Time PCR System. β-Actin was amplified as internal reference to normalize gene expression. Relative expression of genes was calculated using 2 ΔΔCT method as described previously [29]. Primers were synthesized by Beijing Genomics Institute with the following sequences: ERK8 (forward: 5′-GACCAGAAGCCGTCCAATGT-3′, reverse: 5′-GTATCGGTGCGAAGAGAGCA-3′); Fas (forward: 5′-TGAAGGACATGGCTTAGAAGTG-3′, reverse: 5′-GG TGCAAGGGTCACAGTGTT-3′) and β-actin (forward: 5′-ATGGGTCAGAAGGATTCCTATGTG-3′, reverse: 5′-CTTCATGAGGTAGTCAGTCAGGTC-3′).
Cell lysate preparation and conditions of western blot and immunoprecipitation
After treatment, cells were then washed thrice with ice-cold PBS, scraped into centrifuge tube, and then harvested by centrifugation at 1000× g for 5 min at 4°C. For subcellular protein preparation, the cytoplasmic, nuclear-soluble, and chromatin-bound fractions were prepared using the Subcellular Protein Fractionation Kit for Cultured Cells in accordance with the manufacturer. For western blot analysis, cell pellets were lysed in radioimmunoprecipitation assay buffer according to the protocol as described previously [28]. Equal amounts of proteins were fractionated on a SDS-polyacrylamide gel and transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat dry milk in PBS containing 0.05% Tween 20 and probed with various primary antibodies. After incubation with secondary antibodies, immunoblots were visualized with the enhanced chemiluminescence detection kit (GE Healthcare). The normalized target band signal intensities of one data set among the three independent repeats were expressed as relative ratios and presented as bar graphs. For immunoprecipitation, cells were harvested at 80 to 90% confluence and proteins were extracted in NP40 cell lysis buffer [50 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 0.5% NP40] with freezing and thawing. Equal amounts of proteins were subjected to immunoprecipitation followed by western blot analysis.
In vitro kinase assay
To detect IκBα phosphorylation, GST-IκBα proteins (1 μg) were mixed with purified recombinant active or kinase dead His-ERK8, 0.2 μM ATP, and 1× kinase buffer and incubated at 30°C for 30 min. The reaction was stopped by adding 5× SDS sample buffer. The samples were boiled and then separated by SDS-PAGE and visualized by western blot analyses, or Coomassie Blue staining.
Reporter gene assay
The 5× κB luciferase reporter plasmid construct which contains the 5× NF-κB binding site was used. NF-κB activity was analyzed by transfection of the 5× κB luciferase reporter plasmid into ERK8-overexpressing or -knockdown cells. Cells were disrupted with lysis buffer (dual-luciferase reporter assay system, Promega) at room temperature for 30 min by gentle shaking, and then firefly luciferase activity was measured. The NF-κB-luciferase activity was normalized against Renilla luciferase activity (pRL-SV40P).
Statistical analysis
Statistical analysis was done by using two-tailed Student’s t-test. A P value of < 0.05 was considered significant. Except for western blot analyses, all other quantitative results were expressed as the mean ± SD of triplicate samples. The reproducibility of experimental results was confirmed in three separate experiments and representative data set was shown.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the grants from the National Natural Science Foundation of China (Nos. 31271445 and 31170785), the Science and Technology Planning Project of Guangdong Province of China (No. 2016A020215144), the Guangdong Natural Science Foundation of China (No. S2012030006289), “Thousand, hundred, and ten” project of The Department of Education of Guangdong Province (No. 124), and the Department of Education, Guangdong Government under the Top-tier University Development Scheme for Research and Control of Infectious Diseases. We would like to thank members of the Lau And Xu laboratory for critical reading of this manuscript.
CONFLICTS OF INTEREST
The authors declare they have no competing interests.
REFERENCES
1. Abe MK, Saelzler MP, Espinosa R 3rd, Kahle KT, Hershenson MB, Le Beau MM, Rosner MR. ERK8, a new member of the mitogen-activated protein kinase family. J Biol Chem. 2002; 277:16733–16743.
2. Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011; 75:50–83.
3. Abe MK, Kuo WL, Hershenson MB, Rosner MR. Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol Cell Biol. 1999; 19:1301–1312.
4. Klevernic IV, Stafford MJ, Morrice N, Peggie M, Morton S, Cohen P. Characterization of the reversible phosphorylation and activation of ERK8. Biochem J. 2006; 394:365–373.
5. Abe MK, Kahle KT, Saelzler MP, Orth K, Dixon JE, Rosner MR. ERK7 is an autoactivated member of the MAPK family. J Biol Chem. 2001; 276:21272–21279.
6. Iavarone C, Acunzo M, Carlomagno F, Catania A, Melillo RM, Carlomagno SM, Santoro M, Chiariello M. Activation of the Erk8 mitogen-activated protein (MAP) kinase by RET/PTC3, a constitutively active form of the RET proto-oncogene. J Biol Chem. 2006; 281:10567–10576.
7. Klevernic IV, Martin NM, Cohen P. Regulation of the activity and expression of ERK8 by DNA damage. FEBS Lett. 2009; 583:680–684.
8. Xu YM, Zhu F, Cho YY, Carper A, Peng C, Zheng D, Yao K, Lau AT, Zykova TA, Kim HG, Bode AM, Dong Z. Extracellular signal-regulated kinase 8-mediated c-Jun phosphorylation increases tumorigenesis of human colon cancer. Cancer Res. 2010; 70:3218–3227.
9. Jin DH, Lee J, Kim KM, Kim S, Kim DH, Park J. Overexpression of MAPK15 in gastric cancer is associated with copy number gain and contributes to the stability of c-Jun. Oncotarget. 2015; 6:20190–20203. doi: 10.18632/oncotarget.4171.
10. Groehler AL, Lannigan DA. A chromatin-bound kinase, ERK8, protects genomic integrity by inhibiting HDM2-mediated degradation of the DNA clamp PCNA. J Cell Biol. 2010; 190:575–586.
11. Colecchia D, Strambi A, Sanzone S, Iavarone C, Rossi M, Dall'Armi C, Piccioni F, Verrotti di Pianella A, Chiariello M. MAPK15/ERK8 stimulates autophagy by interacting with LC3 and GABARAP proteins. Autophagy. 2012; 8:1724–1740.
12. Murgo AJ. Clinical trials of arsenic trioxide in hematologic and solid tumors: overview of the National Cancer Institute Cooperative Research and Development Studies. Oncologist. 2001; 6:22–28.
13. Miller WH Jr, Schipper HM, Lee JS, Singer J, Waxman S. Mechanisms of action of arsenic trioxide. Cancer Res. 2002; 62:3893–3903.
14. Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray, F. GLOBOCAN2012 v1.0, Cancer incidence and mortality worldwide. IARC CancerBase. 2013; 11.
15. Kim ES. Chemotherapy Resistance in Lung Cancer. Adv Exp Med Biol. 2016; 893:189–209.
16. Lam SK, Mak JC, Zheng CY, Li YY, Kwong YL, Ho JC. Downregulation of thymidylate synthase with arsenic trioxide in lung adenocarcinoma. Int J Oncol. 2014; 44:2093–2102.
17. Seo SK, Kim JH, Choi HN, Choe TB, Hong SI, Yi JY, Hwang SG, Lee HG, Lee YH, Park IC. Knockdown of TWIST1 enhances arsenic trioxide- and ionizing radiation-induced cell death in lung cancer cells by promoting mitochondrial dysfunction. Biochem Biophys Res Commun. 2014; 449:490–495.
18. Yang MH, Zang YS, Huang H, Chen K, Li B, Sun GY, Zhao XW. Arsenic trioxide exerts anti-lung cancer activity by inhibiting angiogenesis. Curr Cancer Drug Targets. 2014; 14:557–566.
19. Suzuki T, Ishibashi K, Yumoto A, Nishio K, Ogasawara Y. Utilization of arsenic trioxide as a treatment of cisplatin-resistant non-small cell lung cancer PC-9/CDDP and PC-14/CDDP cells. Oncol Lett. 2015; 10:805–809.
20. Chang KJ, Yang MH, Zheng JC, Li B, Nie W. Arsenic trioxide inhibits cancer stem-like cells via down-regulation of Gli1 in lung cancer. Am J Transl Res. 2016; 8:1133–1143.
21. Cheepsattayakorn A, Cheepsattayakorn R. Lung cancer chemotherapy, new treatment and related patents. Recent Pat Anticancer Drug Discov. 2014; 9:372–381.
22. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008; 132:344–362.
23. Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol. 1999;19:2098–2108.
24. Singh NP, Nagarkatti M, Nagarkatti PS. Role of dioxin response element and nuclear factor-kappaB motifs in 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated regulation of Fas and Fas ligand expression. Mol Pharmacol. 2007;71:145–157.
25. Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997; 278:860–866.
26. Peng C, Cho YY, Zhu F, Xu YM, Wen W, Ma WY, Bode AM, Dong Z. RSK2 mediates NF-{kappa}B activity through the phosphorylation of IkappaBalpha in the TNF-R1 pathway. FASEB J. 2010; 24:3490–3499.
27. Wu X, Qi J, Bradner JE, Xiao G, Chen LF. Bromodomain and extraterminal (BET) protein inhibition suppresses human T cell leukemia virus 1 (HTLV-1) Tax protein-mediated tumorigenesis by inhibiting nuclear factor κB (NF-kB) signaling. J Biol Chem. 2013; 288:36094–36105.
28. Xu YM, Zhou Y, Chen DJ, Huang DY, Chiu JF, Lau ATY. Proteomic analysis of cadmium exposure in cultured lung epithelial cells: evidence for oxidative stress-induced cytotoxicity. Toxicol Res. 2013; 2:280–287.
29. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–408.