
INTRODUCTION
The immune system has the capacity to produce a vast repertoire of immunoglobulins, (Ig) in response to the wide number of existing antigens, by processes such as V(D)J recombination (it occurs during B-cell maturation) [1, 2], somatic hypermutation (generated during B-cell affinity maturation) [3, 4] and antigen selection (during B-cell activation) [5].
The Ig structure comprises four chains: two identical heavy chains and two identical light chains linked by disulfide bonds. Heavy chains are classified according to the heavy chain into 5 groups: α, δ, ε, γ, and μ (for IgA, IgD, IgE, IgG, and IgM, respectively). Concerning light chains, there are two types: kappa (κ) and lambda (λ). In turn, each heavy and light chain presents a constant (C) and a variable (V) region. The effector function of the Ig is located in the C region of the heavy chain. The Igs also comprise fragment antigen binding (Fab) and fragment crystallizable (Fc) regions, where Fab region is the part which binds to antigens (including the variable domain of each heavy and light chain) and presents a set of complementary determining regions (CDR), also known as hypervariable regions, located between stable regions named frameworks (FR). In turn, the Fc region is the binding site for endogenous receptors (presented in macrophages, dendritic cells, and other cells of the immune system) and complement system proteins [6].
Igs are present as soluble proteins in proximal body fluids (serum, synovial fluid, and saliva, among others) or membrane-bound proteins attached to the B-cells (similar structure to IgM) for regulating the immune system and performing the function of B-cell receptors (BCR), respectively. Specifically, the disrupted activation of the BCR appears as the responsible of the chronic lymphocytic leukemia (CLL) [7, 8]. This disease, characterized by its high heterogeneity, is the most common human blood cancer in Western countries and it usually shows a monoclonal expansion of an aberrant B-cell clone [9, 10].
Determining the protein profiles of B-CLL cells could have a great impact on disease knowledge, progression, origin, and identification of new drug targets; particularly referring to immune-system proteins, antibodies and immunoglobulins. In this sense, high-throughput DNA sequencing has been the approach of choice for extracting the most Ig biological information [11]. In fact, Next Generation Sequencing (NGS) allows the characterization of millions of BCR sequences in a single experiment [12]. However, despite the remarkable advances made in the field, the peptide sequence information is required for the complete description of the Ig sequences as the cellular state is finally defined by the translated genes (i.e., proteins) – everything that is transcribed (genomics) may not be translated (proteomics)-. Henceforth, the integration of genomics and proteomics data sets for Ig sequencing might provide complementary information about evidence of gene expression at the protein level [13, 14, 15].
The determination of the length and peptide sequence of the H-chain CDR-H3 region (related to antibody specificities) as well as the IGHV and IGHJ gene patterns and the VH:VL pairing [16, 17] are paramount for antibody clonotyping. In fact, the alterations in these sequences could be specific and characteristic of the pathology highlighting the importance of their determination as previously reported by Henriques and colleagues [18]. Moreover, the evaluation of immune repertoires is coming up as a useful approach for the identification of antigen-specific BCR, and evaluation of the efficacy of immune checkpoint blockers and cancer immune therapy, among others [19, 20].
Also, these described approaches are becoming quite critical in the characterization of pharmaceutical IgGs, such as anti-CD20, trastuzumab, and cetuximab [21]. Related to this, the production of biosimilars (biologic medical products which are almost identical copies of original products), which are considered as good alternatives to improve healthcare access and outcomes at reduced costs, could be greatly improved if disease-related antibody sequences are determined (e.g., monoclonal antibodies, cytokines…) [22, 23].
Generally, Ig-sequencing (soluble Ig) studies have been performed by using MS/MS approaches in serum samples [24], whereas BCR-sequencing (membrane-bound Ig) has been addressed from a genomic point of view using B-cells as a sample [25]. Thus, their combination might have a tremendous importance to understand the humoral antibody responses.
Peptide analysis of BCR sequences remains a challenge mainly due to the drawbacks in its processing and purification (e.g., low BCR levels). Thus, NGS approaches have been the usually selected strategies for the amplification of the BCR and its sequencing. However, a library including the mass spectra for BCR-related peptides may be of great interest for the full characterization of BCR production, Ig secretion, and immune responses. Since little variations in antibody sequences (even one single amino acid) could lead to modified specific antigen bindings; then, MS/MS strategies seem a good alternative for providing confidence in peptide sequencing [25].
To this end, we have evaluated three approaches for peptide sequencing of BCR as well as B-cell proteins associated to BCR (MHC-I, MHC-II, CD20, CD79b, among others) and also related to the immune system using an MS/MS approach. Moreover, an in-house algorithm has been designed for the comparison of sequences obtained by MS/MS and molecular sequencing (Figure 1) to demonstrate the potential of MS/MS approaches in B cells to characterize Ig sequences (426 unique peptide sequences have been identified in this study) and correlate them with DNA sequence libraries.
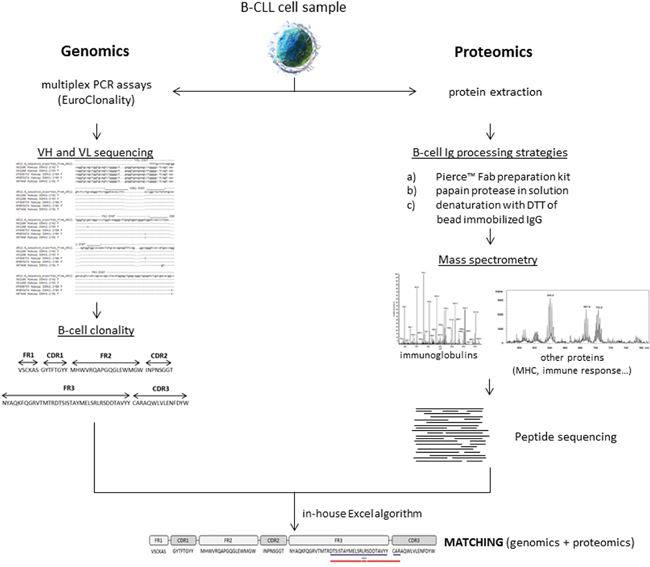
Figure 1: General workflow of the study.
RESULTS
Sample processing for BCR profiling by MS/MS assays
As mentioned in the Introduction section, cellular Igs use to be characterized by DNA-sequencing strategies whereas soluble Igs (due to their high concentration in serum) are easier to be characterized by MS approaches. The key point is the isolation and enrichment of BCR (membrane-bound Ig) in order to increase the peptide coverage that facilitates the peptide sequencing of the BCR. With this purpose, three different approaches (termed Fab preparation kit (a), papain (b), and DTT (c) in Materials and Methods section) were tested to evaluate the utility and robustness of MS/MS assays for Ig peptide identification.
While the first approach (Fab purification kit) uses an available commercial kit for the isolation of Fab fragments from protein samples employing affinity columns, the second and third approaches (papain and DTT, respectively) include an Ig-enrichment step – based on protein G microbeads - previous to the addition of papain or DTT for the isolation of Fab fragments or the breakage of disulfide bonds, respectively. With all these strategies, it is expected to purify the B-cell Igs in order to increase the peptide coverage and the subsequent BCR-peptide characterization.
In Supplementary Information 1, the proteins and the corresponding peptides for each approach are collected. As it has been depicted in Table 1, CLL samples have been randomly distributed among the 3 suggested approaches. Additionally, human IgG protein (whole molecule) was processed using the Fab preparation kit to use it as a reference.
Table 1: Patient characteristics
Sample ID |
Agea (yr) |
Gender |
Diagnosisb % clonal population (from WBC)b V(D)J rearrangementc |
MSc Rai stage |
Light chainb |
Molecular biology sequence |
MS/MS processing approachd Gel section digested |
|---|---|---|---|---|---|---|---|
1 |
83 |
F |
CLL 88.1 |
UM-H |
kappa+dim |
Avail. |
DTT |
2 |
58 |
F |
CLL 21.4 |
M-H |
kappa+dim |
N.A. |
DTT |
3 |
62 |
F |
CLL 22.1 |
UM-H |
kappa+dim |
Avail. |
DTT |
4 |
48 |
M |
CLL 90.0 |
M-H |
kappa+dim |
Avail. |
DTT |
5 |
72 |
F |
CLL 70.1 |
M-H |
kappa+dim |
N.A. |
DTT |
6 |
49 |
M |
CLL 80.4 |
M-H |
kappa+dim |
N.A. |
Fab preparation kit |
7 |
65 |
M |
CLL 87.5 |
UM-H |
kappa+dim |
N.A. |
Papain |
8 |
70 |
M |
CLL 64.9 |
M-H |
kappa+dim |
N.A. |
Papain |
9 |
56 |
M |
CLL 83.2 |
UM-H |
lambda+dim |
N.A. |
DTT |
Clinical characteristics of the samples used in this study, including the age, gender, diagnosis, V(D)J rearrangement, molecular biology features, and MS/MS processing approach applied to each sample.
a Age at time of sample collection. b From phenotypic studies. c From molecular biology studies. d DTT for denaturation of bead-immobilized immunoglobulins (Ig); papain for isolation of Fab fragments from bead-immobilized Ig; Fab preparation kit for isolation of Fab fragments using a commercial kit.
WBC, white blood cells. MS, mutational status (UM-H, unmutated; M-H, mutated). F, female. M, male. CLL, chronic lymphocytic leukemia. Avail., available. N.A., information not available.
A total of 31 different peptides related to Ig were identified when processing the human IgG sample, corresponding to 20 different proteins. As expected, the approach concerning Fab preparation kit allowed the isolation of these peptides from a purified Ig sample. However, its application on a complex sample (purified B-CLL cells, sample 6) reported 22 different proteins (related to BCR and Ig) in comparison with the control sample. Nevertheless, when looking for the 20 human IgG proteins mentioned above in the samples processed with papain and DTT, the overlapping was increased up to 40% (8/20) and 70% (14/20), respectively.
Considering as reference these results, the denaturation approach provides better results when dealing with high complex samples. Nonetheless, and due to this complexity, the purification and enrichment of the sample may become a key step during the processing.
Our results show that the number of identified proteins related to the immune system is increased as the processing strategy is simplified. Thus, 40, 54, and 171 immune system proteins were reported in CLL samples for Fab preparation, papain, and DTT approaches, respectively (Supplementary Information 1). Therefore, for MS/MS characterization of cellular Ig, two main considerations are needed: i) enrichment step for the isolation and purification of Ig-related proteins, and ii) simple post-processing for minimizing loss of sample.
Regarding the distribution of immune system related proteins reported by the three approaches (Figure 2), papain approach was the most efficient for the identification of specific Ig proteins (40.8%), followed by Fab preparation and DTT approaches. Furthermore, proteins belonging to the human leukocyte antigen (HLA) system were better detected by the DTT approach (20.5%) in comparison to papain and Fab preparation strategies (11.1 and 2.5%, respectively). The remaining protein groups (B-cell proteins, antigens, leukocyte/lymphocyte proteins, and others) presented a similar distribution.
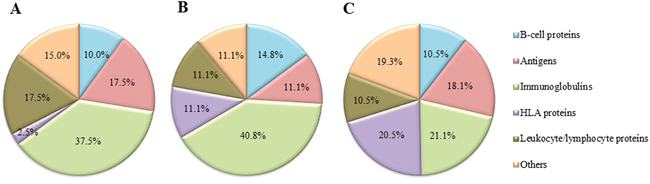
Figure 2: Distribution of proteins related to the immune system detected by shotgun MS/MS after processing the samples by three different approaches. (A) Fab preparation kit for isolation of Fab fragments using a commercial kit; (B) papain for isolation of Fab fragments from bead-immobilized immunoglobulins (Ig); (C) DTT for denaturation of bead-immobilized Ig). The MS/MS analysis was performed using Peptide Shaker search engine (neXtProt database, 2016).
Characterization of Ig peptide sequences by MS/MS
The construction of peptide sequence libraries for soluble Igs and BCRs is still a challenge. As mentioned before, several improvements have been done in serum-Ig sequencing, but profiling cellular Ig without using DNA-approaches is really difficult mainly due to the low amount of membrane-bound Ig present in the cell (compared to the rest of cell proteins).
In our study, a total of 60, 98, and 426 different unique peptides were identified across the three approaches (Fab preparation, papain, and DTT, respectively) establishing the beginning of a CLL peptide sequence library exclusively obtained by MS/MS technique. Only 15 peptides out of the total identified were in common among the studied strategies, whereas the approach concerning denaturation of Ig enriched samples reported 362 unique peptides not identified by the other two techniques (data not shown). These results suggest the importance of perfectly well-defined workflow when processing complex samples for Ig sequencing.
As Figure 3 shows, the peptide sequence length is distributed from 6 to 23 amino acids in the samples. This distribution is more uniform for peptides obtained with the DTT approach. However, the Fab preparation kit has generated a low number of long unique peptides which could be a drawback for the identification of cellular Ig.
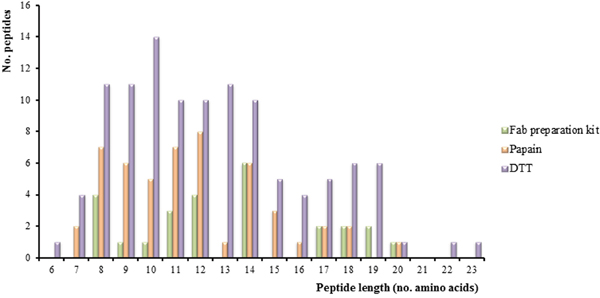
Figure 3: Peptide length distribution. Graphic showing the number of peptides corresponding to immunoglobulin (Ig) proteins detected by the three approaches (Fab preparation kit for the isolation of Fab fragments using a commercial kit, papain for isolation of Fab fragments from bead-immobilized Ig, and DTT for denaturation of bead-immobilized Ig) using Peptide Shaker as search engine and neXtProt (release February 2016) as database.
Regarding peptide sequence variability, the combination of different proteases (in addition to trypsin, which has been used in this study) might be an important improvement. In this way, peptide sequence libraries could increase in size providing a better Ig characterization.
Matching genomic and proteomic data sets for BCR profiling at peptide level
Sequencing of membrane-bound Ig of clonal B-cell populations belonging to CLL patients is routinely done. This sequencing is performed by using molecular biology techniques and allows the stratification of the patients, being also crucial for determining the patient treatment. Despite multiplex PCR assays are performed to determine the B-cell clonality (following the guidelines of the EuroClonality European Consortium, http://www.euroclonality.org/); this approach presents a limitation because it is not possible to characterize the whole set of proteins related to the immune system in a single assay. In this regard, proteomics is able to achieve the Ig characterization at peptide level with the same processing speed and robustness as genomics, but with the gain of sequencing thousands of peptides at once (including Ig or proteins related to the immune response, among others). This is of great relevance given the importance of surrounding proteins that determine the final behavior of B-CLL cells.
In fact, both disciplines must be considered as complementary instead of competitive or encountered methodologies. With the purpose of integrating the results from genomics and proteomics, an in-house algorithm in Excel software (Supplementary Information 2) was designed. This tool allows the identification of sequences overlapping between the peptide and DNA-translated providing information about which specific Ig region (CDR or FR) presents the overlapping and the coverage percentage for each sample.
In this study, information about the Ig sequences from three samples (samples ID 1, 3, and 4) are reported and compared to our MS/MS peptide sequences (from the same samples) using the mentioned algorithm.
As Ig sequences are characterized by their high variability, the common databases used for MS/MS searching do not include all of them (hence specific databases only focused on Igs are dramatically needed). Thus, two different search engines (i.e., Mascot and PeptideShaker) were selected for evaluating their capability on detecting non-assigned peptides (i.e., amino acid sequences belonging to highly variable proteins). On one side, PeptideShaker only presents as result sequences matching with annotated unique peptides and their corresponding proteins; whereas the Mascot search engine reports well-defined peptides although they do not correspond to annotated proteins. This comparison will highlight the relevance of the appropriate search engine (independently of the sample preparation) according to the sample features.
The peptides identified by both search engines are collected in Supplementary Information 3 for the three samples mentioned above (“List of peptides” sheet). For Mascot results, “annotated” and “unknown” proteins are displayed in different columns. These peptide sequences were compared to the corresponding molecular biology sequence by using the in-house Excel algorithm. The results of these comparisons are also depicted in Supplementary Information 3.
Figure 4 summarizes the overlapping between both genomics and proteomics and both Mascot and PeptideShaker. As indicated in this Figure 4, FR regions present the greater overlap between genomics and proteomics. Regarding the CDR3 region, 3 amino acids (CAR) have been properly detected (high ion score) within an 11-amino acids length peptide constituting a 20% coverage of the mentioned region.

Figure 4: IGH gene sequences. The framework (FR) and complementary determining regions (CDR) of IGH genes were analyzed and translated into amino acids after DNA-sequencing using the IMGT tool. (A) sample ID 1; (B) sample ID 3; (C) sample ID 4. Violet lines depict the sequence coverage by both DNA-sequencing and MS/MS sequencing using Mascot search engine (UniProt database). Blue lines correspond to sequence coverage by both DNA-sequencing and MS/MS sequencing using Peptide Shaker search engine (neXtProt database). Red lines correspond to predicted tryptic peptides determined by PeptideCutter tool (ExPASy Bioinformatics Resource Portal).
Regarding the search engine employed, Mascot seems to be more efficient than PeptideShaker. In Table 2, it is reported the coverage percentage for each sample and Ig region. Although PeptideShaker coverage for sample 4 is good enough, Mascot is more homogeneous along the three samples, possibly due to the fact previously mentioned.
Table 2: Sequence coverage of IGH genes after DNA-sequencing and MS/MS sequencing and analysis by Mascot and Peptide Shaker search engines
Sample IDa |
Search engineb |
Regionc |
Region coveraged (%) |
Total coveragee (%) |
|---|---|---|---|---|
1 |
Mascot |
FR3 |
23/37 (62.1%) |
26/91 (28.6%) |
CDR3 |
3/15 (20.0%) |
|||
Peptide Shaker |
FR3 |
2/37 (5.4%) |
2/91 (2.2%) |
|
3 |
Mascot |
FR2 |
6/17 (35.3%) |
17/104 (16.3%) |
FR3 |
11/37 (29.7%) |
|||
Peptide Shaker |
FR3 |
2/37 (5.4%) |
2/104 (1.9%) |
|
4 |
Mascot |
FR2 |
6/17 (35.3%) |
9/109 (8.3%) |
FR3 |
3/37 (8.1%) |
|||
Peptide Shaker |
FR1 |
4/14 (28.6%) |
13/109 (11.9%) |
|
FR3 |
9/37 (24.3%) |
a Sample ID information is available at Table 1. b Mascot search engine was used with UniProt database, whereas Peptide Shaker was tested with the neXtProt database. c It is referred to the framework (FR) or complementary determining region (CDR) of the IGH gene sequence. d Region coverage was calculated considering the number of amino acids in common between DNA-sequencing and MS/MS sequencing in relation to the region (FR or CDR) length. e Total coverage was calculated considering the total number of amino acids in common between DNA-sequencing and MS/MS sequencing of the whole IGH gene in relation to the IGH gene length.
DISCUSSION
The high diversity present in gene sequences requires tailor-made DNA sequence databases, particularly in encoding genes for Ig and histocompatibility complexes. Next-generation sequencing approaches (genomics) have allowed a significant improvement in this area (BCR- and Ig-sequencing), but the progress in proteomics for the BCR-sequencing is still in their beginnings. Whereas the sequencing of soluble Ig is perfectly addressed by MS/MS strategies, the peptide profiling of membrane-bound Ig is still a great challenge.
In this study, we have characterized Ig attached to the B-CLL cells surface at peptide level by performing three different sample preparation approaches (i.e., usage of a Fab preparation commercial kit, and usage of papain in solution and DTT in Ig-enriched agarose beads from CLL protein samples for the isolation of Fab and denaturation of the Ig, respectively). Our results have indicated that the strategy including denaturation with DTT is the most efficient approach. Bearing in mind these results, several guidelines could be described about peptide sequencing of membrane-bound Ig: i) including an Ig-enrichment step; ii) reducing the complexity of the procedure (with regard to the number of steps) to avoid the loss of the sample (membrane-bound Ig are less abundant than soluble ones); iii) using a suitable MS/MS search engine for the identification of peptides corresponding to uncharacterized or unassigned proteins; and iv) integrating MS/MS and molecular biology results to get a wide and comprehensive view of Ig and associated immune system proteins expressed in response to a specific disease or particular cellular condition.
In summary, all these results indicate that the integration of genomics and proteomics could provide a full characterization of Ig sequences, BCR and the proteins related to the immune system in response to a specific disease or particular cell situation. Nonetheless, further assays will be required to identify specific patterns in BCR sequences which could be of interest in pathological situations such as CLL, autoimmunity or immunodeficiency, among others.
MATERIALS AND METHODS
Figure 5 depicts a general overview of the procedure described below.
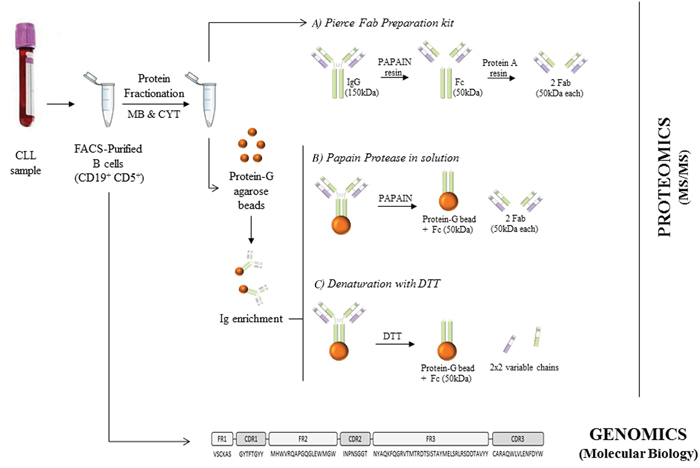
Figure 5: Overview of the procedures performed in this study. B cells were purified from CLL peripheral blood using FACSAria. These purified cells were directly employed for BCR-sequencing by molecular biology and also processed to obtain membrane (MB) and cytoplasmic (CYT) proteins. These isolated proteins were used for Fab purification using Pierce Fab preparation kit (A). Moreover, they were Ig-enriched for applying papain protease in solution (B) and denaturation with DTT (C) for the BCR-sequencing by LC-MS/MS.
Sample collection
Peripheral blood (PB) B-lymphocytes (CD19+ CD5+) from 9 CLL patients (Table 1) were systematically purified by FACSAria II flow cytometer (BD, San Jose/CA, USA) (at a purity >95%) at General Service of Cytometry of University of Salamanca [26]. High-sensitive multi-parameter flow cytometry was performed for the immunophenotyping of clonal B-cell populations, according to previously described procedures [18]. All cases presented a clonal (8 kappa CLL clone, 1 lambda CLL clone) and aberrant CD5+ CLL-like B-cell population.
PB samples were obtained with informed consent and according to the guidelines of the local ethics committees of the University Hospital of Salamanca, in accordance with the Helsinki Declaration of 1975, as revised in 2008.
Molecular studies: sequencing of immunoglobulin human domains
Each clonal B-cell population – purified by FACSAria – was screened for its patterns of rearrangement of the IGHV genes (Table 1) following genomic DNA preparation, PCR amplification, sequencing, and analysis of V, (D), J genes, as described elsewhere [27, 28]. Specifically, the B-cell clonality was determined following the guidelines described by the EuroClonality Consortium, http://www.euroclonality.org/, which combine the usage of IGH and IGK multiplex PCR assays for the assessment of Ig gene rearrangements [29, 30]. The sequences of the V(D)J fragments of these samples have been widely reported in a previous study performed by our group [31]. Resulting sequences were aligned with germline sequences using the IMGT database and tools (http://www.imgt.org). Sequences were considered as somatically mutated if they contained 2% deviation from the germline sequence.
Subcellular protein fractionation
FACS-purified B cells were snap-frozen in liquid nitrogen after adding lysis solution (5 mM HEPES, 10 mM MgCl2, 140 mM NaCl, 0.1% Tween 20, and 1% protease inhibitor mix. All purchased from Sigma, St. Louis/MO, USA). Then, cells were thawed on ice and centrifuged at 15,000 g for 15 min; henceforth, supernatant and pellet were treated separately, although processing stepwise in an identical manner. 10 % (w/v) octyl-β-D-glucopyranoside (Sigma) was added followed by sonication (3 pulses for 3 seconds each). Samples were incubated for 30 min and centrifuged at 15,000 g for 15 min. Cytoplasmic proteins were collected from the supernatant, whereas membrane proteins were collected from the pellet.
Immunoglobulin enrichment from cell extract
Samples were IgG-enriched by using protein-G-agarose beads (GE Healthcare, Buckinghamshire, UK), according to manufacturer’s instructions, and stored at -80°C until used.
B-Cells immunoglobulin processing strategies
Three different approaches were performed to achieve the peptide sequencing of B-cell Igs: a) Pierce™ Fab preparation kit (Thermo Scientific, Waltham/MA, USA), based on the usage of immobilized papain protease for IgG digestion and protein A agarose for purification; b) papain protease in solution (Worthington, Biochemical Corporation, Lakewood/NJ, USA), for the digestion of bead immobilized IgG and the obtaining of Fab fragments [32]; c) denaturation with DTT of bead immobilized IgG for the breakage of their disulfide bonds and the obtaining of heavy and light chains.
Approach a) was performed following the manufacturer’s indications. For approach b), papain was activated with a digestion buffer (5 mM L-Cys-HCl, 1 mM EDTA and PBS; all from Sigma) at pH=7 for 30 min at 37°C. The activated papain was incubated with the sample at 1:50 ratio (papain:sample) for 2 h at 37°C with gently mixing. Afterward, samples were centrifuged at 12,000 g for 3 min and the supernatant was collected. The supernatant contained the Fab released from the IgG, whereas the pellet contained the beads and Fc remaining fractions. Papain digestion was stopped with SDS sample buffer. The second step of papain digestion was performed with the pellet at the same conditions indicated above and joined to the one obtained in the first digestion. Finally, approach c) was performed incubating the samples with 40 mM DTT for 5 min at 99°C.
In all these performed approaches (a, b, and c), human IgG protein (Abcam, Cambridge/UK) was used as a reference.
SDS-PAGE separation
Each processed sample was separated on a 10% SDS-PAGE gel under non-reducing conditions for approaches a) and b), and reducing conditions for approach c). After electrophoresis, gels were stained in a solution of 2.5 g/L silver nitrate and stored at 4 °C in an aqueous solution containing 1% (v/v) acetic acid, until analysis (Supplementary Figure 1).
nUPLC-MS/MS analysis
Each gel lane was cut into fragments and digested with trypsin following the method of Shevchenko et al. [33] with slight modifications. Briefly, gel pieces were destained with 15 mM potassium ferrocyanide and 50 mM sodium thiosulfate. Protein reduction and alkylation were performed with 10 mM DTT at 56 °C for 45 min, and with 55 mM IAA at room temperature for 30 min, respectively. Proteins were digested with trypsin (6.25 ng/mL) at 37 °C for 18 h. The peptide solution was acidified with FA and desalted by using C18-Stage-Tips columns [34]. The samples were partially dried and stored at −20 °C until they were analyzed by LC-MS/MS.
A nUPLC system (nanoAcquity, Waters Corp., Milford/MA, USA) coupled to a LTQ-Velos-Orbitrap mass spectrometer (Thermo Fisher Scientific, San Jose/CA, USA) via a nanoelectrospray ion source (NanoSpray flex, Proxeon, Thermo) was used for reversed-phase LC-MS/MS analysis. Peptides were dissolved in 0.5% FA/3% ACN and loaded onto a trapping column (nanoACQUITY UPLC 2G-V/M Trap Symmetry 5 μm particle size, 180 μm × 20 mm C18 column, Waters Corp., Milford/MA, USA). Peptides were separated on a nanoACQUITY UPLC BEH 1.7 μm, 130 Å, 75 μm × 250 mm C18 column (Waters Corp., Milford/MA, USA) with a linear gradient from 7% to 35% solvent B (ACN/0.1% FA) at a flow rate of 250 nL/min over 120 min.
The nUPLC- LTQ-Orbitrap Velos was operated in the positive ion mode by applying a data-dependent automatic switch between survey MS scan and tandem mass spectra (MS/MS) acquisition. Survey scans were acquired in the mass range of m/z 400 to 1600 with a 60 000 resolution at m/z 400 with lock mass option enabled for the 445.120025 ion [35].
The 20 most intense peaks having ≥2 charge state and above the 500 intensity threshold were selected in the ion trap for fragmentation by collision-induced dissociation with 35% normalized energy, 10 ms activation time, q = 0.25, ± 2 m/z precursor isolation width and wideband activation. Maximum injection time was 1000 and 50 ms for survey and MS/MS scans, respectively. AGC was 1 × 106 for MS and 5 × 103 for MS/MS scans. Dynamic exclusion was enabled for 90 s.
Database search
Raw data were translated to mascot general file (.mgf) format using Thermo Scientific Proteome Discoverer software (v. 1.4.1.14). The searches were conducted using both Mascot and Peptide Shaker search engines. For the first approach, the MASCOT [36] algorithm was used to search for the acquired MS/MS spectra, using Thermo Scientific Proteome Discoverer software (v. 1.4.1.14) against a custom database of all human-reviewed sequences downloaded from the UniProt database (February 2014) and common contaminant sequences (e.g., human keratins, trypsin, BSA). Search parameters were as follows: fully-tryptic digestion with up to two missed cleavages; 10 ppm, and 0.8 Da mass tolerances for precursor and product ions, respectively; carbamidomethylation of cysteines as fixed modification and oxidation of methionine and n-terminus acetylation as variable modifications were considered. Peptides having MASCOT ion scores of <20 were not considered for analysis. A 1% false discovery rate (FDR) using Percolator [37] was employed for peptide validation. For Peptide Shaker approach, the search was conducted using SearchGUI version 1.30.1 [38]. Protein identification was conducted against a concatenated target/decoy version of all human-reviewed sequences downloaded from the neXtProt database (release of September 2015, 20066 protein entries) and common contaminant sequences (e.g., human keratins, trypsin). The decoy sequences were created by reversing the target sequences in SearchGUI. The identification settings were the same fixed for the MASCOT searching. Peptides and proteins were inferred from the spectrum identification results using PeptideShaker version 0.41.1 [39]. Peptide-Spectrum Matches (PSMs), peptides, and proteins were validated at a 1% FDR estimated using the decoy hit distribution.
The MS data along with the identification results have been deposited to the ProteomeXchange Consortium via the PRIDE [40] partner repository with the dataset identifier PXD004466.
Peptide mass spectra and DNA sequences interpretation
An in-house Excel algorithm was designed for the integration of results obtained after DNA sequencing and MS/MS analysis. The basis of this Excel macro was the searching of homologies between the translated DNA sequences and the peptides identified by nUPLC-MS/MS. This tool looks for homology of 2 or more consecutive amino acids corresponding to the CDR or FR regions. For reviewing purposes, the Excel algorithm is provided as Supplementary Information 2.
Abbreviations
BCR, B-cell receptor; CDR, complementary determining region; CLL, chronic lymphocytic leukemia; Fab, fragment antigen binding; FR, framework; HLA, human leukocyte antigen; Ig, immunoglobulin; MS, mass spectrometry; NGS, Next-Generation Sequencing; nUPLC-MS/MS, nano-ultra-performance liquid chromatography tandem MS; PB, peripheral blood; PSM, peptide-spectrum match.
ACKNOWLEDGMENTS AND FUNDING
We gratefully acknowledge financial support from the Spanish Health Institute Carlos III (ISCIII) for the grants: FIS PI11/02114 and FIS PI114/01538. We also acknowledge Fondos FEDER (EU) and Junta Castilla-León (grant BIO/SA07/15). This work has been also sponsored by Fundación Solórzano (FS/23-2015). The Proteomics Unit belongs to ProteoRed, PRB2-ISCIII, supported by grant PT13/0001, of the PE I+D+I 2013-2016, funded by ISCIII and FEDER. The authors would like to thank all the clinicians and technicians in the Cytometry and Cell Purification Services of the University of Salamanca, the Spanish National DNA Bank (Banco Nacional de DNA Carlos III, University of Salamanca) and the Genomic Unit of Cancer Research Centre (IBMCC, USAL-CSIC) for their support in the data collection for the preparation of this manuscript. P.D. is supported by a JCYL-EDU/346/2013 Ph.D. scholarship.
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
REFERENCES
1. Schmidt S, Rainer J, Riml S, Ploner C, Jesacher S, Achmüller C, Presul E, Skvortsov S, Crazzolara R, Fiegl M, Raivio T, Jänne OA, Geley S, et al. Utilization of Ig heavy chain variable, diversity, and joining gene segments in children with B-lineage acute lymphoblastic leukemia: implications for the mechanisms of VDJ recombination and for pathogenesis. Blood. 2006; 91:4602-4609.
2. Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes and Development. 2004. 18:1-11.
3. Tonegawa S. Somatic generation of antibody diversity. Nature. 1983; 302:575-581.
4. Odegard VH, Schatz DG. Targeting of somatic hypermutation. Nat Rev Immunol. 2006; 6:573-583.
5. Jordan M, Baxter AG. Quantitative and qualitative approaches to GOD: the first 10 years of the clonal selection theory. Immunol Cell Biol. 2008; 86:72-79.
6. Schroeder HW Jr, Cavacini L. Structure and function of immunoglobulins. J Allergy Clin Immunol. 2010; 125:S41-52.
7. Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends in Immunology. 2013; 34:592-601.
8. Díez P, Góngora R, Orfao A, Fuentes M. Functional proteomics insights in B-cell chronic lymphocytic leukemia. Expert Review of Proteomics. 2016; 14:137-146.
9. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005; 352:804-815.
10. Lopez C, Delgado J, Costa D, Villamor N, Navarro A, Cazorla M, Gomez C, Arias A, Munoz C, Cabezas S, Baumann T, Rozman M, Aymerich M, et al. Clonal evolution in chronic lymphocytic leukemia: analysis of correlations with IGHV mutational status, NOTCH1 mutations and clinical significance. Genes Chromosomes Cancer. 2013; 52:920-927.
11. Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014; 32:158-168.
12. Brown JR, Fernandes SM, Kasar S, Hoang K, Moorhead M, Carlton V, Faham M, Miklos DB, Logan AC. Next-Generation Sequencing Reveals Clonal Evolution at the Immunoglobulin Loci in Chronic Lymphocytic Leukemia. Blood. 2014; 124:3302.
13. Woo S, Cha SW, Merrihew G, He Y, Castellana N, Guest C, Maccoss M, Bafna V. Proteogenomic database construction driven from large scale RNA-seq data. J Proteome Res. 2014; 13:21-28.
14. Castellana N, Bafna V. Proteogenomics to discover the full coding content of genomes: A computational perspective. Journal of Proteomics. 2010. 73:2124-35.
15. Díez P, Fuentes M. Proteogenomics for the comprehensive analysis of human cellular and serum antibody repertoires. Proteogenomics, Advances in Experimental Medicine and Biology. 2016; 926:153-162.
16. Lavinder JJ, Horton AP, Georgiou G, Ippolito GC. Next-generation sequencing and protein mass spectrometry for the comprehensive analysis of human cellular and serum antibody repertoires. Current Opinion in Chemical Biology. 2015; 24:112-120.
17. Xu JL, Davis MM. Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity. 2000; 13:37-45.
18. Henriques A, Rodriguez-Caballero A, Criado I, Langerak AW, Nieto WG, Lecrevisse Q, Gonzalez M, Cortesao E, Paiva A, Almeida J, Orfao A. Molecular and cytogenetic characterization of expanded B-cell clones from multiclonal versus monoclonal B-cell chronic lymphoproliferative disorders. Haematologica. 2014; 99:897-907.
19. Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014; 32:158-168.
20. Robins H. Immunosequencing: applications of immune repertoire deep sequencing. Curr Opin Immunol. 2013; 25:646-652.
21. Beck A, Sanglier-Cianférani S, Van Dorsselaer A. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 2012; 84:4637-4646.
22. Dorvignit D, Palacios JL, Merino M, Hernández T, Sosa K, Casacó A, López-Requena A, Mateo de Acosta C. Expression and biological characterization of an anti-CD20 biosimilar candidate antibody. MAbs. 2012; 4:488-496.
23. Wooding KM, Peng W, Mechref Y. Characterization of pharmaceutical IgG and biosimilars using miniaturized platforms and LC-MS/MS. Curr Pharm Biotechnol. 2016; 17:788-801.
24. Boutz DR, Horton AP, Wine Y, Lavinder JJ, Georgiou G, Marcotte EM. Proteomic identification of monoclonal antibodies from serum. Anal Chem. 2014; 86:4758-4766.
25. Cheung WC, Beausoleil SA, Zhang X, Sato S, Schieferl SM, Wieler JS, Beaudet JG, Ramenani RK, Popova L, Comb MJ, Rush J, Polakiewicz RD. A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat Biotechnol. 2012; 1080:1-8.
26. Nieto WG, Almeida J, Romero A, Teodosio C, Lopez A, Henriques AF, Sanchez ML, Jara-Acevedo M, Rasillo A, Gonzalez M, Fernandez-Navarro P, Vega T, Orfao A, et al. Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood. 2009; 114:33-37.
27. González D, Balanzategui A, García-Sanz R, Gutiérrez N, Seabra C, van Dongen JJM, González M, San Miguel JF. Incomplete DJH rearrangements of the IgH gene are frequent in multiple myeloma patients: immunobiological characteristics and clinical implications. Leukemia. 2003; 17:1398-1403.
28. van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, van Krieken JH, Droese J, Gonzalez D, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003; 17:2257-2317.
29. Evans PA, Pott Ch, Groenen PJ, Salles G, Davi F, Berger F, Garcia JF, van Krieken JH, Pals S, Kluin P, Schuuring E, Spaargaren M, Boone E, et al. Significantly improved PCR-based clonality testing in B-cell malignancies by use of multiple immunoglobulin gene targets. Report of the BIOMED-2 Concerted Action BHM4-CT98-3936. Leukemia. 2007; 21:207-14.
30. Wren D, Walker BA, Brüggemann M, Catherwood MA, Pott C, Stamatopoulos K, Langerak AW, Gonzalez D. Comprehensive translocation and clonality detection in lymphoproliferative disorders by next-generation sequencing. Haematologica. 2017; 102:e57-e60.
31. Henriques A, Rodríguez-Caballero A, Nieto WG, Langerak AW, Criado I, Lécrevisse Q, González M, Pais ML, Paiva A, Almeida J, Orfao A. Combined Patterns of IGHV Repertoire and Cytogenetic/Molecular Alterations in Monoclonal B Lymphocytosis versus Chronic Lymphocytic Leukemia. PLoS One. 2013; 8:e67751.
32. Andrew SM, Titus JA. Fragmentation of immunoglobulin G. Curr Protoc Immunol. 2003; chapter 16:unit 16.4.
33. Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006; 1:2856-2560.
34. Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007; 2:1896-1906.
35. Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005; 4:2010-2021.
36. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999; 20:3551-3567.
37. Brosch M, Yu L, Hubbard T, Choudhary J. Accurate and sensitive peptide identification with mascot percolator. J Proteome Res. 2009; 8:3176-3181.
38. Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L. SearchGUI: An open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteomics. 2011; 11:996-999.
39. Vaudel M, Burkhart JM, Zahedi RP, Oveland E, Berven FS, Sickmann A, Martens L, Barsnes H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat Biotechnol. 2015; 33:22-24.
40. Vizcaíno JA, Csordas A, Del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, Xu QW, Wang R, Hermjakob H. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016; 44:D447-456.